
Characterization of Water in Wood by Time-Domain Nuclear Magnetic Resonance Spectroscopy (TD-NMR): A Review


Abstract
:1. Introduction
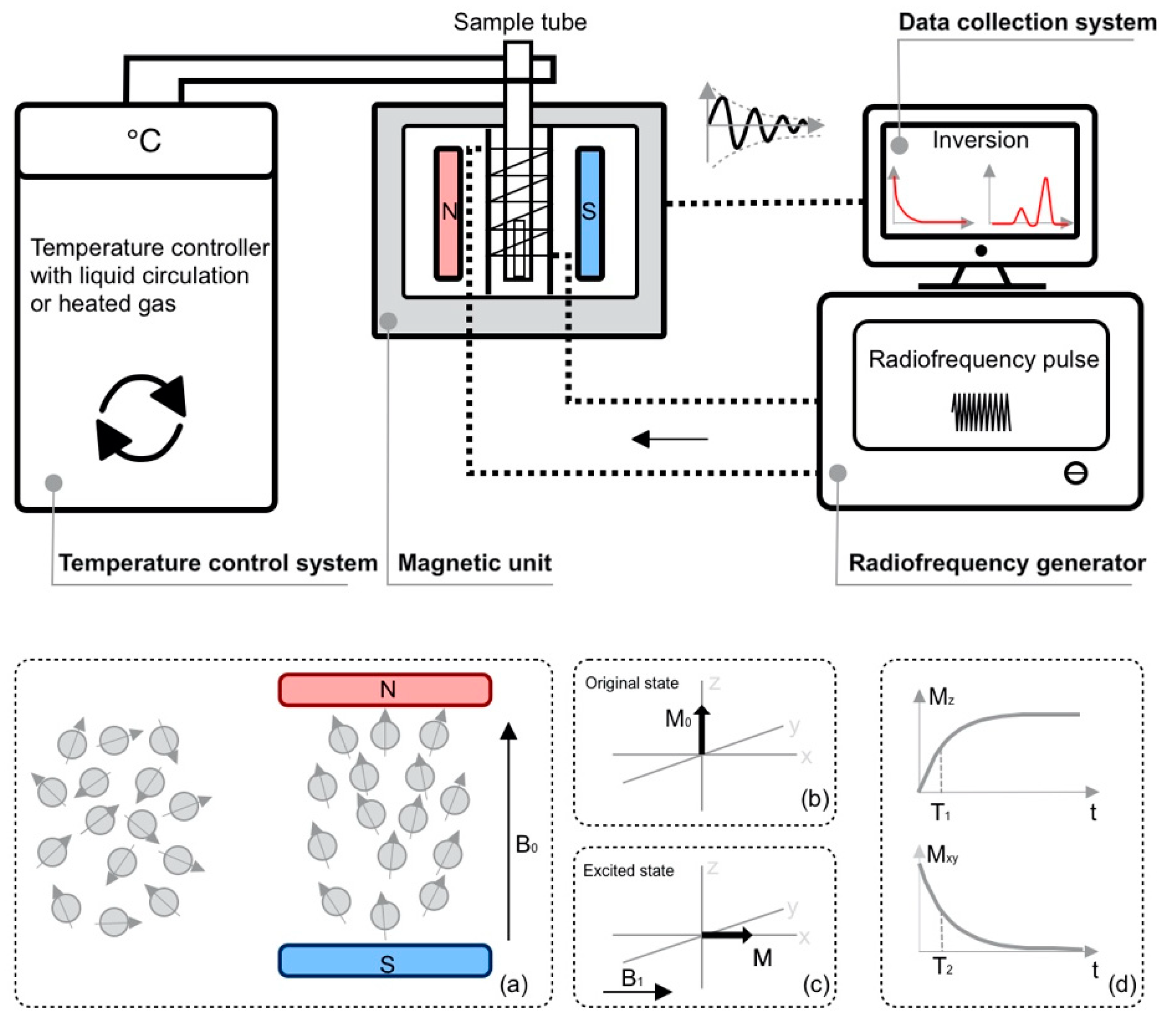
1.1. Basic Principles of TD-NMR Spectroscopy
1.2. The Overview of TD-NMR Applications for Studying Wood-Water Relations
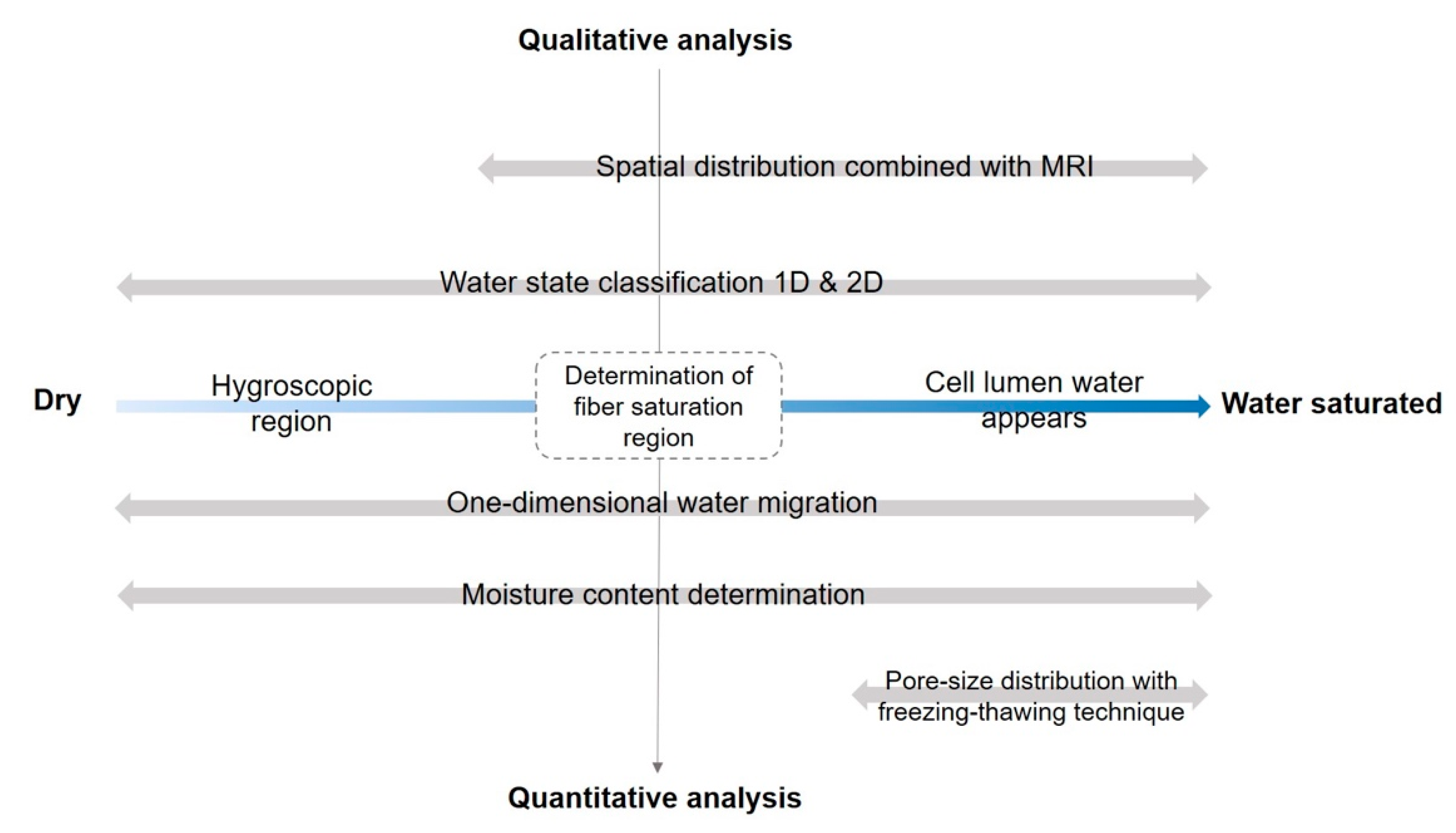
2. The TD-NMR Applications in Wood-Water Study
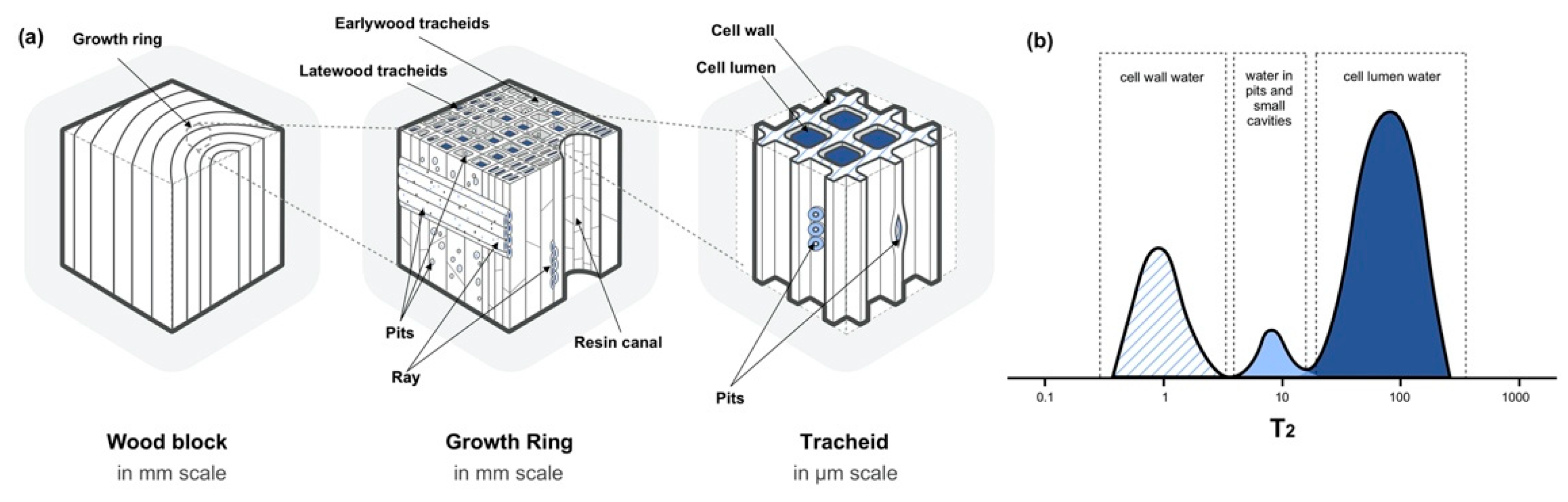
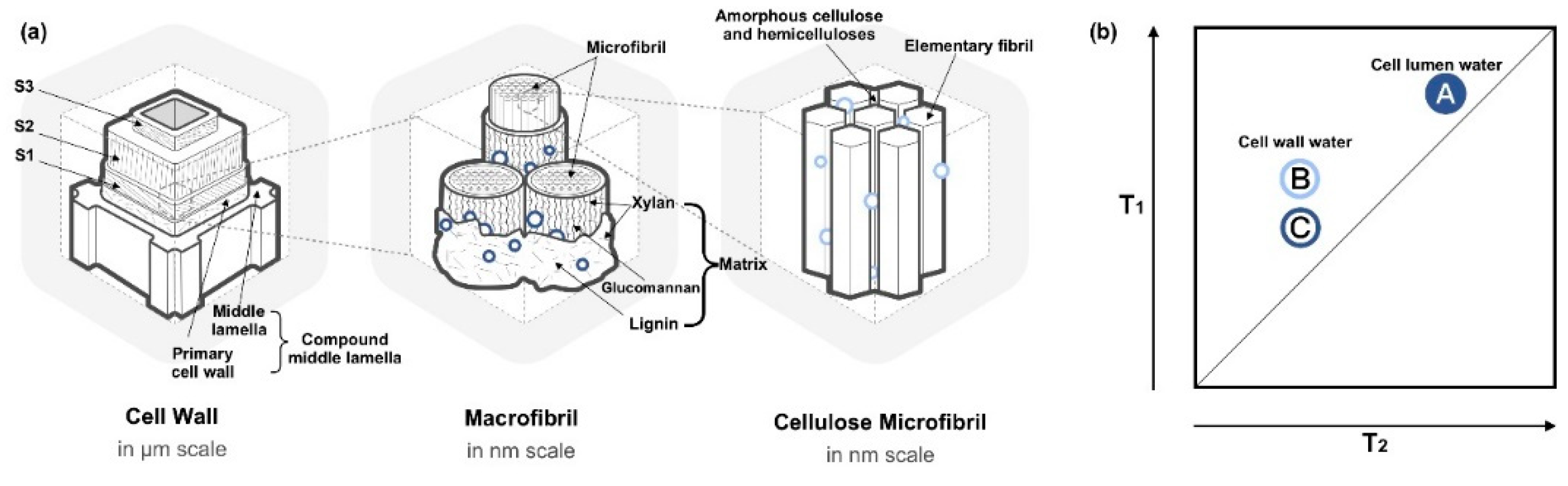
2.1. States of Water in the Wood
2.2. Determining the MC and Fiber Saturation State
2.3. Pore Size and Cell Wall Water Distribution
2.4. MRI and Water Transport
3. Remaining Issues
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Glass, S.V.; Zelinka, S.V. Moisture Relations and Physical Properties of Wood. In Wood Handbook: Wood as an Engineering Material; Forest Products Laboratory, Forest Service, U.S. Department of Agriculture: Madison, WI, USA, 2010; p. 80. [Google Scholar]
- Skaar, C. Wood-Water Relations; Springer Science & Business Media: Berlin/Heidelberg, Germany, 1988; p. 42. [Google Scholar]
- Engelund, E.T.; Thygesen, L.G.; Svensson, S.; Hill, C.A.S. A Critical Discussion of the Physics of Wood-Water Interactions. Wood Sci. Technol. 2013, 47, 141–161. [Google Scholar] [CrossRef] [Green Version]
- Siau, J.F. Transport Processes in Wood; Springer: Berlin/Heidelberg, Germany; New York, NY, USA; Tokyo, Japan, 1984; p. 1. [Google Scholar]
- Thybring, E.E.; Kymäläinen, M.; Rautkari, L. Experimental Techniques for Characterising Water in Wood Covering the Range from Dry to Fully Water-Saturated. Wood Sci. Technol. 2018, 52, 297–329. [Google Scholar] [CrossRef] [Green Version]
- Nasir, V.; Nourian, S.; Zhou, Z.; Rahimi, S.; Avramidis, S.; Cool, J. Classification and Characterization of Thermally Modified Timber Using Visible and near-Infrared Spectroscopy and Artificial Neural Networks: A Comparative Study on the Performance of Different Nde Methods and Anns. Wood Sci. Technol. 2019, 53, 1093–1109. [Google Scholar] [CrossRef]
- Fathi, H.; Kazemirad, S.; Nasir, V. A Nondestructive Guided Wave Propagation Method for the Characterization of Moisture-Dependent Viscoelastic Properties of Wood Materials. Mater. Struct. 2020, 53, 1–14. [Google Scholar] [CrossRef]
- Wang, W.; Chen, J.; Cao, J. Using Low-Field NMR and MRI to Characterize Water Status and Distribution in Modified Wood During Water Absorption. Holzforschung 2019, 73, 997–1004. [Google Scholar] [CrossRef]
- Zhou, B. The Applications of NMR Relaxometry, NMR Cryoporometry, and FFC NMR to Nanoporous Structures and Dynamics in Shale at Low Magnetic Fields. Energy Fuels 2018, 32, 8897–8904. [Google Scholar] [CrossRef]
