Complete Chloroplast Genome of Japanese Larch (Larix kaempferi): Insights into Intraspecific Variation with an Isolated Northern Limit Population

Abstract
:1. Introduction
2. Materials and Methods
3. Results
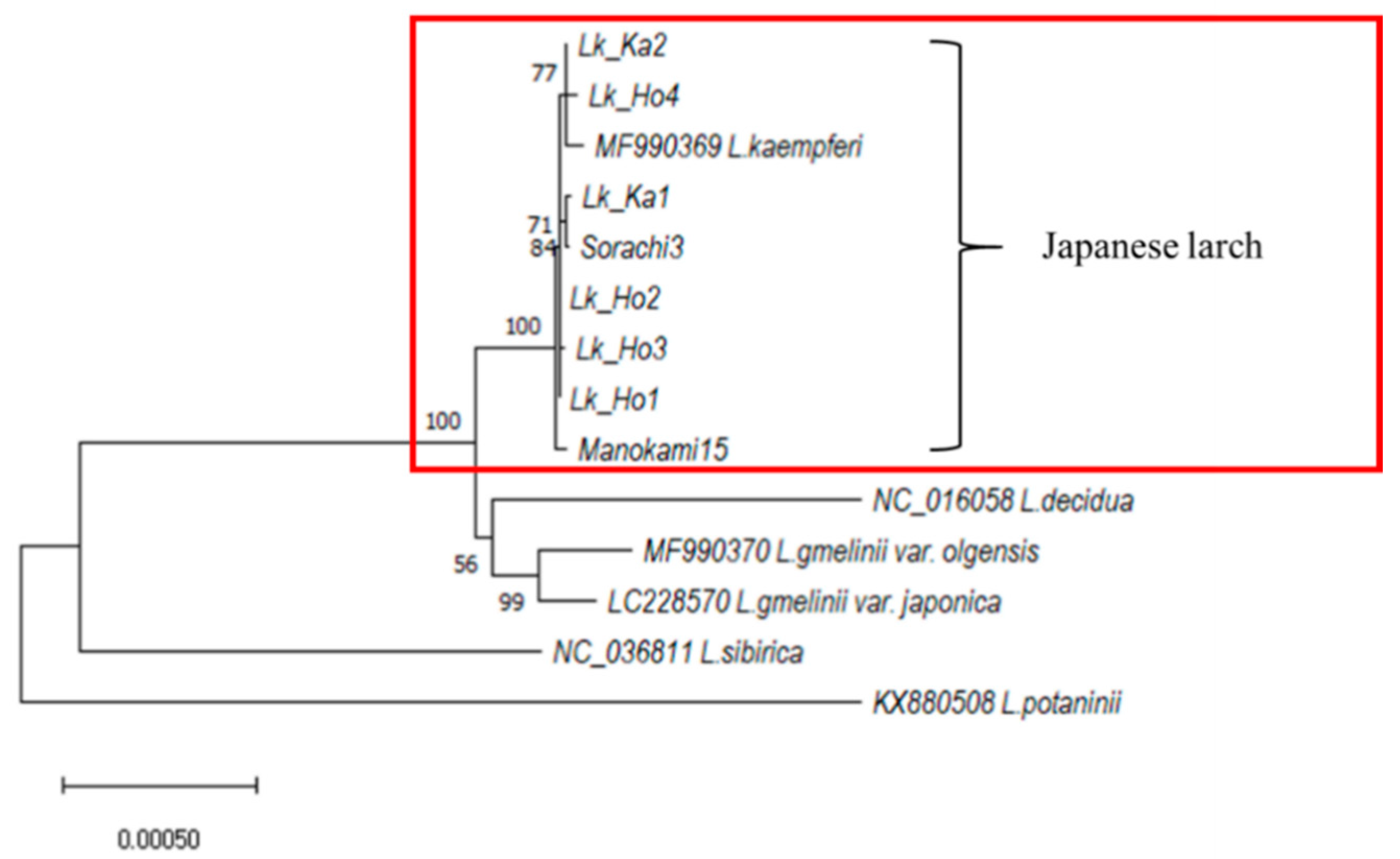
3.1. Phylogenetic Analysis
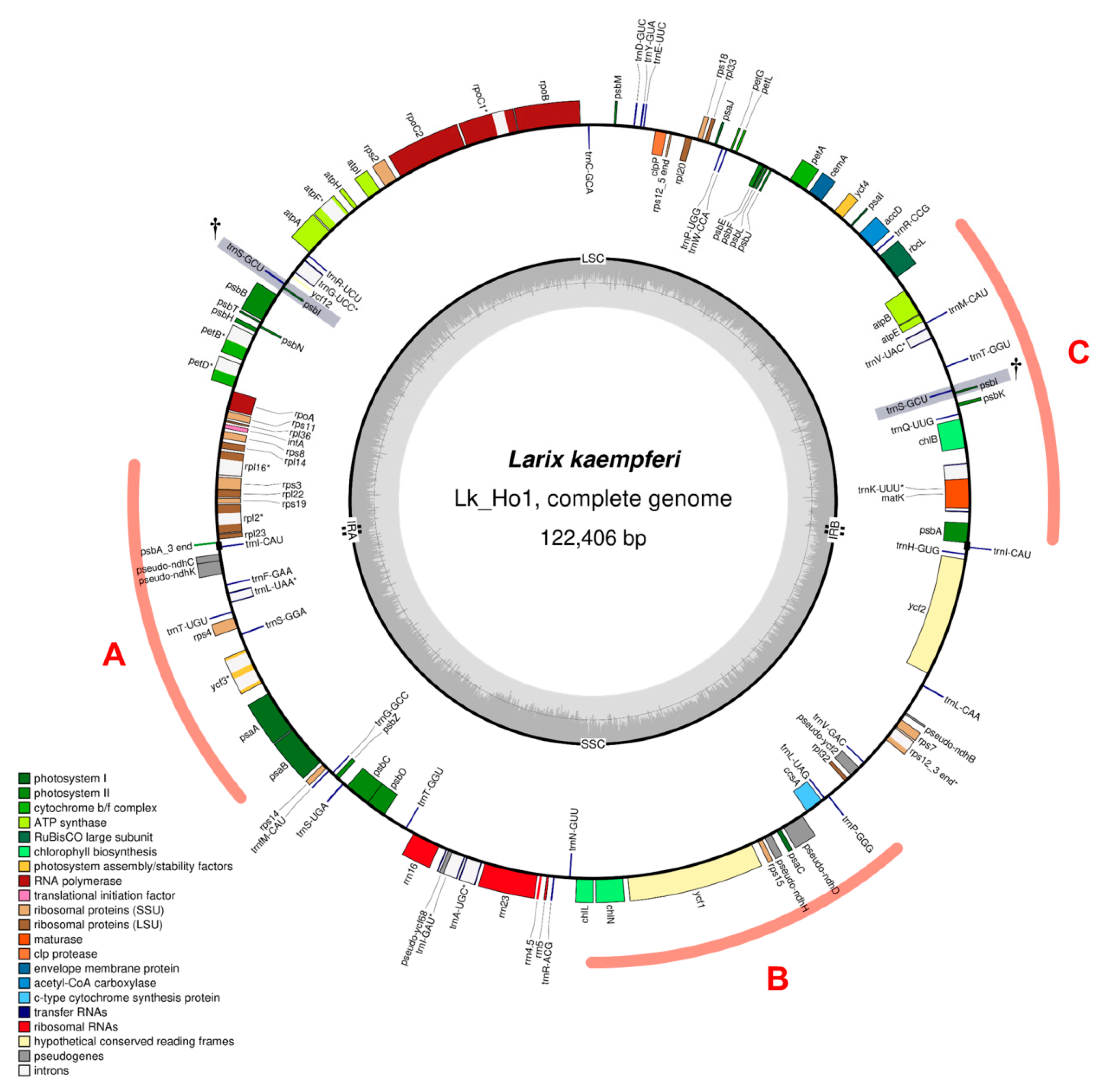
3.2. Characteristics of the Japanese Larch Chloroplast Genome
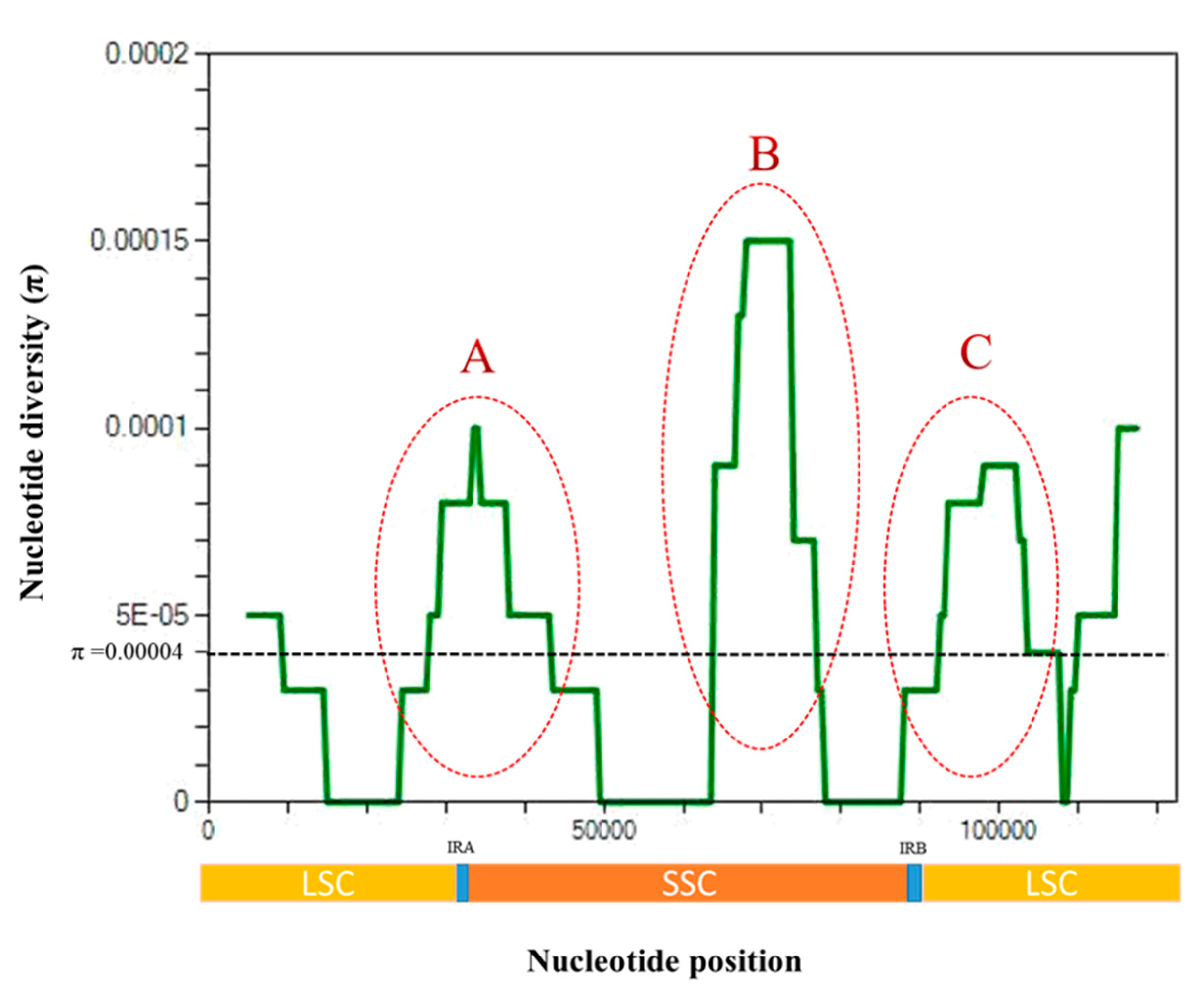
3.3. Nucleotide Diversity Analysis
3.4. Repeat Sequence Analysis of the Japanese Larch Chloroplast Genome
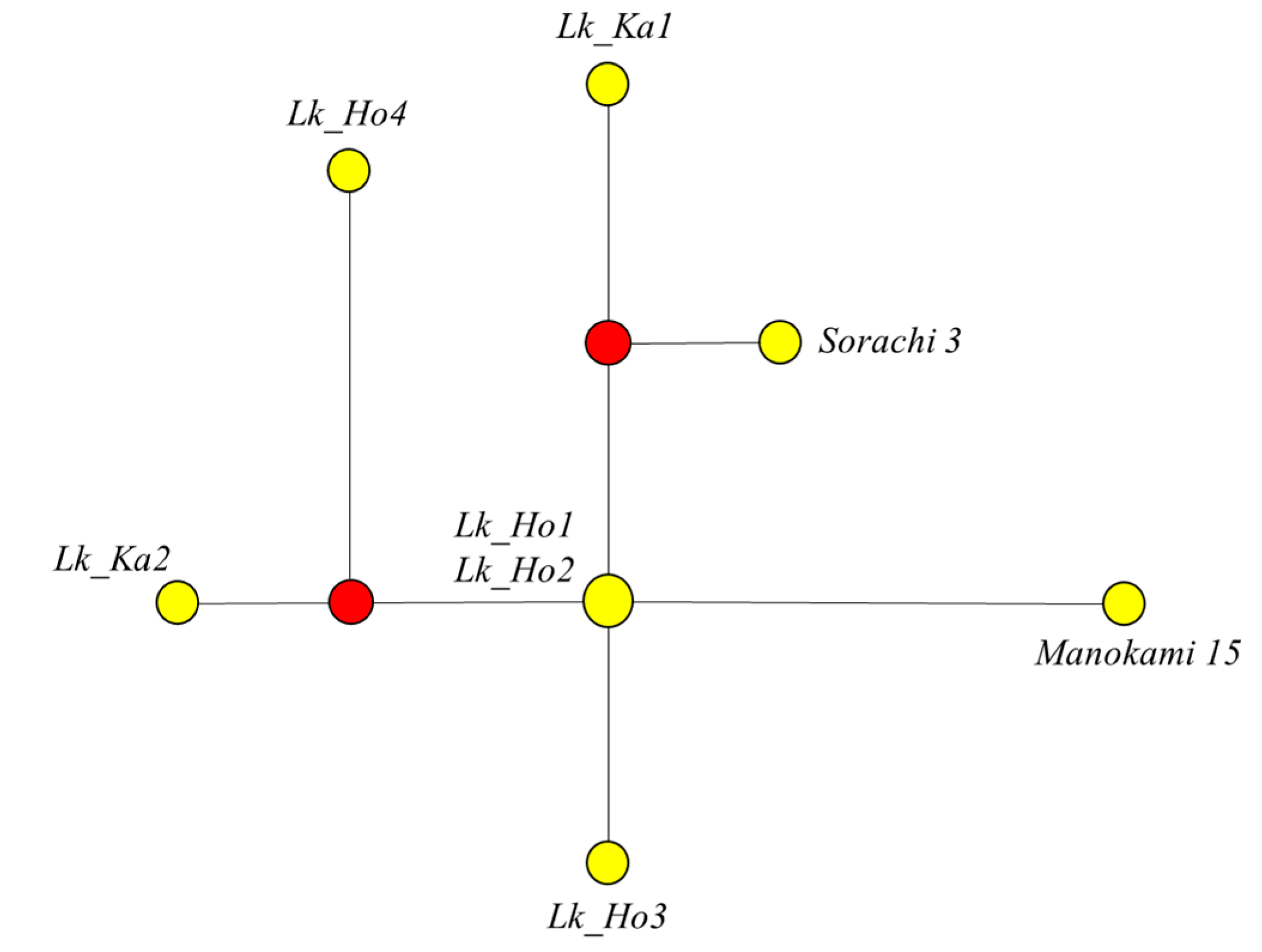
3.5. Genetic Variation among Japanese Larch Chloroplast Genomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wolfe, K.H.; Li, W.-H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.H.; Kan, D.P.; Lee, S.B.; Daniell, H.; Lee, Y.W.; Lin, C.C.; Lin, N.S.; Lin, C.S. Complete nucleotide sequence of Dendrocalamus latiflorus and Bambusa oldhamii chloroplast genomes. Tree Physiol. 2009, 29, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Nock, C.J.; Waters, D.L.E.; Edwards, M.A.; Bowen, S.G.; Rice, N.; Cordeiro, G.M.; Henry, R.J. Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 2011, 9, 328–333. [Google Scholar] [CrossRef]
- Wagner, D.B.; Furnier, G.R.; Saghai-Maroof, M.A.; Williams, S.M.; Dancik, B.P.; Allard, R.W. Chloroplast DNA polymorphisms in lodgepole and jack pines and their hybrids. Proc. Natl. Acad. Sci. USA 1987, 84, 2097. [Google Scholar] [CrossRef] [Green Version]
- Tsumura, Y.; Suyama, Y.; Taguchi, H.; Ohba, K. Geographical cline of chloroplast DNA variation in Abies mariesii. Theor. Appl. Genet. 1994, 89, 922–926. [Google Scholar] [CrossRef]
