SNP Genotyping with Target Amplicon Sequencing Using a Multiplexed Primer Panel and Its Application to Genomic Prediction in Japanese Cedar, Cryptomeria japonica (L.f.) D.Don

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Primer Panel Design
2.2. Panel Evaluation Via Actual Genotyping
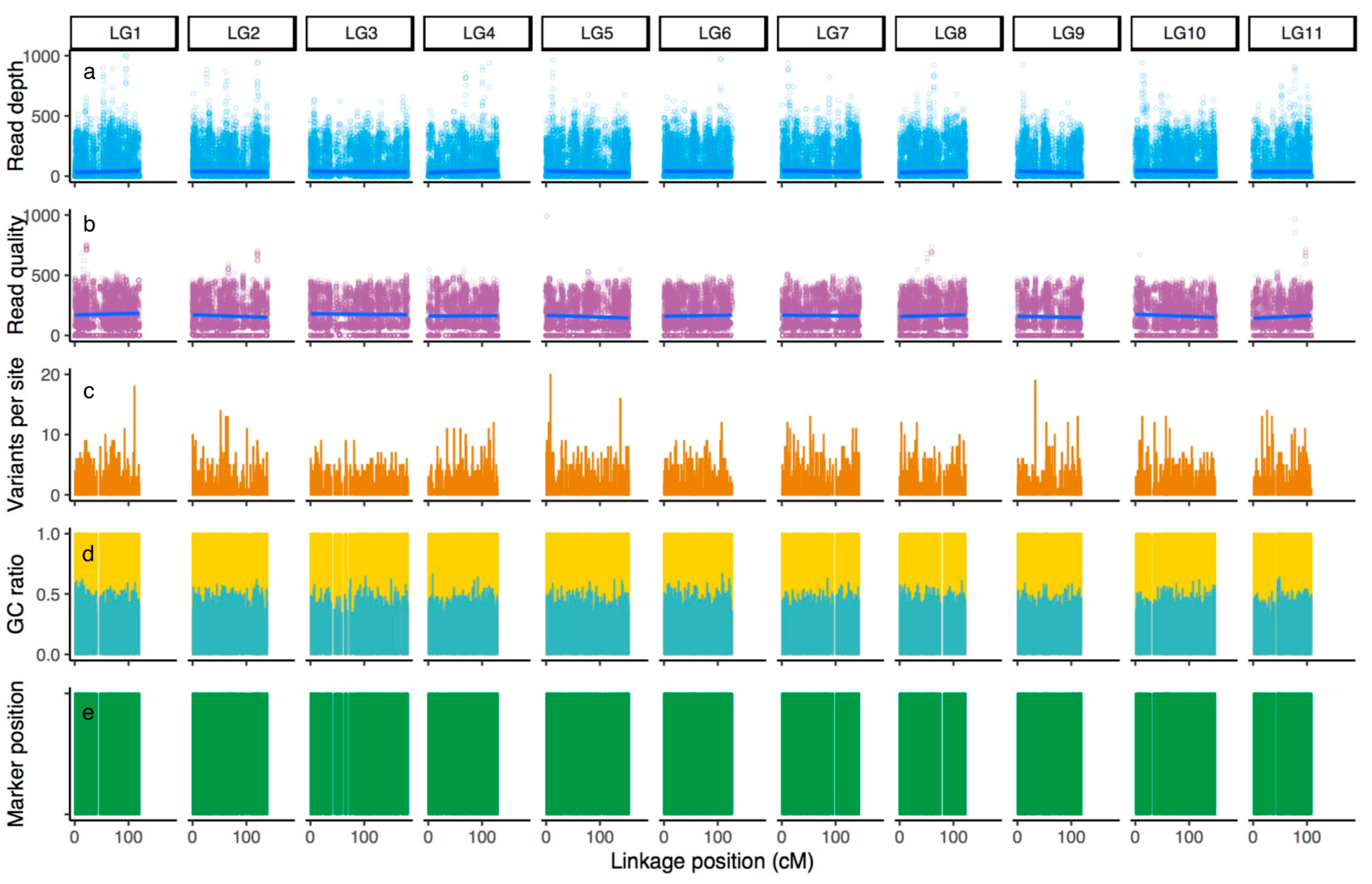
2.3. Data Processing and Visualization
2.4. In Silico Panel Evaluation
2.5. Genotyping a F1 Population
2.6. Phenotypic Data
2.7. Genomic Prediction Within the F1 Population
3. Results
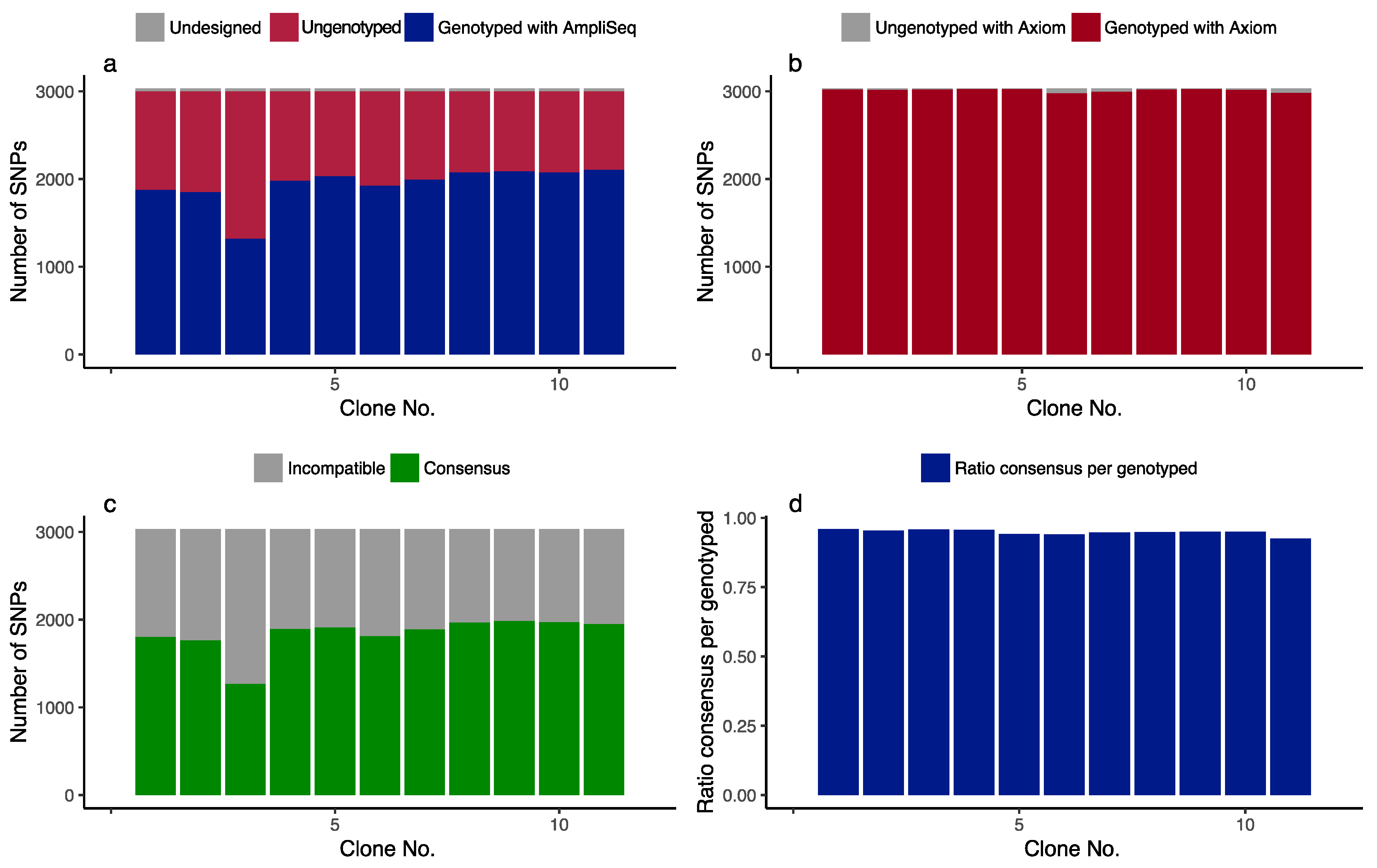
3.1. Primary Evaluation of the Multiplex Primer Panel
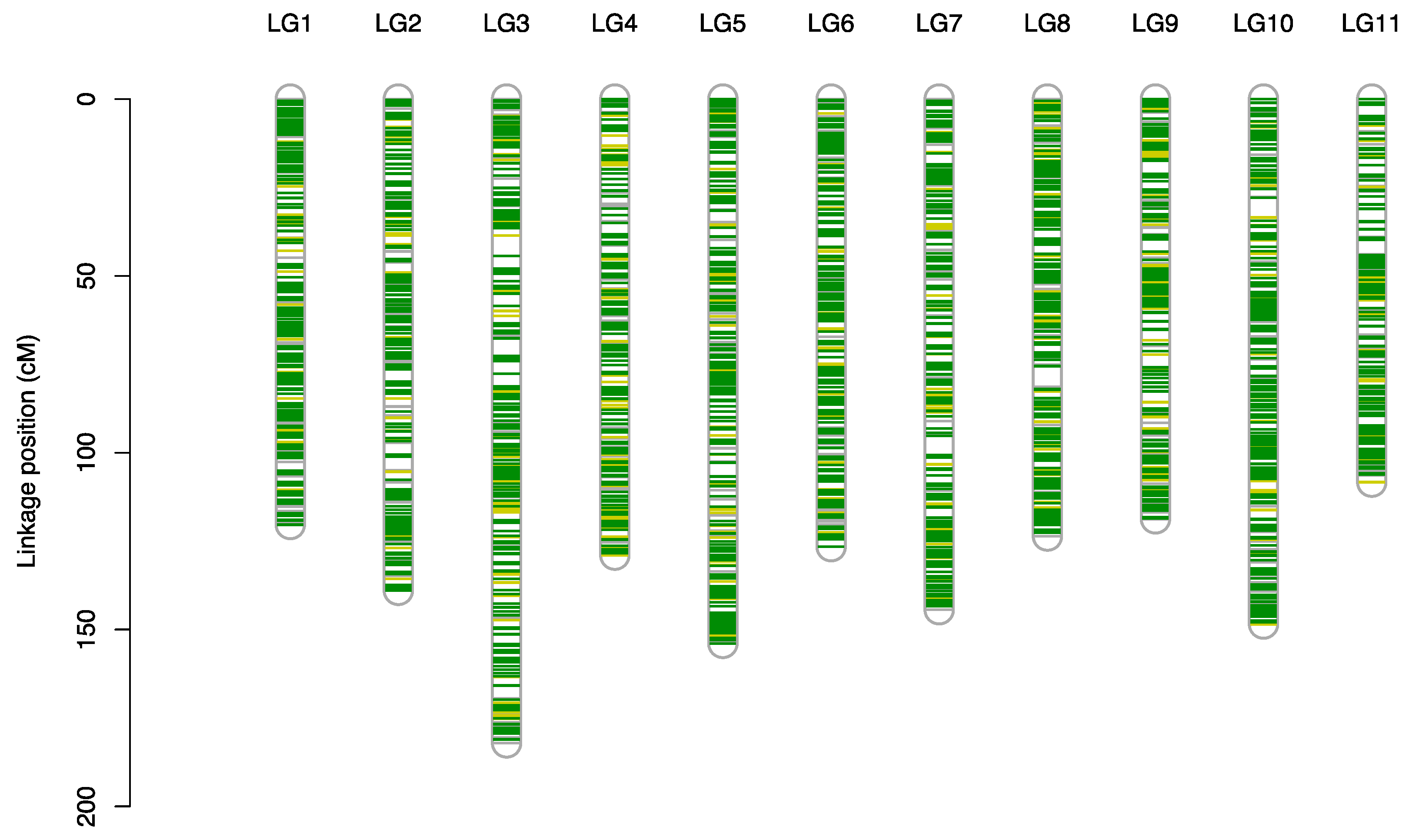
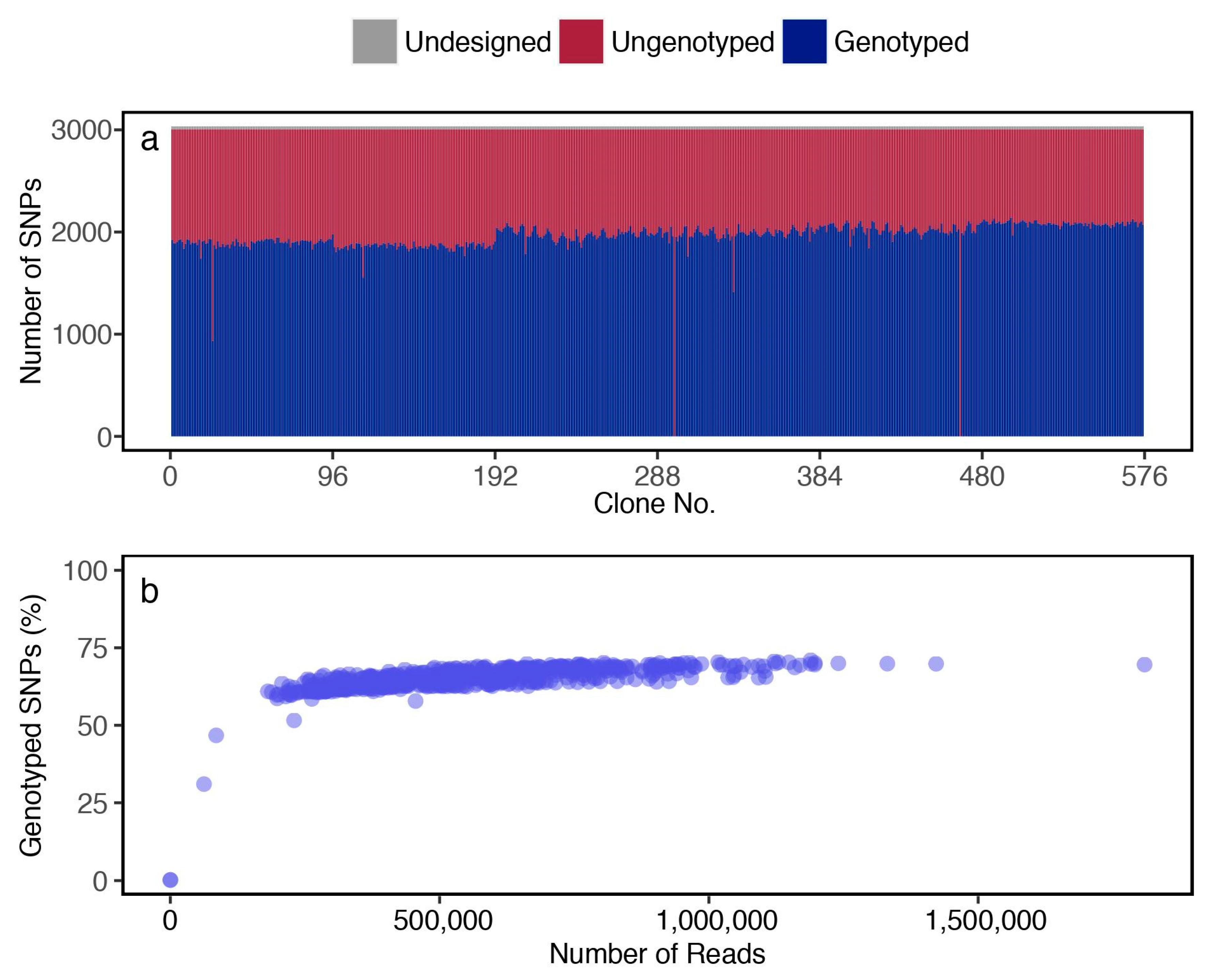
3.2. Genotyping of the F1 Population
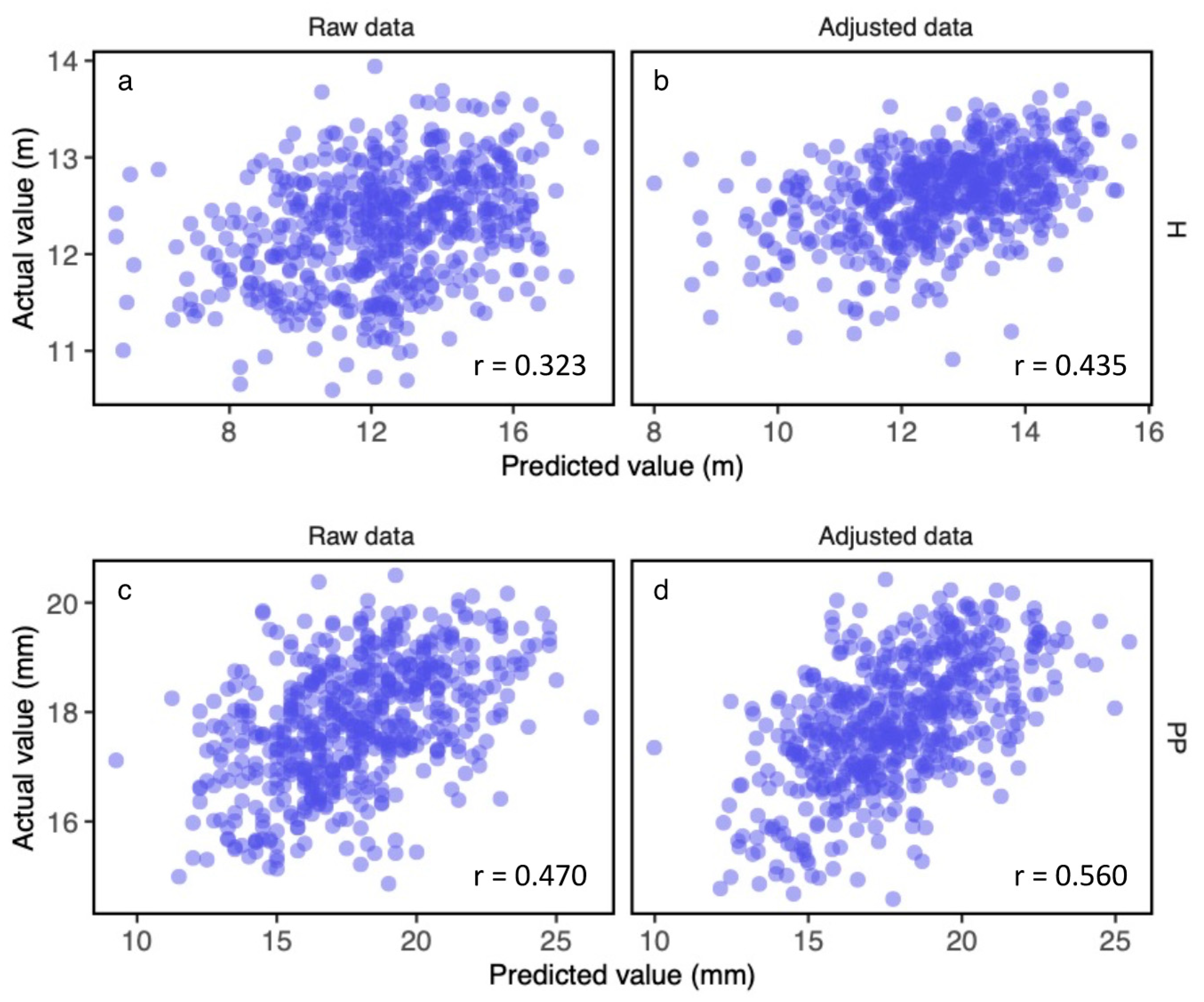
3.3. Construction of the Genomic Prediction Models with the F1 Population
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquin, D.; Campos, G.; Burgueño, J.; Camacho-González, J.; Perez-Elizalde, S.; Beyene, Y.; et al. Genomic selection in plant breeding: Methods, models, and perspectives. Trends Plant Sci. 2017, 22, 961–997. [Google Scholar] [CrossRef]
- Lenaerts, B.; Collard, B.C.Y.; Demont, M. Review: Improving global food security through accelerated plant breeding. Plant Sci. 2019, 287, 110207. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar]
- Lin, Z.; Hayes, B.J.; Daetwyler, H.D. Genomic selection in crops, trees and forages: A review. Crop Pasture Sci. 2014, 65, 1177–1191. [Google Scholar] [CrossRef]
- Desta, Z.A.; Ortiz, R. Genomic Selection: Genome-wide prediction in plant improvement. Trends Plant Sci. 2014, 19, 592–601. [Google Scholar] [CrossRef]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef]