- Berman, P.; Leshem, A.; Etziony, O.; Levi, O.; Parmet, Y.; Saunders, M.; Wiesman, Z. Novel 1H Low Field Nuclear Magnetic Resonance Applications for the Field of Biodiesel. Biotechnol. Biofuels 2013, 6, 55–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamason, C.; MacMillan, B.; Balcom, B.; Leblon, B.; Pirouz, Z. Examination of Water Phase Transitions in Black Spruce by Magnetic Resonance and Magnetic Resonance Imaging. Wood Fiber Sci. 2014, 46, 423–436. [Google Scholar]
- Song, Y.Q.; Venkataramanan, L.; Hurlimann, M.D.; Flaum, M.; Frulla, P.; Straley, C. T1-T2 Correlation Spectra Obtained Using a Fast Two-Dimensional Laplace Inversion. J. Magn. Reson. 2002, 154, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; McDonald, P.J.; Gardiner, B.A. A Study of Water Exchange in Wood by Means of 2D NMR Relaxation Correlation and Exchange. Holzforschung 2010, 64, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Telkki, V.-V. Hyperpolarized Laplace NMR. Magn. Reson. Chem. 2018, 56, 619–632. [Google Scholar] [CrossRef]
- Bernin, D.; Topgaard, D. NMR Diffusion and Relaxation Correlation Methods: New Insights in Heterogeneous Materials. Curr. Opin. Colloid Interface Sci. 2013, 18, 166–172. [Google Scholar] [CrossRef]
- Carr, H.Y.; Purcell, E.M. Effects of Diffusion on Free Precession in Nuclear Magnetic Resonance Experiments. Phys. Rev. 1954, 94, 630–638. [Google Scholar] [CrossRef]
- Meiboom, S.; Gill, D. Modified Spin--Echo Method for Measuring Nuclear Relaxation Times. Rev. Sci. Instrum. 1958, 29, 688–691. [Google Scholar] [CrossRef] [Green Version]
- Thygesen, L.G.; Elder, T. Moisture in Untreated, Acetylated, and Furfurylated Norway Spruce Studied during Drying Using Time Domain NMR. Wood Fiber Sci. 2008, 40, 309–320. [Google Scholar] [CrossRef]
- Menon, R.S.; MaCkay, A.L.; Hailey, J.R.T.; Bloom, M.; Burgess, A.E.; Swanson, J.S. An NMR Determination of the Physiological Water Distribution in Wood during Drying. J. Appl. Polym. Sci. 1987, 33, 1141–1155. [Google Scholar] [CrossRef]
- Mitchell, J.; Stark, S.C.; Strange, J.H. Probing Surface Interactions by Combining Nmr Cryoporometry and NMR Relaxometry. J. Phys. D 2005, 38, 1950–1958. [Google Scholar] [CrossRef]
- Pedersen, H.T.; Bro, R.; Engelsen, S.B. Towards Rapid and Unique Curve Resolution of Low-Field NMR Relaxation Data: Trilinear Slicing Versus Two-Dimensional Curve Fitting. J. Magn. Reson. 2002, 157, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, M.; Thygesen, L.G. The States of Water in Norway Spruce (Picea abies (L.) Karst.) Studied by Low-Field Nuclear Magnetic Resonance (LFNMR) Relaxometry: Assignment of Free-Water Populations Based on Quantitative Wood Anatomy. Holzforschung 2017, 71, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Whittall, K.P.; Bronskill, M.J.; Henkelman, R.M. Investigation of Analysis Techniques for Complicated Nmr Relaxation Data. J. Magn. Reson. 1991, 95, 221–234. [Google Scholar] [CrossRef]