- Neale, D.B.; Sederoff, R.R. Paternal inheritance of chloroplast DNA and maternal inheritance of mitochondrial DNA in loblolly pine. Theor. Appl. Genet. 1989, 77, 212–216. [Google Scholar] [CrossRef]
- Vendramin, G.G.; Lelli, L.; Rossi, P.; Morgante, M. A set of primers for the amplification of 20 chloroplast microsatellites in Pinaceae. Mol. Ecol. 1996, 5, 595–598. [Google Scholar] [CrossRef]
- Sugiura, M. The chloroplast chromosomes in land plants. Annu. Rev. Cell Biol. 1989, 5, 51–70. [Google Scholar] [CrossRef]
- Wakasugi, T.; Tsudzuki, J.; Ito, S.; Nakashima, K.; Tsudzuki, T.; Sugiura, M. Loss of all ndh genes as determined by sequencing the entire chloroplast genome of the black pine Pinus thunbergii. Proc. Natl. Acad. Sci. USA 1994, 91, 9794–9798. [Google Scholar] [CrossRef] [Green Version]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef] [Green Version]
- Ishizuka, W.; Tabata, A.; Ono, K.; Fukuda, Y.; Hara, T. Draft chloroplast genome of Larix gmelinii var. japonica: Insight into intraspecific divergence. J. For. Res. 2017, 22, 393–398. [Google Scholar] [CrossRef]
- Branca, A.; Paape, T.D.; Zhou, P.; Briskine, R.; Farmer, A.D.; Mudge, J.; Bharti, A.K.; Woodward, J.E.; May, G.D.; Gentzbittel, L.; et al. Whole-genome nucleotide diversity, recombination, and linkage disequilibrium in the model legume. Proc. Natl. Acad. Sci. USA 2011, 108, E864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngamitsu, T.; Nagasaka, K.; Yoshimaru, H.; Tsumura, Y. Provenance tests for survival and growth of 50-year-old Japanese larch (Larix kaempferi) trees related to climatic conditions in central Japan. Tree Genet. Genomes 2014, 10, 87–99. [Google Scholar] [CrossRef]
