
Novel Post-Glacial Haplotype Evolution in Birch—A Case for Conserving Local Adaptation



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. DNA Extraction
2.3. Chloroplast DNA Sequencing and Polymorphism Discovery
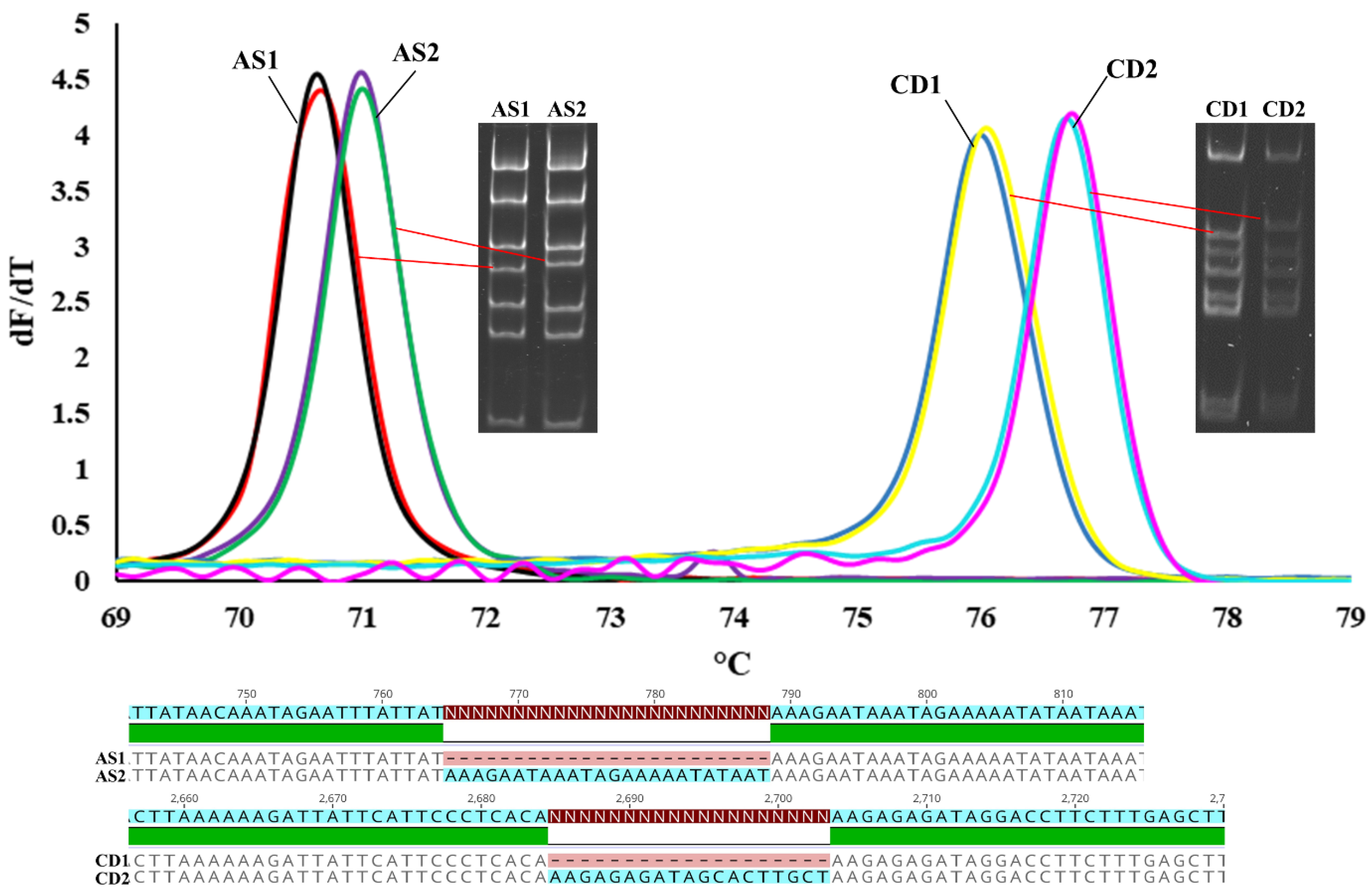
2.4. HRM Experiments