- Provencher, S.W. CONTIN: A General Purpose Constrained Regularization Program for Inverting Noisy Linear Algebraic and Integral Equations. Comput. Phys. Commun. 1982, 27, 229–242. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Z. Time Domain-NMR Studies of Average Pore Size of Wood Cell Walls during Drying and Moisture Adsorption. Wood Sci. Technol. 2020, 54, 1241–1251. [Google Scholar] [CrossRef]
- Sharp, A.R.; Riggin, M.T.; Kaiser, R.; Schneider, M. Determination of Moisture Content of Wood by Pulsed Nuclear Magnetic Resonance. Wood Fiber Sci. 1978, 10, 74–81. [Google Scholar]
- Riggin, M.T.; Sharp, A.R.; Kaiser, R.; Schneider, M.H. Transverse NMR Relaxation of Water in Wood. J. Appl. Polym. Sci. 1979, 23, 3147–3154. [Google Scholar] [CrossRef]
- Araujo, C.D.; MacKay, A.L.; Hailey, J.R.T.; Whittall, K.P.; Le, H. Proton Magnetic Resonance Techniques for Characterization of Water in Wood: Application to White Spruce. Wood Sci. Technol. 1992, 26, 101–113. [Google Scholar] [CrossRef]
- Merela, M.; Oven, P.; Serša, I.; Mikac, U. A Single Point Nmr Method for an Instantaneous Determination of the Moisture Content of Wood. Holzforschung 2009, 63, 348–351. [Google Scholar] [CrossRef]
- Menon, R.S.; Mackay, A.L.; Flibotte, S.G.; Hailey, J.R.T. Quantitative Separation of NMR Images of Water in Wood on the Basis of T2. J. Magn. Reson. 1989, 82, 205–210. [Google Scholar] [CrossRef]
- Flibotte, S.; Menon, R.S.; Mackay, A.L.; Hailey, J.R.T. Proton Magnetic Resonance of Western Red Cedar. Wood Fiber Sci. 1990, 22, 362–376. [Google Scholar]
- Araujo, C.D.; Avramidis, S.; MacKay, A.L. Behaviour of Solid Wood and Bound Water as a Function of Moisture Content. A Proton Magnetic Resonance Study. Holzforschung 1994, 48, 69–74. [Google Scholar] [CrossRef]
- Hartley, I.D.; Avramidis, S.; MacKay, A.L. H-NMR Studies of Water Interactions in Sitka Spruce and Western Hemlock: Moisture Content Determination and Second Moments. Wood Sci. Technol. 1996, 30, 141–148. [Google Scholar] [CrossRef]
- Xu, Y.; Araujo, C.D.; MacKay, A.L.; Whittall, K.P. Proton Spin–Lattice Relaxation in Wood—T1 Related to Local Specific Gravity Using a Fast-Exchange Model. J. Magn. Reson. 1996, 110, 55–64. [Google Scholar] [CrossRef]
- Labbé, N.; Jéso, B.D.; Lartigue, J.-C.; Daudé, G.; Pétraud, M.; Ratier, M. Moisture Content and Extractive Materials in Maritime Pine Wood by Low Field 1H NMR. Holzforschung 2002, 56, 25–31. [Google Scholar] [CrossRef]
- Labbé, N.; De Jéso, B.; Lartigue, J.-C.; Daudé, G.; Pétraud, M.; Ratier, M. Time-Domain 1H NMR Characterization of the Liquid Phase in Greenwood. Holzforschung 2006, 60, 265–270. [Google Scholar] [CrossRef]
- Almeida, G.; Gagné, S.; Hernández, R.E. A NMR Study of Water Distribution in Hardwoods at Several Equilibrium Moisture Contents. Wood Sci. Technol. 2006, 41, 293–307. [Google Scholar] [CrossRef]