- Grattapaglia, D.; Resende, M.D.V. Genomic selection in forest tree breeding. Tree Genet. Genomes 2011, 7, 241–255. [Google Scholar] [CrossRef]
- Iwata, H.; Hayashi, T.; Tsumura, Y. Prospects for genomic selection in conifer breeding: A simulation study of Cryptomeria japonica. Tree Genet. Genomes 2011, 7, 747–758. [Google Scholar] [CrossRef]
- Uchiyama, K.; Iwata, H.; Moriguchi, Y.; Ujino-Ihara, T.; Ueno, S.; Taguchi, Y.; Tsubomura, M.; Mishima, K.; Iki, T.; Watanabe, A.; et al. Demonstration of genome-wide association studies for identifying markers for wood property and male strobili traits in Cryptomeria japonica. PLoS ONE 2013, 8, e79866. [Google Scholar] [CrossRef] [Green Version]
- Chagné, D.; Brown, G.; Lalanne, C.; Madur, D.; Pot, D.; Neale, D.; Plomion, C. Comparative genome and qtl mapping between maritime and loblolly pines. Mol. Breed. 2003, 12, 185–195. [Google Scholar] [CrossRef]
- Neale, D.B.; Wegrzyn, J.L.; Stevens, K.A.; Zimin, A.V.; Puiu, D.; Crepeau, M.W.; Cardeno, C.; Koriabine, M.; Holtz-Morris, A.E.; Liechty, J.D.; et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 2014, 15, R59. [Google Scholar] [CrossRef] [Green Version]
- Neale, D.B.; Savolainen, O. Association genetics of complex traits in conifers. Trends Plant Sci. 2004, 9, 325–330. [Google Scholar] [CrossRef]
- Plomion, C.; Chancerel, E.; Endelman, J.; Lamy, J.-B.; Mandrou, E.; Lesur, I.; Ehrenmann, F.; Isik, F.; Bink, M.C.A.M.; van Heerwaarden, J.; et al. Genome-wide distribution of genetic diversity and linkage disequilibrium in a mass-selected population of maritime pine. BMC Genomics 2014, 15, 171. [Google Scholar] [CrossRef]
