
Genome-Wide Identification of miRNAs and Its Downstream Transcriptional Regulatory Network during Seed Maturation in Tilia tuan

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Construction of RNA Library and Transcriptome Sequencing
2.3. Construction of Small RNA Library and High-Throughput Sequencing
2.4. Sequence Data Analysis
2.5. Identification of Known and Novel miRNAs
2.6. Expression Analysis of miRNA
2.7. Target Prediction of miRNA and Function Enrichment Analysis
3. Results
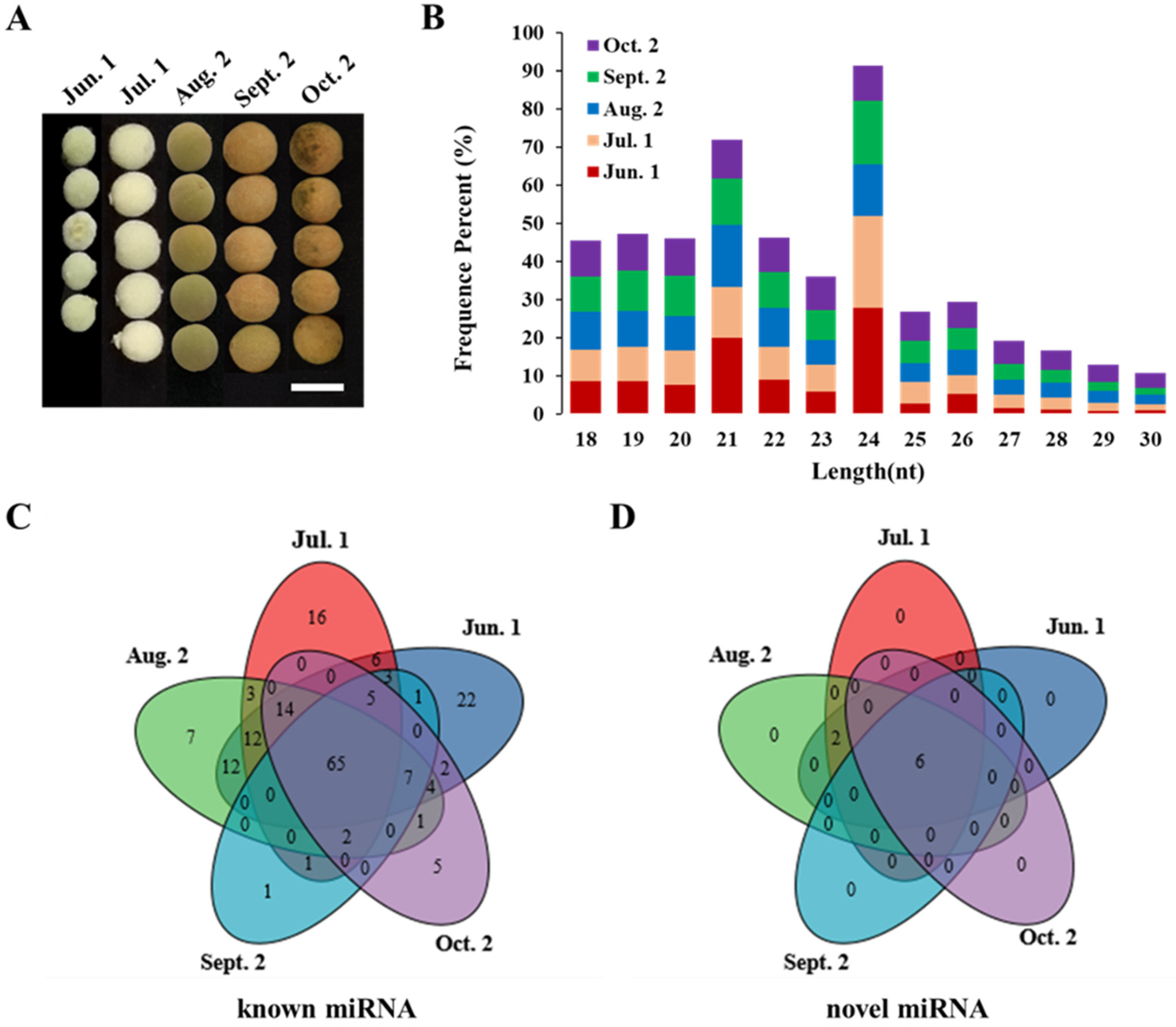