- Beck, G.; Thybring, E.E.; Thygesen, L.G.; Hill, C. Characterization of Moisture in Acetylated and Propionylated Radiata Pine Using Low-Field Nuclear Magnetic Resonance (LFNMR) Relaxometry. Holzforschung 2018, 72, 225–233. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, M.; Xiao, Z.; Chen, H.; Xie, Y. Vaporization Heat of Bound Water in Wood Chemically Modified via Grafting and Crosslinking Patterns by DSC and NMR Analysis. Holzforschung 2018, 72, 1043–1049. [Google Scholar] [CrossRef]
- Li, J.; Ma, E.; Yang, T. Differences between Hygroscopicity Limit and Cell Wall Saturation Investigated by LF-NMR on Southern Pine (Pinus spp.). Holzforschung 2019, 73, 911–921. [Google Scholar] [CrossRef]
- Cai, C.; Javed, M.A.; Komulainen, S.; Telkki, V.-V.; Haapala, A.; Heräjärvi, H. Effect of Natural Weathering on Water Absorption and Pore Size Distribution in Thermally Modified Wood Determined by Nuclear Magnetic Resonance. Cellulose 2020, 27, 4235–4247. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, M.; Courtier-Murias, D.; Faure, P.; Rodts, S.; Care, S. NMR Determination of Sorption Isotherms in Earlywood and Latewood of Douglas Fir. Identification of Bound Water Components Related to Their Local Environment. Holzforschung 2017, 71, 481–490. [Google Scholar] [CrossRef]
- Rostom, L.; Courtier-Murias, D.; Rodts, S.; Care, S. Investigation of the Effect of Aging on Wood Hygroscopicity by 2D 1H NMR Relaxometry. Holzforschung 2019, 74, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Rostom, L.; Caré, S.; Courtier--Murias, D. Analysis of Water Content in Wood Material through 1D and 2D 1H NMR Relaxometry: Application to the Determination of the Dry Mass of Wood. Magn. Reson. Chem. 2021, 59, 614–627. [Google Scholar] [CrossRef]
- Hiltunen, S.; Mankinen, A.; Javed, M.A.; Ahola, S.; Venäläinen, M.; Telkki, V.-V. Characterization of the Decay Process of Scots Pine Caused by Coniophora Puteana Using NMR and MRI. Holzforschung 2020, 74, 1021–1032. [Google Scholar] [CrossRef]
- Telkki, V.-V.; Yliniemi, M.; Jokisaari, J. Moisture in Softwoods: Fiber Saturation Point, Hydroxyl Site Content, and the Amount of Micropores as Determined from NMR Relaxation Time Distributions. Holzforschung 2013, 67, 291–300. [Google Scholar] [CrossRef]
- Kekkonen, P.M.; Ylisassi, A.; Telkki, V.-V. Absorption of Water in Thermally Modified Pine Wood as Studied by Nuclear Magnetic Resonance. J. Phys. Chem. C 2014, 118, 2146–2153. [Google Scholar] [CrossRef]
- Gao, X.; Zhuang, S.; Jin, J.; Cao, P. Bound Water Content and Pore Size Distribution in Swollen Cell Walls Determined by NMR Technology. Bioresources 2015, 10, 8202–8224. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, X.-M.; Chen, Z.-J. Water States and Migration in Xinjiang Poplar and Mongolian Scotch Pine Monitored by TD-NMR During Drying. Holzforschung 2018, 72, 113–123. [Google Scholar] [CrossRef]