- Japan Forest Tree Breeding Association. Characteristics and conservation of isolated and northernmost natural population of larch in Japan-observing the population and trying to reveal what it really is like. In Proceedings of the 1st Forest Genetics Seminar, Miyagi, Japan, July 1994; p. 97. NAID: 10015777891. (In Japanese). [Google Scholar]
- Shiraishi, S.; Isoda, K.; Watanabe, A.; Kawasaki, H. DNA systematical study on the Larix relicted at Mt.Manokami, the Zao Mountains. J. Jpn. For. Soc. 1996, 78, 175–182. [Google Scholar]
- Oda, H. The northern natural Japanese larch forest at Manokami, the Zao Mountaions. For. Sci. 2013, 38, 52–58. (In Japanese) [Google Scholar]
- Tozawa, T. Ecological studies on the natural forest of the Japanese larch in Tohoku districts (Northeast of Honshu Island, Japan), 1: Natural forest of the now as its northern limit in Japan. Univ. For. Rep. Iwate Univ. 1975, 6, 1–18. [Google Scholar]
- Han, K.; Li, J.; Zeng, S.; Liu, Z.-L. Complete chloroplast genome sequence of Chinese larch (Larix potaninii var. chinensis), an endangered conifer endemic to China. Conserv. Genet. Resour. 2017, 9, 111–113. [Google Scholar]
- Bondar, E.I.; Putintseva, Y.A.; Oreshkova, N.V.; Krutovsky, K.V. Siberian larch (Larix sibirica Ledeb.) chloroplast genome and development of polymorphic chloroplast markers. BMC Bioinform. 2019, 20, 38. [Google Scholar] [CrossRef]