2.5. Chloroplast Microsatellites
2.6. Data Analysis
3. Results
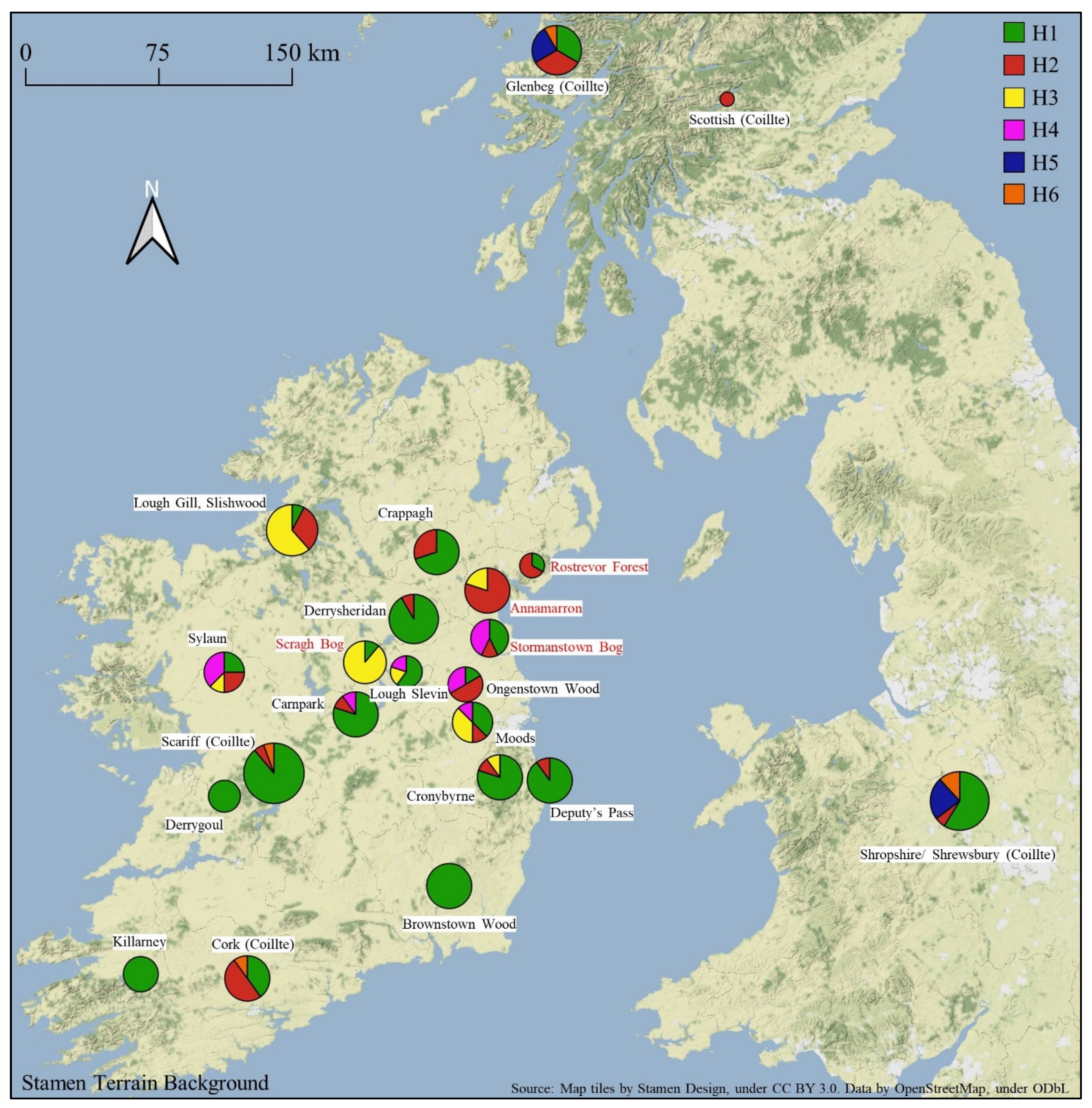
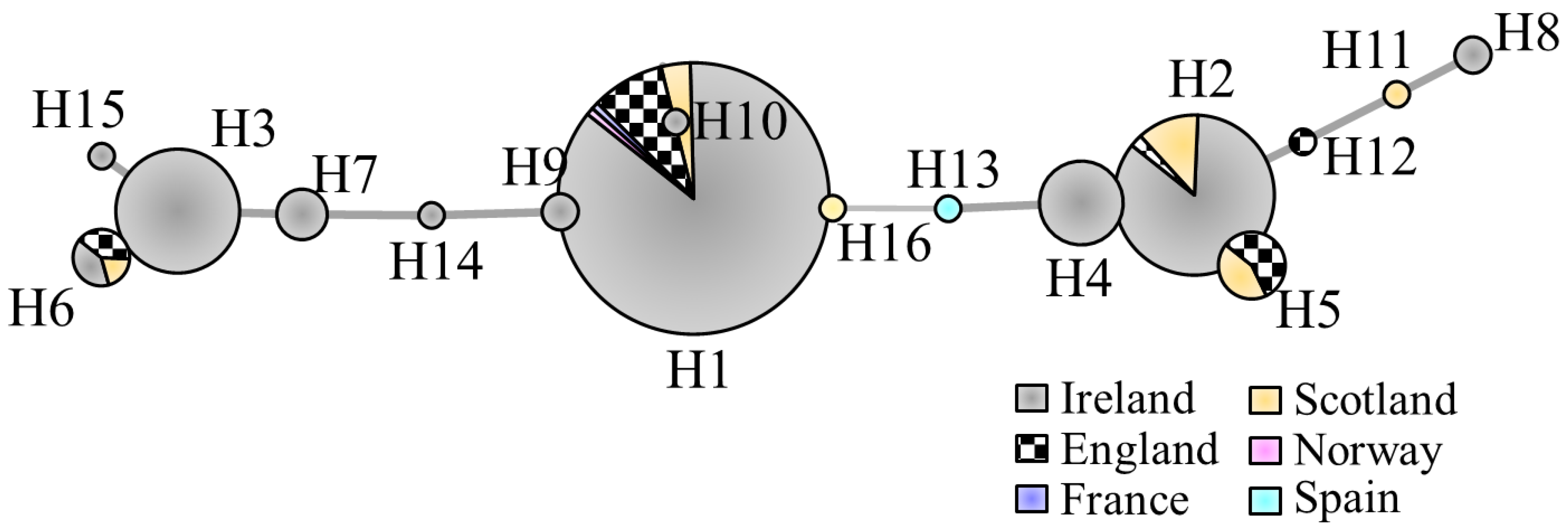
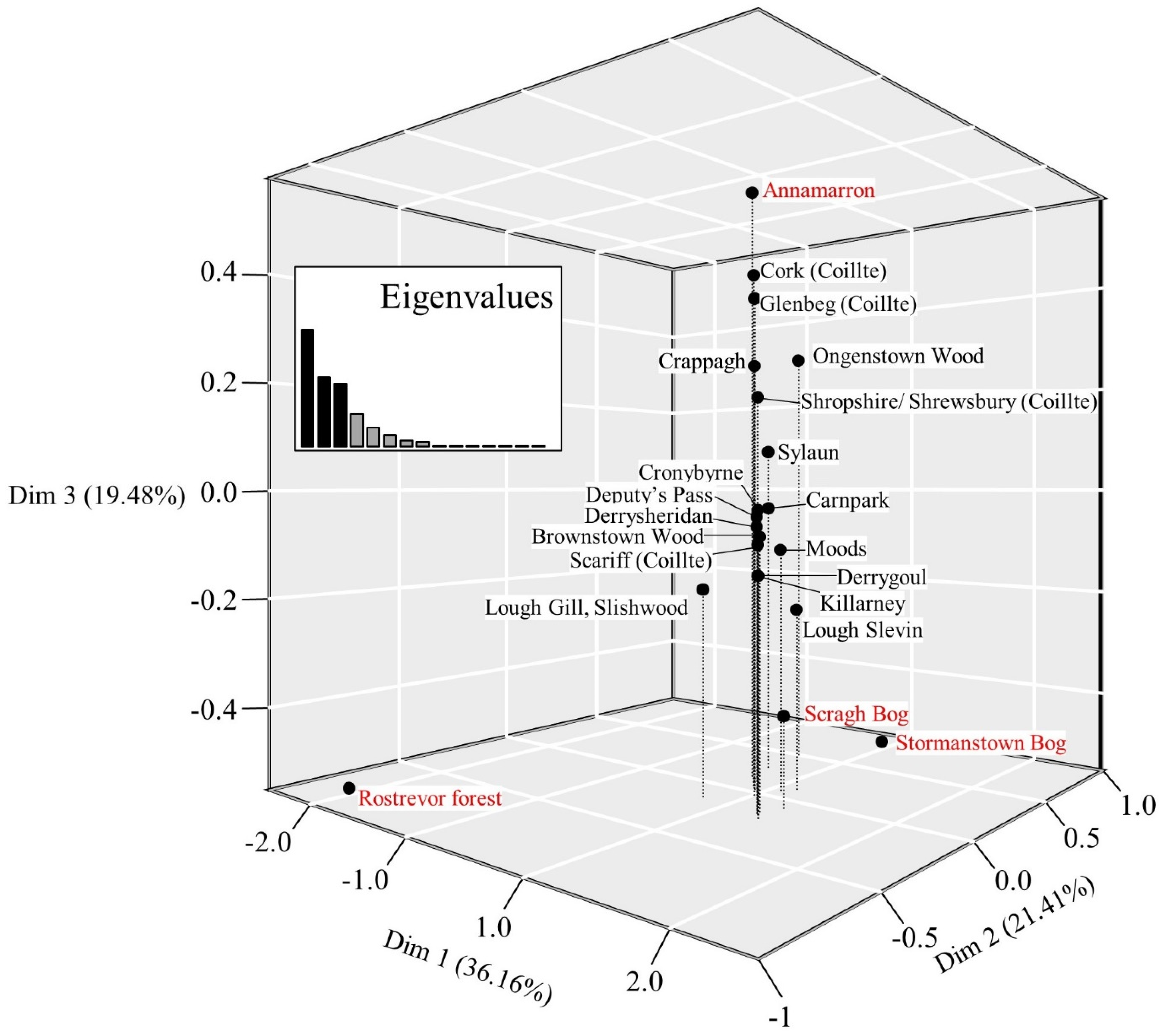
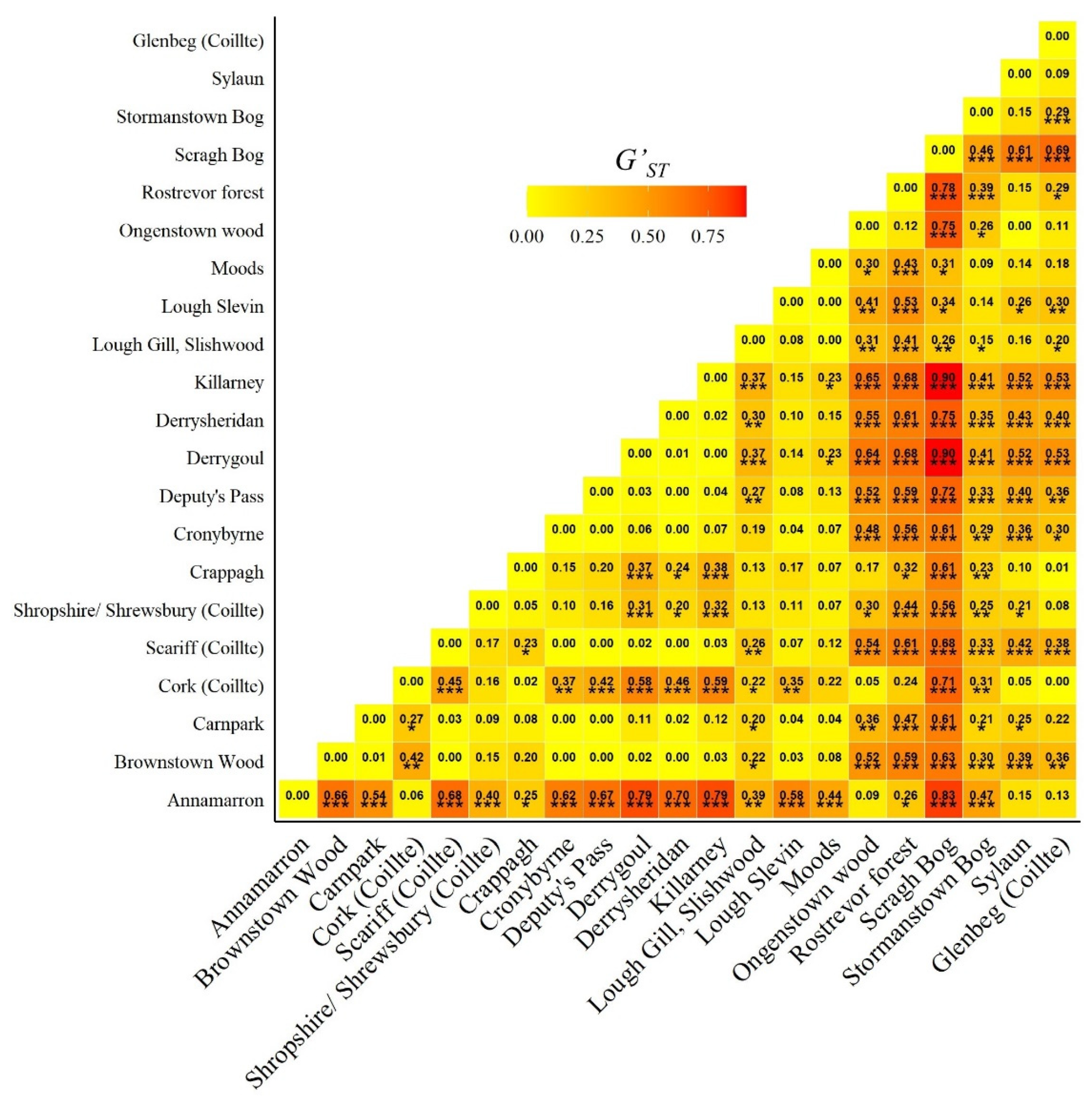
3.1. Chloroplast DNA Variation