- Javed, M.A.; Kekkonen, P.M.; Ahola, S.; Telkki, V.-V. Magnetic Resonance Imaging Study of Water Absorption in Thermally Modified Pine Wood. Holzforschung 2015, 69, 899–907. [Google Scholar] [CrossRef]
- Passarini, L.; Malveau, C.; Hernández, R.E. Distribution of the Equilibrium Moisture Content in Four Hardwoods below Fiber Saturation Point with Magnetic Resonance Microimaging. Wood Sci. Technol. 2015, 49, 1251–1268. [Google Scholar] [CrossRef]
- Maunu, S. NMR Studies of Wood and Wood Products. Prog. Nucl. Magn. Reson. Spectrosc. 2002, 40, 151–174. [Google Scholar] [CrossRef]
- Rodin, V.V. One- and Two-Dimensional NMR in Studying Wood–Water Interaction at Moisturizing Spruce. Anisotropy of Water Self-Diffusion. Colloids Interfaces 2019, 3, 54. [Google Scholar] [CrossRef] [Green Version]
- Almeida, G.; Hernandez, R.E. Changes in Physical Properties of Yellow Birch Below and above the Fiber Saturation Point. Wood Fiber Sci. 2006, 38, 74–83. [Google Scholar]
- Almeida, G.; Hernández, R.E. Changes in Physical Properties of Tropical and Temperate Hardwoods Below and above the Fiber Saturation Point. Wood Sci. Technol. 2006, 40, 599–613. [Google Scholar] [CrossRef]
- Passarini, L.; Malveau, C.; Hernández, R.E. Water State Study of Wood Structure of Four Hardwoods below Fiber Saturation Point with Nuclear Magnetic Resonance. Wood Fiber Sci. 2014, 46, 480–488. [Google Scholar]
- Wiedenhoeft, A. Structure and Function of Wood. In Wood Handbook: Wood as an Engineering Material; Forest Products Laboratory, Forest Service, U.S. Department of Agriculture: Madison, WI, USA, 2010; p. 72. [Google Scholar]
- Elder, T.; Labbe, N.; Harper, D.; Rials, T. Time Domain-Nuclear Magnetic Resonance Study of Chars from Southern Hardwoods. Biomass Bioenergy 2006, 30, 855–862. [Google Scholar] [CrossRef]
- Thygesen, L.G.; Elder, T. Moisture in Untreated, Acetylated, and Furfurylated Norway Spruce Monitored During Drying Below Fiber Saturation Using Time Domain NMR. Wood Fiber Sci. 2009, 41, 194–200. [Google Scholar]
- Elder, T.; Houtman, C. Time-Domain NMR Study of the Drying of Hemicellulose Extracted Aspen (Populus Tremuloides Michx.). Holzforschung 2012, 67, 405–411. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, X.; Gazo, R. Water States in Yellow Poplar During Drying Studied by Time-Domain Nuclear Magnetic Resonance. Wood Fiber Sci. 2013, 45, 423–428. [Google Scholar]
- Felby, C.; Thygesen, L.G.; Kristensen, J.B.; Jørgensen, H.; Elder, T. Cellulose–Water Interactions during Enzymatic Hydrolysis as Studied by Time Domain NMR. Cellulose 2008, 15, 703–710. [Google Scholar] [CrossRef]
- Thygesen, L.G.; Tang Engelund, E.; Hoffmeyer, P. Water Sorption in Wood and Modified Wood at High Values of Relative Humidity. Part I: Results for Untreated, Acetylated, and Furfurylated Norway Spruce. Holzforschung 2010, 64, 315–323. [Google Scholar] [CrossRef]