- Kim, S.-C.; Lee, J.-W.; Lee, M.-W.; Baek, S.-H.; Hong, K.-N. The complete chloroplast genome sequences of Larix kaempferi and Larix olgensis var. koreana (Pinaceae). Mitochondrial DNA Part B 2018, 3, 36–37. [Google Scholar] [CrossRef] [Green Version]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW: A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Kuraku, S.; Zmasek, C.M.; Nishimura, O.; Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013, 41, W22–W28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Templeton, A.R.; Crandall, K.A.; Sing, C.F. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram Estim. Genet. 1992, 132, 619. [Google Scholar]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-S.; Lin, C.-P.; Hsu, C.-Y.; Wang, R.-J.; Chaw, S.-M. Comparative chloroplast genomes of Pinaceae: Insights into the mechanism of diversified genomic organizations. Genome Biol. Evol. 2011, 3, 309–319. [Google Scholar] [CrossRef]
- Kang, H.-I.; Lee, H.O.; Lee, I.H.; Kim, I.S.; Lee, S.-W.; Yang, T.J.; Shim, D. Complete chloroplast genome of Pinus densiflora Siebold & Zucc. and comparative analysis with five pine trees. Forests 2019, 10, 600. [Google Scholar]
- Drescher, A.; Ruf, S.; Calsa, T., Jr.; Carrer, H.; Bock, R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 2000, 22, 97–104. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firetti, F.; Zuntini, A.R.; Gaiarsa, J.W.; Oliveira, R.S.; Lohmann, L.G.; Van Sluys, M.