- Isik, F.; Bartholomé, J.; Farjat, A.; Chancerel, E.; Raffin, A.; Sanchez, L.; Plomion, C.; Bouffier, L. Genomic selection in maritime pine. Plant Sci. 2016, 242, 108–119. [Google Scholar] [CrossRef]
- Ritland, K.; Krutovsky, K.V.; Tsumura, Y.; Pelgas, B.; Isabel, N.; Bousquet, J. Genetic Mapping in Conifers. In Genetics, Genomics and Breeding of Conifers; Plomion, C., Bousquet, J., Eds.; CRC Press: Boca Raton, FL, USA, 2011; pp. 196–238. [Google Scholar]
- Echt, C.S.; May-Marquardt, P.; Hseih, M.; Zahorchak, R. Characterization of microsatellite markers in eastern white pine. Genome 1996, 39, 1102–1108. [Google Scholar] [CrossRef] [Green Version]
- Soranzo, N.; Provan, J.; Powell, W. Characterization of microsatellite loci in Pinus sylvestris L. Mol. Ecol. 1998, 7, 1260–1261. [Google Scholar]
- Elsik, C.G.; Minihan, V.T.; Hall, S.E.; Scarpa, A.M.; Williams, C.G. Low-copy microsatellite markers for Pinus Taeda L. Genome 2000, 43, 550–555. [Google Scholar] [CrossRef]
- Mariette, S.; Chagne, D.; Decroocq, S.; Giovanni Giuseppe, V.; Lalanne, C.; Madur, D.; Plomion, C. Microsatellite markers for Pinus Pinaster Ait. Ann. For. Sci. 2001, 58. [Google Scholar] [CrossRef] [Green Version]
- Paglia, G.P.; Olivieri, A.M.; Morgante, M. Towards second-generation STS (sequence-tagged sites) linkage maps in conifers: A genetic map of Norway spruce (Picea Abies K.). Mol. Gen. Genet. 1998, 258, 466–478. [Google Scholar] [CrossRef]
- Scotti, I.; Magni, F.; Paglia, G.; Morgante, M. Trinucleotide microsatellites in Norway spruce (Picea abies): Their features and the development of molecular markers. Theor. Appl. Genet. 2002, 106, 40–50. [Google Scholar] [CrossRef]