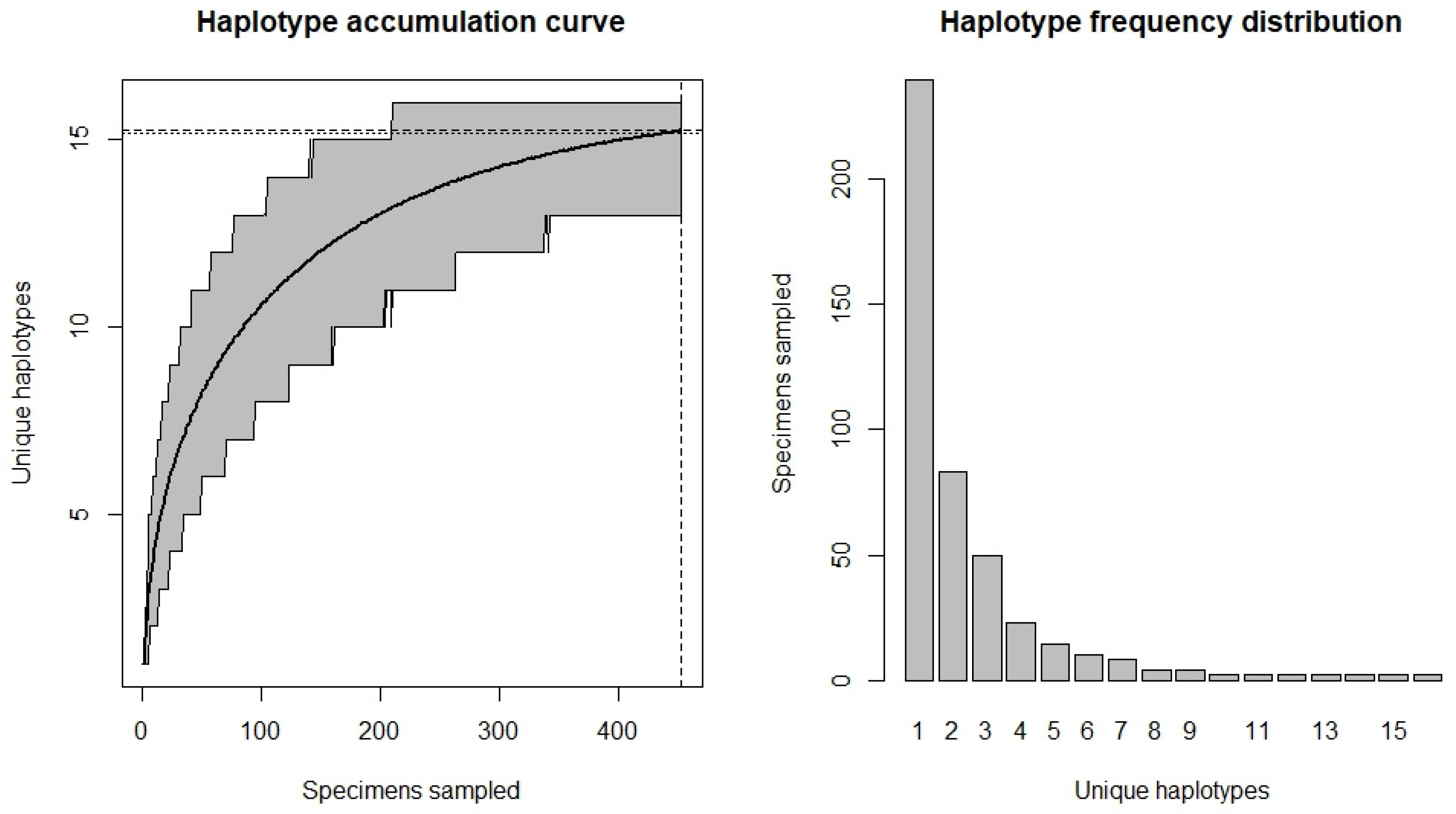
3.2. Sufficiency of Haplotype Capture in a European Context
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Atkinson, M.D. Betula pendula Roth (B. Verrucosa Ehrh.) and B. pubescens Ehrh. J. Ecol. 1992, 80, 837–870. [Google Scholar] [CrossRef]
- Perrin, P.; Martin, J.; Barron, S.; O’Neill, F.; NcNutt, K.; Delaney, A. National Survey of Native Woodlands 2003–2008; National Parks and Wildlife Service, Department of the Environment, Heritage and Local Goverment: Dublin, Ireland, 2008; Volume 1, p. 177.