- Fredriksson, M.; Johansson, P. A Method for Determination of Absorption Isotherms at High Relative Humidity Levels: Measurements on Lime-Silica Brick and Norway Spruce (Picea abies (L.) Karst.). Dry. Technol. 2015, 34, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Thybring, E.E.; Fredriksson, M.; Ma, E.; Cao, J.; Digaitis, R.; Thygesen, L.G. Effects of Changes in Biopolymer Composition on Moisture in Acetylated Wood. Forests 2020, 11, 719. [Google Scholar] [CrossRef]
- Yang, T.; Cao, J.; Ma, E. How Does Delignification Influence the Furfurylation of Wood? Ind. Crop. Prod. 2019, 135, 91–98. [Google Scholar] [CrossRef]
- Thybring, E.E.; Fredriksson, M. Wood Modification as a Tool to Understand Moisture in Wood. Forests 2021, 12, 372. [Google Scholar] [CrossRef]
- Salmén, L.; Burgert, I. Cell Wall Features with Regard to Mechanical Performance. A Review Cost Action E35 2004–2008: Wood Machining–Micromechanics and Fracture. Holzforschung 2009, 63, 121–129. [Google Scholar] [CrossRef]
- Jakes, J.E.; Hunt, C.G.; Zelinka, S.L.; Ciesielski, P.N.; Plaza, N.Z. Effects of Moisture on Diffusion in Unmodified Wood Cell Walls: A Phenomenological Polymer Science Approach. Forests 2019, 10, 1084. [Google Scholar] [CrossRef] [Green Version]
- Jeoh, T.; Karuna, N.; Weiss, N.D.; Thygesen, L.G. Two-Dimensional 1H-Nuclear Magnetic Resonance Relaxometry for Understanding Biomass Recalcitrance. ACS Sustain. Chem. Eng. 2017, 5, 8785–8795. [Google Scholar] [CrossRef]
- Plaza, R.N.Z. Neutron Scattering Studies of Nano-Scale Wood-Water Interactions; University of Washington: Madison, WI, USA, 2017. [Google Scholar]
- Wiesman, Z.; Linder, C.; Resende, M.T.; Ayalon, N.; Levi, O.; Bernardinelli, O.D.; Colnago, L.A.; Mitre, C.I.N.; Jackman, R. 2D and 3D Spectrum Graphics of the Chemical-Morphological Domains of Complex Biomass by Low Field Proton NMR Energy Relaxation Signal Analysis. Energy Fuels 2018, 32, 5090–5102. [Google Scholar] [CrossRef]
- Hartley, I.D.; Kamke, F.A.; Peemoeller, H. Cluster Theory for Water Sorption in Wood. Wood Sci. Technol. 1992, 26, 83–99. [Google Scholar] [CrossRef]
- Hartley, I.D.; Avramidis, S. Analysis of the Wood Sorption Isotherm Using Clustering Theory. Holzforschung 1993, 47, 163–167. [Google Scholar] [CrossRef]
- Li, X.; Gao, Y.; Zhang, M.; Wang, X.; Wei, X. Water Migration in Poplar Wood During Microwave Drying Studied by Time Domain Nuclear Magnetic Resonance (TD-NMR). Holzforschung 2017, 71, 881–887. [Google Scholar] [CrossRef]
- Xu, K.; Lu, J.; Gao, Y.; Wu, Y.; Li, X. Determination of Moisture Content and Moisture Content Profiles in Wood During Drying by Low-Field Nuclear Magnetic Resonance. Dry. Technol. 2017, 35, 1909–1918. [Google Scholar] [CrossRef]
- Tiemann, H.D. Effect of Moisture upon the Strength and Stiffness of Wood; Forest Service, US Department of Agriculture: Madison, WI, USA, 1906; pp. 83–84.