- Roschanski, A.M.; Csilléry, K.; Liepelt, S.; Oddou-Muratorio, S.; Ziegenhagen, B.; Huard, F.; Ullrich, K.; Postolache, D.; Giovanni Giuseppe, V.; Fady, B. Evidence of divergent selection for drought and cold tolerance at landscape and local scales in Abies alba Mill. in the French Mediterranean Alps. Mol. Ecol. 2016, 25, 776–794. [Google Scholar] [CrossRef]
- Hirao, T.; Matsunaga, K.; Hirakawa, H.; Shirasawa, K.; Isoda, K.; Mishima, K.; Tamura, M.; Watanabe, A. construction of genetic linkage map and identification of a novel major locus for resistance to pine wood nematode in Japanese black pine (Pinus thunbergii). BMC Plant Biol. 2019, 19. [Google Scholar] [CrossRef] [Green Version]
- Pavy, N.; Gagnon, F.; Rigault, P.; Blais, S.; Deschênes, A.; Boyle, B.; Pelgas, B.; Deslauriers, M.; Clément, S.; Lavigne, P.; et al. Development of high-density SNP genotyping arrays for white spruce (Picea glauca) and transferability to subtropical and Nordic congeners. Mol. Ecol. Res. 2013, 13, 324–336. [Google Scholar] [CrossRef]
- Howe, G.T.; Jayawickrama, K.; Kolpak, S.E.; Kling, J.; Trappe, M.; Hipkins, V.; Ye, T.; Guida, S.; Cronn, R.; Cushman, S.A.; et al. An Axiom SNP genotyping array for douglas-fir. BMC Genom. 2020, 21, 9. [Google Scholar] [CrossRef] [Green Version]
- Parchman, T.L.; Jahner, J.P.; Uckele, K.A.; Galland, L.M.; Eckert, A.J. RADseq Approaches and applications for forest tree genetics. Tree Genet. Genomes 2018, 14, 39. [Google Scholar] [CrossRef]
- Suyama, Y.; Matsuki, Y. MIG-seq: An effective PCR-Based method for genome-wide single-nucleotide polymorphism genotyping using the next-generation sequencing platform. Sci. Rep. 2015, 5, 16963. [Google Scholar] [CrossRef] [Green Version]
- Tsumura, Y.; Ohba, K. Allozyme variation of five natural populations of Cryptomeria japonica in western Japan. Jpn. J. Genet. 1992, 67, 299–308. [Google Scholar] [CrossRef]