- Praeger, R.L. Hybrids in the Irish Flora: A Tentative List. Proc. R. Ir. Acad. B 1951, 54, 1–14. [Google Scholar]
- Palme, A.E.; Su, Q.; Palsson, S.; Lascoux, M. Extensive Sharing of Chloroplast Haplotypes among European Birches Indicates Hybridization among Betula pendula, B. pubescens and B. nana. Mol. Ecol. 2004, 13, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Birks, H.J.B. Holocene Isochrone Maps and Patterns of Tree-Spreading in the British Isles. J. Biogeogr. 1989, 16, 503–540. [Google Scholar] [CrossRef]
- Mitchell, F.J.G. Where Did Ireland’s Trees Come From? Biol. Environ. Proc. R. Ir. Acad. 2006, 106B, 251–259. [Google Scholar] [CrossRef]
- Hewitt, G.M. Genetic Consequences of Climatic Oscillations in the Quaternary. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 183–195; discussion 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petit, R.J.; Csaikl, U.M.; Bordács, S.; Burg, K.; Coart, E.; Cottrell, J.; van Dam, B.; Deans, J.D.; Dumolin-Lapègue, S.; Fineschi, S.; et al. Chloroplast DNA Variation in European White Oaks: Phylogeography and Patterns of Diversity Based on Data from over 2600 Populations. For. Ecol. Manag. 2002, 156, 5–26. [Google Scholar] [CrossRef]
- Kelleher, C.T.; Hodkinson, T.R.; Kelly, D.L.; Douglas, G.C. Characterisation of Chloroplast DNA Haplotypes to Reveal the Provenance and Genetic Structure of Oaks in Ireland. For. Ecol. Manag. 2004, 189, 123–131. [Google Scholar] [CrossRef]
- Bennett, K.D.; Tzedakis, P.C.; Willis, K.J. Quaternary Refugia of North European Trees. J. Biogeogr. 1991, 18, 103–115. [Google Scholar] [CrossRef]
- Lascoux, M.; Palmé, A.E.; Cheddadi, R.; Latta, R.G. Impact of Ice Ages on the Genetic Structure of Trees and Shrubs. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Maliouchenko, O.; Palmé, A.E.; Buonamici, A.; Vendramin, G.G.; Lascoux, M. Comparative Phylogeography and Population Structure of European Betula Species, with Particular Focus on B. pendula and B. pubescens. J. Biogeogr. 2007, 34, 1601–1610. [Google Scholar] [CrossRef]
- Palmé, A.E.; Su, Q.; Rautenberg, A.; Manni, F.; Lascoux, M. Postglacial Recolonization and cpDNA Variation of Silver Birch, Betula pendula. Mol. Ecol. 2003, 12, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.M. Some Genetic Consequences of Ice Ages, and Their Role in Divergence and Speciation. Biol. J. Linn. Soc. 1996, 58, 247–276. [Google Scholar] [CrossRef]
- Tsuda, Y.; Semerikov, V.; Sebastiani, F.; Vendramin, G.G.; Lascoux, M. Multispecies Genetic Structure and Hybridization in the Betula Genus across Eurasia. Mol. Ecol. 2017, 26, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Binney, H.A.; Willis, K.J.; Edwards, M.E.; Bhagwat, S.A.; Anderson, P.M.; Andreev, A.A.; Blaauw, M.; Damblon, F.; Haesaerts, P.; Kienast, F.; et al. The Distribution of Late-Quaternary Woody Taxa in Northern Eurasia: Evidence from a New Macrofossil Database. Quat. Sci. Rev. 2009, 28, 2445–2464. [Google Scholar] [CrossRef] [Green Version]
- Kelleher, C.T. A National Forest Tree Gene Conservation Strategy and Action Plan for Ireland. Ir. For. J. 2020, 77, 7–32. [Google Scholar]
- Dang, X.D.; Kelleher, C.T.; Howard-Williams, E.; Meade, C.V. Rapid Identification of Chloroplast Haplotypes Using High Resolution Melting Analysis. Mol. Ecol. Resour. 2012, 12, 894–908. [Google Scholar] [CrossRef] [Green Version]
- Cubry, P.; Gallagher, E.; O’Connor, E.; Kelleher, C.T. Phylogeography and Population Genetics of Black Alder (Alnus glutinosa (L.) Gaertn.) in Ireland: Putting It in a European Context. Tree Genet. Genomes 2015, 11, 99. [Google Scholar] [CrossRef]
- Guido, N.; Starostina, E.; Leake, D.; Saaem, I. Improved PCR Amplification of Broad Spectrum GC DNA Templates. PLoS ONE 2016, 11, e0156478. [Google Scholar] [CrossRef] [Green Version]
- Demesure, B.; Sodzi, N.; Petit, R.J. A Set of Universal Primers for Amplification of Polymorphic Non-Coding Regions of Mitochondrial and Chloroplast DNA in Plants. Mol. Ecol. 1995, 4, 129–131. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Liu, W.; Miller, J.; Siripun, K.C.; Winder, C.T.; Schilling, E.E.; Small, R.L. The Tortoise and the Hare II: Relative Utility of 21 Noncoding Chloroplast DNA Sequences for Phylogenetic Analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taberlet, P.; Gielly, L.; Pautou, G.; Bouvet, J. Universal Primers for Amplification of Three Non-Coding Regions of Chloroplast DNA. Plant. Mol. Biol. 1991, 17, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Taberlet, P.; Coissac, E.; Pompanon, F.; Gielly, L.; Miquel, C.; Valentini, A.; Vermat, T.; Corthier, G.; Brochmann, C.; Willerslev, E. Power and Limitations of the Chloroplast trnL (UAA) Intron for Plant DNA Barcoding. Nucleic Acids Res. 2007, 35, e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weising, K.; Gardner, R.C. A Set of Conserved PCR Primers for the Analysis of Simple Sequence Repeat Polymorphisms in Chloroplast Genomes of Dicotyledonous Angiosperms. Genome 1999, 42, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Petit, R.; Aguinagalde, I.; de Beaulieu, J.L.; Bittkau, C.; Brewer, S.; Cheddadi, R.; Ennos, R.; Fineschi, S.; Grivet, D.; Lascoux, M.; et al. Glacial Refugia: Hotspots but Not Melting Pots of Genetic Diversity. Science 2003, 300, 1563–1565. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T. Adegenet: A R Package for the Multivariate Analysis of Genetic Markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T.; Ahmed, I. Adegenet 1.3-1: New Tools for the Analysis of Genome-Wide SNP Data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dray, S.; Dufour, A.-B. The Ade4 Package: Implementing the Duality Diagram for Ecologists. J. Stat. Softw. 2007, 22, 20. [Google Scholar] [CrossRef] [Green Version]
- Goudet, J. Hierfstat, a Package for r to Compute and Test Hierarchical F-Statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef] [Green Version]
- Winter, D.J. Mmod: An R Library for the Calculation of Population Differentiation Statistics. Mol. Ecol. Resour. 2012, 12, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R Package for Genetic Analysis of Populations with Clonal, Partially Clonal, and/or Sexual Reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [Green Version]
- Kamvar, Z.N.; Brooks, J.C.; Grünwald, N.J. Novel R Tools for Analysis of Genome-Wide Population Genetic Data with Emphasis on Clonality. Front. Genet. 2015, 6, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nei, M. Analysis of Gene Diversity in Subdivided Populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedrick, P.W. A Standardized Genetic Differentiation Measure. Evolution 2005, 59, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Altman, D.G.; Bland, J.M. How to Obtain the P Value from a Confidence Interval. BMJ 2011, 343, d2304. [Google Scholar] [CrossRef] [Green Version]
- Slatkin, M. Inbreeding Coefficients and Coalescence Times. Genet. Res. 1991, 58, 167–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousset, F. Genetic Differentiation and Estimation of Gene Flow from F-Statistics Under Isolation by Distance. Genetics 1997, 145, 1219. [Google Scholar] [CrossRef]
- Phillips, J.D.; French, S.H.; Hanner, R.H.; Gillis, D.J. HACSim: An R Package to Estimate Intraspecific Sample Sizes for Genetic Diversity Assessment Using Haplotype Accumulation Curves. PeerJ Comput. Sci. 2020, 6, e243. [Google Scholar] [CrossRef] [Green Version]
- Fussi, B.; Westergren, M.; Aravanopoulos, F.; Baier, R.; Kavaliauskas, D.; Finzgar, D.; Alizoti, P.; Bozic, G.; Avramidou, E.; Konnert, M.; et al. Forest Genetic Monitoring: An Overview of Concepts and Definitions. Env. Monit Assess. 2016, 188, 493. [Google Scholar] [CrossRef] [Green Version]
- Heuertz, M.; Fineschi, S.; Anzidei, M.; Pastorelli, R.; Salvini, D.; Paule, L.; Frascaria-Lacoste, N.; Hardy, O.J.; Vekemans, X.; Vendramin, G.G. Chloroplast DNA Variation and Postglacial Recolonization of Common Ash (Fraxinus excelsior L.) in Europe. Mol. Ecol. 2004, 13, 3437–3452. [Google Scholar] [CrossRef] [PubMed]
- Forest Statistics Ireland 2020; Department of Agriculture, Food and the Marine: Dublin, Ireland, 2020; p. 81.
- Lefèvre, F.; Koskela, J.; Hubert, J.; Kraigher, H.; Longauer, R.; Olrik, D.C.; Schüler, S.; Bozzano, M.; Alizoti, P.; Bakys, R.; et al. Dynamic Conservation of Forest Genetic Resources in 33 European Countries. Conserv. Biol. 2013, 27, 373–384. [Google Scholar] [CrossRef]
- Cahalane, G.; Doody, P.; Douglas, G.; Fennessy, J.; O’Reilly, C.; Pfeifer, A. Sustaining and Developing Ireland’s Forest Genetic Resources. An Outline Strategy; COFORD: Dublin, Ireland, 2007; p. 30. [Google Scholar]
- Perdereau, A.C.; Kelleher, C.T.; Douglas, G.C.; Hodkinson, T.R. High Levels of Gene Flow and Genetic Diversity in Irish Populations of Salix caprea L. Inferred from Chloroplast and Nuclear SSR Markers. BMC Plant. Biol. 2014, 14, 202. [Google Scholar] [CrossRef] [Green Version]