-A. Complete chloroplast genome sequences contribute to plant species delimitation: A case study of the Anemopaegma species complex. Am. J. Bot. 2017, 104, 1493–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohyama, K.; Fukuzawa, H.; Kohchi, T.; Shirai, H.; Sano, T.; Sano, S.; Umesono, K.; Shiki, Y.; Takeuchi, M.; Chang, Z. Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature 1986, 322, 572–574. [Google Scholar] [CrossRef]
- Hiratsuka, J.; Shimada, H.; Whittier, R.; Ishibashi, T.; Sakamoto, M.; Mori, M.; Kondo, C.; Honji, Y.; Sun, C.-R.; Meng, B.-Y.; et al. The complete sequence of the rice (Oryza sativa) chloroplast genome: Intermolecular recombination between distinct tRNA genes accounts for a major plastid DNA inversion during the evolution of the cereals. Mol. Gen. Genet. 1989, 217, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Guisinger, M.M.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Extreme reconfiguration of plastid genomes in the Angiosperm family Geraniaceae: Rearrangements, repeats, and codon usage. Mol. Biol. Evol. 2011, 28, 583–600. [Google Scholar] [CrossRef] [Green Version]
- Weng, M.-L.; Blazier, J.C.; Govindu, M.; Jansen, R.K. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol. Biol. Evol. 1994, 31, 645–659. [Google Scholar] [CrossRef] [Green Version]
- Hirao, T.; Watanabe, A.; Kurita, M.; Kondo, T.; Takata, K. A frameshift mutation of the chloroplast matK coding region is associated with chlorophyll deficiency in the Cryptomeria japonica virescent mutant Wogon-Sugi. Curr. Genet. 2009, 55, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.Y.; Shiraishi, S.; Huang, M.R. Analysis of genetic structure in population of Larix Kaempferi by chloroplast SSR markers. Hereditas 2004, 26, 486–490. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | N | In/Del | SNP | SSR | |||