- Tsumura, Y.; Kado, T.; Takahashi, T.; Tani, N.; Ujino-Ihara, T.; Iwata, H. Genome scan to detect genetic structure and adaptive genes of natural populations of Cryptomeria japonica. Genetics 2007, 176, 2393. [Google Scholar] [CrossRef] [Green Version]
- Moriguchi, Y.; Iwata, H.; Ujino-Ihara, T.; Yoshimura, K.; Taira, H.; Tsumura, Y. Development and characterization of microsatellite markers for Cryptomeria japonica D. Don. Theor. Appl. Genet. 2003, 106, 751–758. [Google Scholar] [CrossRef]
- Tani, N.; Takahashi, T.; Ujino-Ihara, T.; Iwata, H.; Yoshimura, K.; Tsumura, Y. Development and characteristics of microsatellite markers for sugi (Cryptomeria japonica D. Don) derived from microsatellite-enriched libraries. Ann. For. Sci. 2004, 61, 569–575. [Google Scholar] [CrossRef]
- Takahashi, T.; Tani, N.; Taira, H.; Tsumura, Y. Microsatellite markers reveal high variation in natural populations of Cryptomeria japonica near refugial areas of the last glacial period. J. Plant Res. 2005, 118, 83–90. [Google Scholar] [CrossRef]
- Moriguchi, Y.; Ueno, S.; Higuchi, Y.; Miyajima, D.; Itoo, S.; Futamura, N.; Shinohara, K.; Tsumura, Y. Establishment of a microsatellite panel covering the sugi (Cryptomeria japonica) genome, and its application for localization of a male-sterile gene (ms-2). Mol. Breed. 2014, 33, 315–325. [Google Scholar] [CrossRef]
- Moriguchi, Y.; Tani, N.; Itoo, S.; Kanehira, F.; Tanaka, K.; Yomogida, H.; Taira, H.; Tsumura, Y. Gene flow and mating system in five Cryptomeria japonica D. Don seed orchards as revealed by analysis of microsatellite markers. Tree Genet. Genomes 2005, 1, 174–183. [Google Scholar] [CrossRef]