- Eidesen, P.B.; Alsos, I.G.; Brochmann, C. Comparative Analyses of Plastid and AFLP Data Suggest Different Colonization History and Asymmetric Hybridization between Betula pubescens and B. nana. Mol. Ecol. 2015, 24, 3993–4009. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.M.; Dick, C.W.; Dayanandan, S. A Similar Phylogeographical Structure among Sympatric North American Birches (Betula) Is Better Explained by Introgression than by Shared Biogeographical History. J. Biogeogr. 2015, 42, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Borrell, J.S.; Bodles, W.J.; Kuttapitiya, A.; Nichols, R.A.; Buggs, R.J. Molecular Footprints of the Holocene Retreat of Dwarf Birch in Britain. Mol. Ecol. 2014, 23, 2771–2782. [Google Scholar] [CrossRef] [Green Version]
- Currat, M.; Ruedi, M.; Petit, R.J.; Excoffier, L. The Hidden Side of Invasions: Massive Introgression by Local Genes. Evolution 2008, 62, 1908–1920. [Google Scholar] [CrossRef]
- Petit, R.J.; Excoffier, L. Gene Flow and Species Delimitation. Trends Ecol. Evol 2009, 24, 386–393. [Google Scholar] [CrossRef]
- Kulju, K.K.M.; Pekkinen, M.; Varvio, S. Twenty-Three Microsatellite Primer Pairs for Betula pendula (Betulaceae). Mol. Ecol. Notes 2004, 4, 471–473. [Google Scholar] [CrossRef]
- Molloy, K.; O’Connell, M. Post-Glaciation Plant Colonisation of Ireland: Fresh Insights from An Loch Mór, Inis Oírr, Western Ireland. Ir. Nat. J. 2014, 33, 66–88. [Google Scholar]
- Thórsson, Æ.T.; Pálsson, S.; Lascoux, M.; Anamthawat-Jónsson, K. Introgression and Phylogeography of Betula nana (Diploid), B. pubescens (Tetraploid) and Their Triploid Hybrids in Iceland Inferred from CpDNA Haplotype Variation. J. Biogeogr. 2010, 37, 2098–2110. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Location | Provenance | Lat., Long. | Samples/Haplotypes | Gene Diversity ± S.D. | Species |
|---|---|---|---|---|---|---|
| Alberes, Pyrenees Orientales | France | Wild | 42.48, 2.95 | 1/NA | 0.0000 ± 0.0000 |  |
| Annamarron | Monaghan, Ireland | Wild | 53.93, −6.66 | 11/2 | 0.1640 ± 0.0334 |  |
| Glenbeg (Coillte) | Scotland | Breeding | 56.69, −5.94 | 13/5 | 0.1982 ± 0.0210 |  |
| Brownstown Wood | Kilkenny, Ireland | Wild | 52.42, −7.03 | 11/2 | 0.0826 ± 0.0313 |  |
| Carnpark | Westmeath, Ireland | Wild | 53.42, −7.82 | 10/3 | 0.1450 ± 0.0373 |  |
| Cork (Coillte) | Cork, Ireland | Breeding | 51.94, −8.73 | 11/3 | 0.2025 ± 0.0229 |  |
| Scariff (Coillte) | Clare, Ireland | Breeding | 52.71, −2.76 | 19/4 | 0.0727 ± 0.0248 |  |
| Scottish (Coillte) | Scotland | Breeding | 52.96, −8.58 | 2/2 | 0.0625 ± 0.0313 |  |
| Shropshire/Shrewsbury (Coillte) | England | Breeding | 56.49, −4.2 | 20/5 | 0.1836 ± 0.0222 |  |
| Crappagh | Monaghan, Ireland | Wild | 54.14, −7.1 | 12/3 | 0.1985 ± 0.0217 |  |
| Cronybyrne | Wicklow, Ireland | Wild | 52.97, −6.25 | 11/3 | 0.1010 ± 0.0333 |  |
| Deputy’s Pass | Wicklow, Ireland | Wild | 52.95, −6.16 | 10/2 | 0.0675 ± 0.0303 |  |
| Derrygoul | Clare, Ireland | Wild | 52.91, −8.85 | 5/1 | 0.0000 ± 0.0000 |  |
| Derrysheridan | Meath, Ireland | Wild | 53.78, -7.33 | 14/2 | 0.0546 ± 0.0252 |  |
| Killarney | Kerry, Ireland | Wild | 51.98, −9.57 | 7/1 | 0.0000 ± 0.0000 |  |
| Lac des Camboux, Lozere, | France | Wild | 44.49, 3.58 | 2/1 | 0.1250 ± 0.0431 |  |
| Lough Gill, Slishwood | Sligo, Ireland | Wild | 54.24, −8.4 | 14/3 | 0.2777 ± 0.0331 |  |
| Lough Slevin | Westmeath, Ireland | Wild | 53.56, −7.32 | 8/4 | 0.1832 ± 0.0437 |  |
| Moods | Kildare, Ireland | Wild | 53.27, −6.79 | 8/4 | 0.2305 ± 0.0426 |  |
| Norway | Norway | Wild | 60.47, 8.47 | 2/1 | 0.0625 ± 0.0312 |  |
| Ongenstown wood | Meath, Ireland | Wild | 53.64, −6.82 | 8/4 | 0.2653 ± 0.0377 |  |
| Rostrevor forest | Down, Ireland | Wild | 54.11, −6.18 | 5/2 | 0.3556 ± 0.0452 |  |
| Scragh Bog | Westmeath, Ireland | Wild | 53.58, −7.36 | 10/2 | 0.0225 ± 0.0172 |  |
| Spain | Spain | Wild | 40.46, 3.75 | 1/1 | 0.0000 ± 0.0000 |  |
| Stormanstown Bog | Louth, Ireland | Wild | 53.88, −6.62 | 12/5 | 0.3212 ± 0.0289 |  |
| Sylaun | Galway, Ireland | Wild | 53.53, −8.93 | 13/5 | 0.3113 ± 0.0320 |  |
| Primer | Region | Location a | Sequence (5′-3′) | Reference |
|---|---|---|---|---|
| trnC-f | trnC-D | 1–20 bp | CCAGTTCAAATCTGGGTGTC | [21] |
| trnC_int_F | trnC-D | 708–734 bp | TCCAGGGGTGTATCTACGTATTTTGCT | This work |
| CD_int_birch_seq1 | trnC-D | 1617–1590 bp | CTTACAATTCGAATTCCTAGAATTTCTG | This work |
| psbMF_Shaw | trnC-D | 2068–2097bp | AGCAATAAATGCGAGAATATTTACTTCCAT | [22] |
| Ag_trnC-D_indel_R | trnC-D | 2237–2215 bp | TCATGATATTGCTCCGATTCGAT | [19] |
| CD_int_birch_seq2 | trnC-D | 3009–2985 bp | CTATACGTTTACAGGAGGCTATACA | This work |
| trnD-M | trnC-D | 3408–3389 bp | GGGATTGTAGTTCAATTGGT | [21] |
| psaA-f | psaA-trnS | 1-22 bp | ACTTCTGGTTCCGGCGAACGAA | [21] |
| Birch_AS_indel1_b-F | psaA-trnS | 892–872 bp | TGGTTGAAGATCACAAGGCGT | This work |
| Birch_AS_SNP3_R | psaA-trnS | 1076–1095 bp | CGGCTCAGCAGTCAATTCTT | This work |