|---|---|---|---|---|---|---|---|
| CDS | IGS | CDS | IGS | CDS | IGS | ||
| Lk_Ho1(Reference) | — | — | — | — | — | — | — |
| Lk_Ho2 | 2 | 2 | |||||
| Lk_Ho3 | 3 | 1 | 1 | 1 | |||
| Lk_Ho4 | 8 | 4 | 2 | 2 | |||
| Lk_Ka1 | 4 | 3 | 1 | ||||
| Lk_Ka2 | 5 | 1 | 3 | 1 | |||
| Sorachi3 | 3 | 3 | |||||
| Manokami15 | 12 | 2 | 2 | 3 | 1 | 4 | |
| Whole sequences | 31 | 4 | 19 | 8 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Ishizuka, W.; Hara, T.; Goto, S. Complete Chloroplast Genome of Japanese Larch (Larix kaempferi): Insights into Intraspecific Variation with an Isolated Northern Limit Population. Forests 2020, 11, 884. https://doi.org/10.3390/f11080884
Chen S, Ishizuka W, Hara T, Goto S. Complete Chloroplast Genome of Japanese Larch (Larix kaempferi): Insights into Intraspecific Variation with an Isolated Northern Limit Population. Forests. 2020; 11(8):884. https://doi.org/10.3390/f11080884
Chicago/Turabian StyleChen, Shufen, Wataru Ishizuka, Toshihiko Hara, and Susumu Goto. 2020. "Complete Chloroplast Genome of Japanese Larch (Larix kaempferi): Insights into Intraspecific Variation with an Isolated Northern Limit Population" Forests 11, no. 8: 884. https://doi.org/10.3390/f11080884
APA StyleChen, S., Ishizuka, W., Hara, T., & Goto, S. (2020). Complete Chloroplast Genome of Japanese Larch (Larix kaempferi): Insights into Intraspecific Variation with an Isolated Northern Limit Population. Forests, 11(8), 884. https://doi.org/10.3390/f11080884