- Miyamoto, N.; Ono, M.; Watanabe, A. Construction of a core collection and evaluation of genetic resources for Cryptomeria japonica (Japanese cedar). J. For. Res. 2015, 20, 186–196. [Google Scholar] [CrossRef]
- Mishima, K.; Hirao, T.; Tsubomura, M.; Tamura, M.; Kurita, M.; Nose, M.; Hanaoka, S.; Takahashi, M.; Watanabe, A. Identification of novel putative causative genes and genetic marker for male sterility in Japanese cedar (Cryptomeria japonica D. Don). BMC Genom. 2018, 19, 277. [Google Scholar] [CrossRef]
- Hiraoka, Y.; Fukatsu, E.; Mishima, K.; Hirao, T.; Teshima, K.M.; Tamura, M.; Tsubomura, M.; Iki, T.; Kurita, M.; Takahashi, M.; et al. Potential of genome-wide studies in unrelated plus trees of a coniferous species, Cryptomeria japonica (Japanese cedar). Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef]
- Croiseau, P.; Legarra, A.; Guillaume, F.; Fritz, S.; Baur, A.; Colombani, C.; Robert-Granié, C.; Boichard, D.; Ducrocq, V. Fine tuning genomic evaluations in dairy cattle through SNP pre-selection with the elastic-net algorithm. Genet. Res. 2011, 93, 409–417. [Google Scholar] [CrossRef]
- Liu, S.; Wang, H.; Zhang, L.; Tang, C.; Jones, L.; Ye, H.; Ban, L.; Wang, A.; Liu, Z.; Lou, F.; et al. Rapid detection of genetic mutations in individual breast cancer patients by next-generation DNA sequencing. Hum. Genom. 2015, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Ogiso-Tanaka, E.; Shimizu, T.; Hajika, M.; Kaga, A.; Ishimoto, M. Highly multiplexed AmpliSeq technology identifies novel variation of flowering time-related genes in soybean (Glycine Max). DNA Res. 2019, 26, 243–260. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria; Available online: https://www.R-project.org/2019 (accessed on 24 May 2019).