- Stamm, A.J. The Fiber-Saturation Point of Wood as Obtained from Electrical Conductivity Measurements. Ind. Eng. Chem. Anal. Ed. 1929, 1, 94–97. [Google Scholar] [CrossRef]
- Stone, J.; Scallan, A. The Effect of Component Removal upon the Porous Structure of the Cell Wall of Wood. Ii. Swelling in Water and the Fiber Saturation Point. Tappi 1967, 50, 496–501. [Google Scholar]
- Hill, C.A.S. The Reduction in the Fibre Saturation Point of Wood Due to Chemical Modification Using Anhydride Reagents: A Reappraisal. Holzforschung 2008, 62, 423–428. [Google Scholar] [CrossRef]
- Stamm, A.J. Review of Nine Methods for Determining the Fiber Saturation Points of Wood and Wood Products. Wood Sci. Technol. 1971, 4, 114–128. [Google Scholar]
- Fredriksson, M.; Thybring, E.E. On Sorption Hysteresis in Wood: Separating Hysteresis in Cell Wall Water and Capillary Water in the Full Moisture Range. PLoS ONE 2019, 14, e0225111. [Google Scholar] [CrossRef]
- Zauer, M.; Kretzschmar, J.; Großmann, L.; Pfriem, A.; Wagenführ, A. Analysis of the Pore-Size Distribution and Fiber Saturation Point of Native and Thermally Modified Wood Using Differential Scanning Calorimetry. Wood Sci. Technol. 2013, 48, 177–193. [Google Scholar] [CrossRef]
- Babiak, M.; Kúdela, J. A Contribution to the Definition of the Fiber Saturation Point. Wood Sci. Technol. 1995, 29, 217–226. [Google Scholar] [CrossRef]
- Berthold, J.; Rinaudo, M.; Salmeń, L. Association of Water to Polar Groups; Estimations by an Adsorption Model for Ligno-Cellulosic Materials. Colloids Surf. A 1996, 112, 117–129. [Google Scholar] [CrossRef]
- Hansen, E.W.; Stöcker, M.; Schmidt, R. Low-Temperature Phase Transition of Water Confined in Mesopores Probed by NMR. Influence on Pore Size Distribution. J. Phys. Chem. 1996, 100, 2195–2200. [Google Scholar] [CrossRef]
- Petrov, O.V.; Furo, I. NMR Cryoporometry: Principles, Applications and Potential. Prog. Nucl. Magn. Reson. Spectrosc. 2009, 54, 97–122. [Google Scholar] [CrossRef]
- Kärenlampi, P.P.; Tynjälä, P.; Ström, P. Phase Transformations of Wood Cell Wall Water. J. Wood Sci. 2005, 51, 118–123. [Google Scholar] [CrossRef]
- Javed, M.A.; Komulainen, S.; Daigle, H.; Zhang, B.; Vaara, J.; Zhou, B.; Telkki, V.-V. Determination of Pore Structures and Dynamics of Fluids in Hydrated Cements and Natural Shales by Various 1H and 129Xe NMR Methods. Microporous Mesoporous Mater. 2019, 281, 66–74. [Google Scholar] [CrossRef]
- Park, S.; Venditti, R.; Jameel, H.; Pawlak, J. Changes in Pore Size Distribution during the Drying of Cellulose Fibers as Measured by Differential Scanning Calorimetry. Carbohydr. Polym. 2006, 66, 97–103. [Google Scholar] [CrossRef]
- Zauer, M.; Pfriem, A.; Wagenführ, A. Toward Improved Understanding of the Cell-Wall Density and Porosity of Wood Determined by Gas Pycnometry. Wood Sci. Technol. 2013, 47, 1197–1211. [Google Scholar] [CrossRef]
- Simpson, L.A.; Barton, A.F.M. Determination of the Fibre Saturation Point in Whole Wood Using Differential Scanning Calorimetry. Wood Sci. Technol. 1991, 25, 301–308. [Google Scholar] [CrossRef]
- Nakamura, K.; Hatakeyama, T.; Hatakeyama, H. Studies on Bound Water of Cellulose by Differential Scanning Calorimetry. Text. Res. J. 1981, 51, 607–613. [Google Scholar] [CrossRef]
- Zelinka, S.L.; Glass, S.V.; Jakes, J.E.; Stone, D.S. A Solution Thermodynamics Definition of the Fiber Saturation Point and the Derivation of a Wood–Water Phase (State) Diagram. Wood Sci. Technol. 2015, 50, 443–462. [Google Scholar] [CrossRef]
- Dvinskikh, S.V.; Henriksson, M.; Berglund, L.A.; Furó, I. A Multinuclear Magnetic Resonance Imaging (MRI) Study of Wood with Adsorbed Water: Estimating Bound Water Concentration and Local Wood Density. Holzforschung 2011, 65, 103–107. [Google Scholar] [CrossRef]
- Nguyen, D.M.; Almeida, G.; Nguyen, T.M.L.; Zhang, J.; Lu, P.; Colin, J.; Perré, P. A Critical Review of Current Imaging Techniques to Investigate Water Transfers in Wood and Biosourced Materials. Transp. Porous Med. 2021, 137, 21–61. [Google Scholar] [CrossRef]
- Almeida, G.; Leclerc, S.; Perre, P. NMR Imaging of Fluid Pathways during Drainage of Softwood in a Pressure Membrane Chamber. Int. J. Multiph. Flow 2008, 34, 312–321. [Google Scholar] [CrossRef]
- Hernández, R.E.; Cáceres, C.B. Magnetic Resonance Microimaging of Liquid Water Distribution in Sugar Maple Wood below Fiber Saturation Point. Wood Fiber Sci. 2010, 42, 259–272. [Google Scholar]