| Birch_AS_SNP3_F | psaA-trnS | 1275–1252 bp | GCTTTATTCTTCTAAAGGTGGGAA | This work |
| Birch_AS_SNP2_F | psaA-trnS | 1845–1826 bp | AGGGCACTAGAACGAAACCC | This work |
| Birch_AS_SNP1_F | psaA-trnS | 2292–2272 bp | TCCTGGAAATTAAGGGGTGCT | This work |
| AS_int_birch_seq_1 | psaA-trnS | 2840–2816 bp | CCCAGATCTCGGATAAATGGAAATT | This work |
| Tab_a | trnT-F | 1–20 bp | CATTACAAATGCGATGCTCT | [23] |
| TF11_Rv | trnT-F | 633–610 bp | GTGTAATTTGAGATACTCGAACGG | This work |
| Tab_b | trnT-F | 968–949 bp | TCTACCGATTTCGCCATATC | [23] |
| trnL(UAA)h | trnT-F | 1155–1134 bp | CCATTGAGTCTCTGCACCTATC | [24] |
| Tab_d | trnT-F | 1399–1380 bp | GGGGATAGAGGGACTTGAAC | [23] |
| Tab_f | trnT-F | 1855–1836 bp | ATTTGAACTGGTGACACGAG | [23] |
| Birch_AS_indel1_F b | psaA-trnS | 885–866 bp | AGATCACAAGGCGTTTCGAA | This work |
| Birch_AS_indel1_R b | psaA-trnS | 693–712 bp | TGGGGACAACAAACAAAACT | This work |
| Birch_CD_indel1_b-F b | trnC-D | 2575–2595 bp | AAGGAGAGTCCGGGTATAAAA | This work |
| Birch_CD_indel1_b-R b | trnC-D | 2746–2725 bp | TCCAAAGAACAAAGAAATGGGA | This work |
| trnC-D | psaA-trnS | ccmp5 | ccmp10 | ccmp6 | ccmp2 | ccmp4 | ccmp7 | Found in: | Frequency a | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All | Pb | Pn | Un | ||||||||||
| H1 | 1 | 1 | 105 | 118 | 100 | 205 | 117 | 147 | Ireland, England, Scotland, France, Norway | 115 | 94 | 20 | 1 |
| H2 | 2 | 2 | 106 | 118 | 100 | 205 | 117 | 147 | Ireland, England, Scotland | 40 | 35 | 3 | 2 |
| H3 | 1 | 1 | 105 | 118 | 100 | 205 | 118 | 147 | Ireland | 24 | 24 | 0 | 0 |
| H4 | 2 | 2 | 108 | 118 | 100 | 205 | 117 | 154 | Ireland | 11 | 11 | 0 | 0 |
| H5 | 2 | 1 | 106 | 118 | 100 | 205 | 117 | 147 | England, Scotland | 7 | 7 | 0 | 0 |
| H6 | 2 | 1 | 105 | 118 | 100 | 205 | 118 | 147 | Ireland, England, Scotland | 5 | 2 | 3 | 0 |
| H7 | 1 | 1 | 105 | 118 | 100 | 205 | 118 | 148 | Ireland | 4 | 4 | 0 | 0 |
| H8 | 2 | 2 | 106 | 118 | 100 | 205 | 118 | 147 | Ireland | 2 | 2 | 0 | 0 |
| H9 | 1 | 1 | 105 | 118 | 100 | 205 | 119 | 147 | Ireland | 2 | 2 | 0 | 0 |
| H10 | 1 | 1 | 104 | 118 | 100 | 205 | 117 | 147 | Ireland | 1 | 0 | 1 | 0 |
| H11 | 2 | 2 | 106 | 118 | 100 | 205 | 119 | 147 | Scotland | 1 | 0 | 1 | 0 |
| H12 | 2 | 2 | 106 | 118 | 100 | 205 | 116 | 147 | England | 1 | 0 | 1 | 0 |
| H13 | 2 | 2 | 105 | 118 | 100 | 205 | 117 | 154 | Spain | 1 | 0 | 0 | 1 |
| H14 | 1 | 1 | 105 | 118 | 100 | 205 | 119 | 148 | Ireland | 1 | 1 | 0 | 0 |
| H15 | 1 | 1 | 105 | 118 | 98 | 205 | 118 | 147 | Ireland | 1 | 1 | 0 | 0 |
| H16 | 1 | 2 | 105 | 118 | 100 | 205 | 117 | 147 | Scotland | 1 | 1 | 0 | 0 |
| hS | hT | GST | GST | |
|---|---|---|---|---|
| All species (n = 236/228 a) | 0.173 | 0.236 | 0.268 [0.214, 0.324] | 0.336 [0.276, 0.397] |
| B. pubescens (n = 193/192 a) | 0.160 | 0.217 | 0.261 [0.205, 0.320] | 0.323 [0.259, 0.387] |
| B. pendula (n = 29/20 a) | 0.136 | 0.144 | 0.056 [−0.042, 0.198] | 0.122 [−0.071, 0.356] |
| Putative hybrid (n = 6/0 a) | NA | NA | NA | NA |
| All Sites. | d.f. | SSD | Variance | Variance (%) | p value | |||||
| Between sites | 20 | 98.56 | 0.33 | 19.16 | 0.015 | |||||
| Species within sites | 8 | 11.94 | 0.03 | 1.58 | 0.301 | |||||
| Within all | 183 | 251.07 | 1.37 | 79.25 | <0.001 | |||||
| Total a | 211 | 361.57 | 1.73 | 100.00 | ||||||
| Irish only | d.f. | SSD | Variance | Variance (%) | p value | |||||
| N-S | E-W | N-S | E-W | N-S | E-W | N-S | E-W | N-S | E-W | |
| Between | 1 | 1 | 15.82 | 0.65 | 0.13 | 0.00 | 7.03 | 0.00 | 0.034 | 1.000 |
| Sites within | 17 | 17 | 74.50 | 89.66 | 0.33 | 0.42 | 18.18 | 23.92 | <0.001 | <0.001 |
| Within all | 162 | 162 | 216.73 | 216.73 | 1.34 | 1.34 | 74.79 | 76.08 | <0.001 | <0.001 |
| Total a | 180 | 180 | 307.04 | 307.04 | 1.79 | 1.76 | 100.00 | 100.00 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belton, S.; Cubry, P.; Fox, E.; Kelleher, C.T. Novel Post-Glacial Haplotype Evolution in Birch—A Case for Conserving Local Adaptation. Forests 2021, 12, 1246. https://doi.org/10.3390/f12091246
Belton S, Cubry P, Fox E, Kelleher CT. Novel Post-Glacial Haplotype Evolution in Birch—A Case for Conserving Local Adaptation. Forests. 2021; 12(9):1246. https://doi.org/10.3390/f12091246
Chicago/Turabian StyleBelton, Samuel, Philippe Cubry, Erica Fox, and Colin T. Kelleher. 2021. "Novel Post-Glacial Haplotype Evolution in Birch—A Case for Conserving Local Adaptation" Forests 12, no. 9: 1246. https://doi.org/10.3390/f12091246
APA StyleBelton, S., Cubry, P., Fox, E., & Kelleher, C. T. (2021). Novel Post-Glacial Haplotype Evolution in Birch—A Case for Conserving Local Adaptation. Forests, 12(9), 1246. https://doi.org/10.3390/f12091246