- Gardner, S.N.; Slezak, T. Simulate_PCR for Amplicon prediction and annotation from multiplex, degenerate primers and probes. BMC Bioinform. 2014, 15, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Sobue, N. Measurement of Young’s modulus by transient longitudinal vibration of wooden beams using a fast fourier transformation spectrum analyzer. Mokuzai Gakkaishi 1986, 32, 744–747, (In Japanese with English Summary). [Google Scholar]
- Panshin, A.J.; de Zeeuw, C. Text Book of Wood Technology; McGraw-Hill Book, Co.: New York, NY, USA, 1980. [Google Scholar]
- Japanese Industrial Standards. Methods of Testing for Woods JIS Z 2101-2009; Japanese Standards Association: Tokyo, Japan, 2009. [Google Scholar]
- Cookrell, R.A. A Comparison of latewood pits, fibril orientation, and shrinkage of normal and compression wood of giant sequoia. Wood Sci. Technol. 1974, 8, 197–206. [Google Scholar] [CrossRef]
- Donaldson, L.A. The Use of pit apertures as windows to measure microfibril angle in chemical pulp fibers. Wood Fiber Sci. 1991, 23, 290–295. [Google Scholar]
- Hirakawa, Y.; Fujisawa, Y. The Relationships between microfibril angles of the S2 layer and latewood tracheid lengths in elite sugi tree (Cryptomeria japonica) Clones. Mokuzai Gakkaishi 1995, 41, 123–131, (In Japanese with English Summary). [Google Scholar]
- Muñoz, F.; Sanchez, L. breedR: Statistical Methods for Forest Genetic Resources Analysis. Available online: https://github.com/famuvie/breedR/2018 (accessed on 1 November 2018).
- Endelman, J. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef] [Green Version]
- Liaw, A.; Wiener, M. Classification and regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Chen, C.; Mitchell, S.E.; Elshire, R.J.; Buckler, E.S.; El-Kassaby, Y.A. Mining conifer’s mega-genome using rapid and efficient multiplexed high-throughput genotyping-by-sequencing (GBS) SNP discovery platform. Tree Genet. Genomes 2013, 9, 1537–1544. [Google Scholar] [CrossRef]
- Huang, Y.; Poland, J.; Wight, C.; Jackson, E.; Tinker, N. Using genotyping-by-sequencing (GBS) for genomic discovery in cultivated oat. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Ueno, S.; Uchiyama, K.; Moriguchi, Y.; Ujino-Ihare, T.; Matsumoto, A.; Wei, F.; Saito, M.; Higuchi, Y.; Futamura, N.; Kanamori, H.; et al. Scanning RNA-Seq and RAD-Seq approach to develop SNP markers closely linked to MALE STERILITY 1 (MS1) in Cryptomeria japonica D. Don. Breed. Sci. 2019, 69, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Tamura, A.; Kurinobu, S.; Fukatsu, E.; Iizuka, K. An investigation on the allocation of selection weight on growth and wood basic density to maximize carbon storage in the stem of sugi (Cryptomeria japonica D. Don) plus-tree clones. J. Jpn. For. Soc. 2006, 88, 15–20. [Google Scholar] [CrossRef]
- Fukatsu, E.; Tamura, A.; Takahashi, M.; Fukuda, Y.; Nakada, R.; Kubota, M.; Kurinobu, S. Efficiency of the indirect selection and the evaluation of the genotype by environment interaction using pilodyn for the genetic improvement of wood density in Cryptomeria japonica. J. For. Res. 2011, 16, 128–135. [Google Scholar] [CrossRef]
- Fujisawa, Y.; Ohta, S.; Nishimura, K.; Toda, T.; Tajima, M. Wood characteristics and genetic variations in sugi (Cryptomeria japonica). 3. Estimation of variance-components of the variation in dynamic modulus of elasticity with plus-tree clones. Mokuzai Gakkaishi 1994, 40, 457–464. [Google Scholar]
- Daetwyler, H.; Kemper, K.; van der Werf, J.; Hayes, B. Components of the accuracy of genomic prediction in a multi-breed sheep population. J. Anim. Sci. 2012, 90, 3375–3384. [Google Scholar] [CrossRef] [Green Version]
- Moritsuka, E.; Hisataka, Y.; Tamura, M.; Uchiyama, K.; Watanabe, A.; Tsumura, Y.; Tachida, H. Extended linkage disequilibrium in noncoding regions in a conifer, Cryptomeria japonica. Genetics 2012, 190, 1145–1148. [Google Scholar] [CrossRef] [Green Version]