- Meder, R.; Codd, S.L.; Franich, R.A.; Callaghan, P.T.; Pope, J.M. Observation of Anisotropic Water Movement in Pinus Radiata D. Don Sapwood above Fiber Saturation Using Magnetic Resonance Micro-Imaging. Holz. Roh. Werkst. 2003, 61, 251–256. [Google Scholar] [CrossRef]
- Quick, J.J.; Hailey, J.R.; MacKay, A.L. Radial Moisture Profiles of Cedar Sapwood during Drying: A Proton Magnetic Resonance Study. Wood Fiber Sci. 2007, 22, 404–412. [Google Scholar]
- Dvinskikh, S.V.; Henriksson, M.; Mendicino, A.L.; Fortino, S.; Toratti, T. NMR Imaging Study and Multi-Fickian Numerical Simulation of Moisture Transfer in Norway Spruce Samples. Eng. Struct. 2011, 33, 3079–3086. [Google Scholar] [CrossRef]
- Gezici-Koç, Ö.; Erich, S.J.F.; Huinink, H.P.; van der Ven, L.G.J.; Adan, O.C.G. Bound and Free Water Distribution in Wood During Water Uptake and Drying as Measured by 1d Magnetic Resonance Imaging. Cellulose 2016, 24, 535–553. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Caré, S.; Courtier-Murias, D.; Faure, P.; Rodts, S.; Coussot, P. Magnetic Resonance Imaging Evidences of the Impact of Water Sorption on Hardwood Capillary Imbibition Dynamics. Wood Sci. Technol. 2018, 52, 929–955. [Google Scholar] [CrossRef]
- Gezici-Koç, Ö.; Erich, S.J.F.; Huinink, H.P.; van der Ven, L.G.J.; Adan, O.C.G. Moisture Content of the Coating Determines the Water Permeability as Measured by 1d Magnetic Resonance Imaging. Prog. Org. Coat. 2019, 130, 114–123. [Google Scholar] [CrossRef]
- Houts, J.H.v.; Wang, S.; Shi, H.; Pan, H.; Kabalka, G.W. Moisture Movement and Thickness Swelling in Oriented Strandboard, Part 1. Analysis Using Nuclear Magnetic Resonance Microimaging. Wood Sci. Technol. 2004, 38, 617–628. [Google Scholar] [CrossRef]
- Žlahtič Zupanc, M.; Mikac, U.; Serša, I.; Merela, M.; Humar, M. Water Distribution in Wood after Short Term Wetting. Cellulose 2018, 26, 703–721. [Google Scholar] [CrossRef]
- Müller, U.; Bammer, R.; Halmschlager, E.; Stollberger, R.; Wimmer, R. Detection of Fungal Wood Decay Using Magnetic Resonance Imaging. Holz. Roh. Werkst. 2001, 59, 190–194. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Species | Wood Type | MC Range (%) | Water Component Assignment and Their T2 (ms) |
|---|---|---|---|---|
| Ref. [37] | One tropical (Robinia coccinea Aublet) and two temperate (Acer saccharum Marsh. and Fagus grandifolia Ehrh.) hardwoods | Diffuse porous woods | Determined at different equilibrium moisture contents during desorption at 25 °C | FastT2 (0–10, bound or cell wall water); Medium T2 (10–100, liquid water in fiber and parenchyma elements); Slow T2 (above 100, liquid water in vessel elements) |
| Refs. [18,59,63] | Norway spruce (Picea abies (L.) Karst.) | Acetylated and furfurylated sapwood | Above fiber saturation point | Lumen water in earlywood tracheids moved from about 80–100 ms to 200–300 ms by both furfurylation and acetylation; Cell wall water reduced from about 1.4 to 0.65 by furfurylation, while acetylation had less effect on this water |
| Ref. [60] | Aspen (Populus tremuloides Michx.) | Hemicellulose-extracted pulp | Vacuum-saturated | Free water: 15–150; Bound water: 1–2; The removal of hemicelluloses results in longer T2 for the bound water |
| Ref. [64] | Norway spruce (Picea abies (L.) Karst.) | Sapwood and heartwood, mature and juvenile | Vacuum-saturated | Bound water: 1.2–2.0; Water in pits: 8.0–31.4; Water in tracheid lumina: 48.8–95.4; Surface water on the specimen: 226.7–1469.4 |
| Ref. [39] | Radiata pine (Pinus radiata D. Don) | Sapwood modified with acetic anhydride and glutaraldehyde | Water-saturated | Two bound water peaks were separated |
| Ref. [65] | Poplar (Populus euramericana Cv.) | Sapwood | Vacuum-saturated | Cell wall water: below 10; Water in small voids or small cavities: 10 to 100; Cell lumen water: above 100; |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Ma, E. Characterization of Water in Wood by Time-Domain Nuclear Magnetic Resonance Spectroscopy (TD-NMR): A Review. Forests 2021, 12, 886. https://doi.org/10.3390/f12070886
Li J, Ma E. Characterization of Water in Wood by Time-Domain Nuclear Magnetic Resonance Spectroscopy (TD-NMR): A Review. Forests. 2021; 12(7):886. https://doi.org/10.3390/f12070886
Chicago/Turabian StyleLi, Jingyu, and Erni Ma. 2021. "Characterization of Water in Wood by Time-Domain Nuclear Magnetic Resonance Spectroscopy (TD-NMR): A Review" Forests 12, no. 7: 886. https://doi.org/10.3390/f12070886
APA StyleLi, J., & Ma, E. (2021). Characterization of Water in Wood by Time-Domain Nuclear Magnetic Resonance Spectroscopy (TD-NMR): A Review. Forests, 12(7), 886. https://doi.org/10.3390/f12070886