- Resende, R.; Resende, M.; Silva, F.; Azevedo, C.; Takahashi, E.; Silva, O.; Grattapaglia, D. Assessing the expected response to genomic selection of individuals and families in Eucalyptus breeding with an additive-dominant model. Heredity 2017, 119, 245–255. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference Templates Used for In Silico PCR Amplification | 3031 Contigs 1 | 34,731 Contigs 2 | ||
|---|---|---|---|---|
| Number of designed primer pairs | 3004 | |||
| Amplified products in in silico PCR 3 (a; a = d + e + f + g) | 3157 | 3395 | ||
| Amplicon size ≤ 300 bp in in silico PCR (b; b = d + h) | 3067 | (97.1) | 3251 | (95.8) |
| Correct pairing (intended pair of forward and reverse primers) (c; c = d + e + f) | 3052 | (96.7) | 3265 | (96.2) |
| Correct amplicon (annealing to correct contig with expected amplicon size) (d) | 3004 | (95.2) | 2747 | (80.9) |
| Off target amplification (unintended primers annealing to the wrong contig) (e) | 6 | (0.2) | 340 | (10.0) |
| Unintended amplicon size (shorter or longer than expected amplicon) (f) | 42 | (1.3) | 178 | (5.2) |
| Mismatched paring (unintended pair of forward and reverse primers) (g) | 105 | (3.3) | 130 | (3.8) |
| Others (possible missing detection)4 (h) | 63 | (2.0) | 504 | (14.8) |
| Model | GBLUP | Random Forest | ||
|---|---|---|---|---|
| Trait | Raw Data | Adjusted Data | Raw Data | Adjusted Data |
| Height | 0.197 ± 0.006 | 0.392 ± 0.003 | 0.302 ± 0.005 | 0.418 ± 0.004 |
| DBH | 0.236 ± 0.005 | 0.408 ± 0.004 | 0.255 ± 0.005 | 0.383 ± 0.004 |
| L* | 0.225 ± 0.004 | 0.241 ± 0.006 | 0.251 ± 0.006 | 0.251 ± 0.007 |
| a* | 0.212 ± 0.005 | 0.236 ± 0.003 | 0.232 ± 0.007 | 0.252 ± 0.006 |
| b* | 0.166 ± 0.005 | 0.185 ± 0.005 | 0.189 ± 0.005 | 0.195 ± 0.004 |
| SWV | 0.445 ± 0.004 | 0.481 ± 0.002 | 0.449 ± 0.003 | 0.487 ± 0.004 |
| PP | 0.436 ± 0.004 | 0.542 ± 0.002 | 0.450 ± 0.003 | 0.555 ± 0.001 |
| DMOE | 0.410 ± 0.004 | 0.445 ± 0.004 | 0.408 ± 0.003 | 0.436 ± 0.003 |
| MOE | 0.283 ± 0.007 | 0.372 ± 0.006 | 0.252 ± 0.010 | 0.324 ± 0.008 |
| MOR | 0.248 ± 0.008 | 0.358 ± 0.006 | 0.228 ± 0.007 | 0.299 ± 0.007 |
| MFA | 0.192 ± 0.015 | 0.246 ± 0.007 | 0.176 ± 0.008 | 0.226 ± 0.008 |
| BD_1 | 0.395 ± 0.004 | 0.521 ± 0.003 | 0.388 ± 0.004 | 0.515 ± 0.002 |
| BD_2 | 0.437 ± 0.003 | 0.516 ± 0.003 | 0.437 ± 0.004 | 0.507 ± 0.004 |
| BD_3 | 0.423 ± 0.003 | 0.505 ± 0.004 | 0.407 ± 0.002 | 0.487 ± 0.002 |
| BD_means | 0.454 ± 0.009 | 0.544 ± 0.004 | 0.409 ± 0.004 | 0.506 ± 0.004 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagano, S.; Hirao, T.; Takashima, Y.; Matsushita, M.; Mishima, K.; Takahashi, M.; Iki, T.; Ishiguri, F.; Hiraoka, Y. SNP Genotyping with Target Amplicon Sequencing Using a Multiplexed Primer Panel and Its Application to Genomic Prediction in Japanese Cedar, Cryptomeria japonica (L.f.) D.Don. Forests 2020, 11, 898. https://doi.org/10.3390/f11090898
Nagano S, Hirao T, Takashima Y, Matsushita M, Mishima K, Takahashi M, Iki T, Ishiguri F, Hiraoka Y. SNP Genotyping with Target Amplicon Sequencing Using a Multiplexed Primer Panel and Its Application to Genomic Prediction in Japanese Cedar, Cryptomeria japonica (L.f.) D.Don. Forests. 2020; 11(9):898. https://doi.org/10.3390/f11090898
Chicago/Turabian StyleNagano, Soichiro, Tomonori Hirao, Yuya Takashima, Michinari Matsushita, Kentaro Mishima, Makoto Takahashi, Taiichi Iki, Futoshi Ishiguri, and Yuichiro Hiraoka. 2020. "SNP Genotyping with Target Amplicon Sequencing Using a Multiplexed Primer Panel and Its Application to Genomic Prediction in Japanese Cedar, Cryptomeria japonica (L.f.) D.Don" Forests 11, no. 9: 898. https://doi.org/10.3390/f11090898
APA StyleNagano, S., Hirao, T., Takashima, Y., Matsushita, M., Mishima, K., Takahashi, M., Iki, T., Ishiguri, F., & Hiraoka, Y. (2020). SNP Genotyping with Target Amplicon Sequencing Using a Multiplexed Primer Panel and Its Application to Genomic Prediction in Japanese Cedar, Cryptomeria japonica (L.f.) D.Don. Forests, 11(9), 898. https://doi.org/10.3390/f11090898