3.1. Small RNA Sequence Statistics
3.2. Identification of Known miRNAs during Seed Maturation in T. tuan
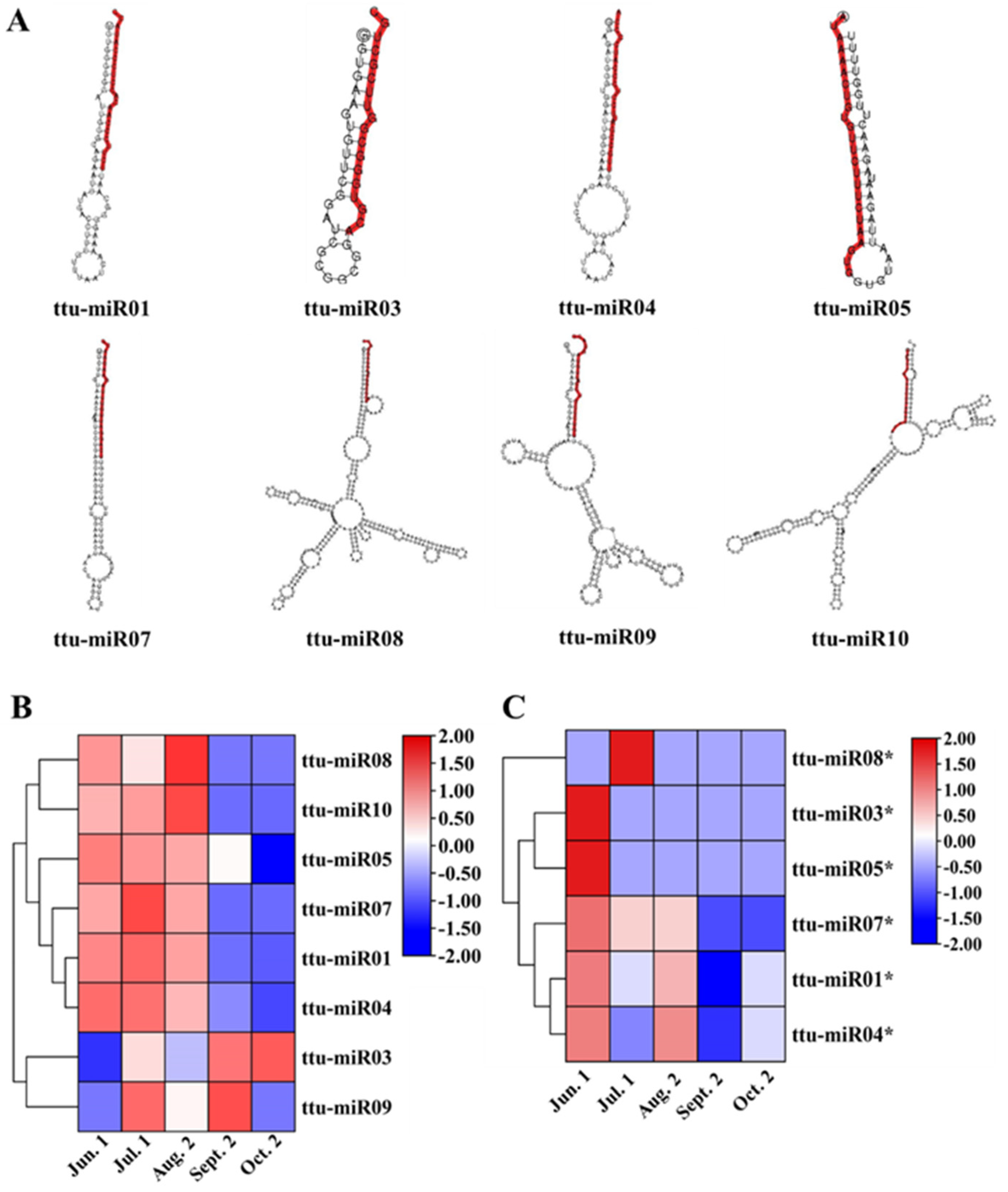
3.3. Identification of Novel miRNAs during Seed Maturation in T. tuan
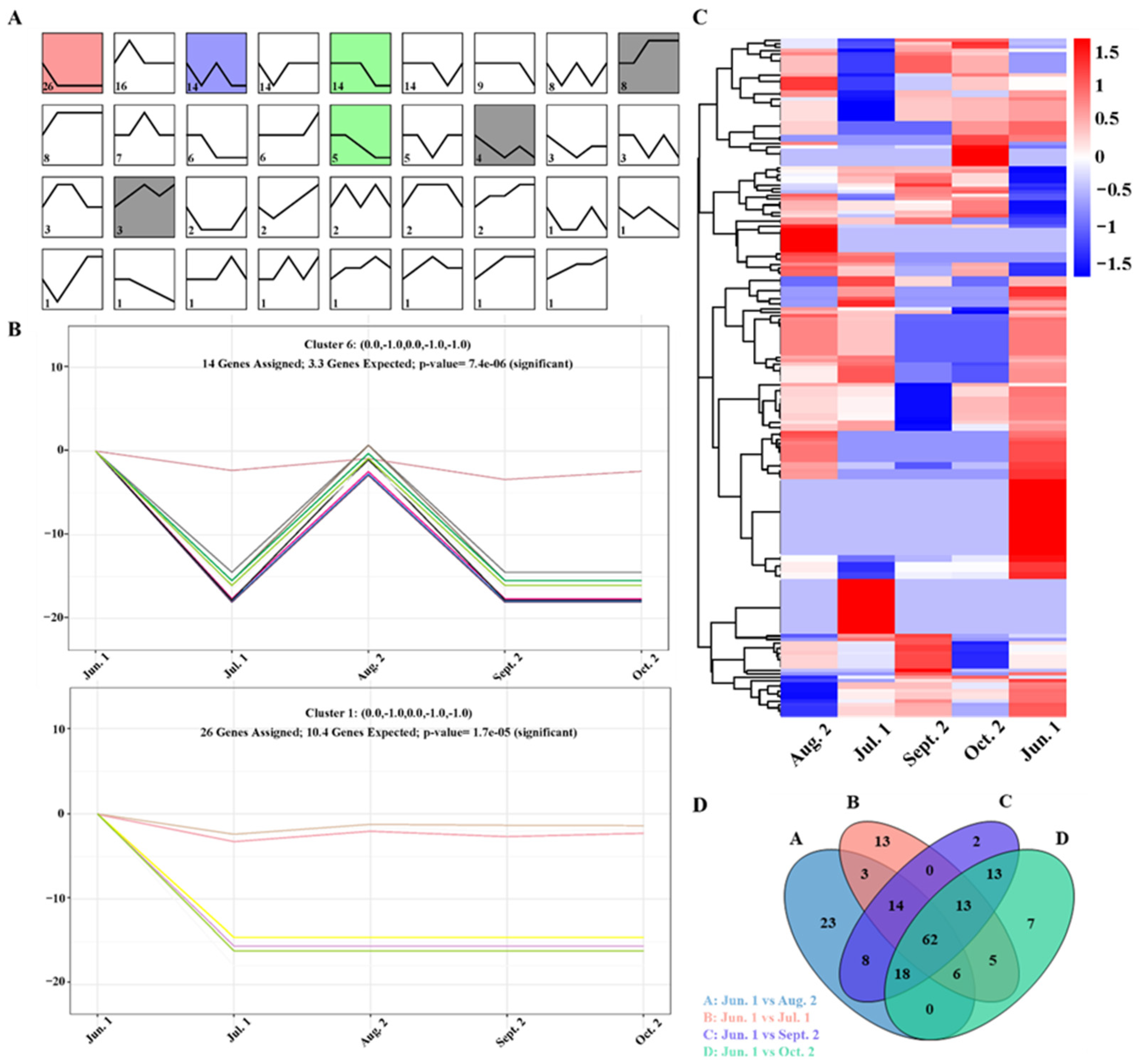
3.4. Expression Analysis of miRNAs
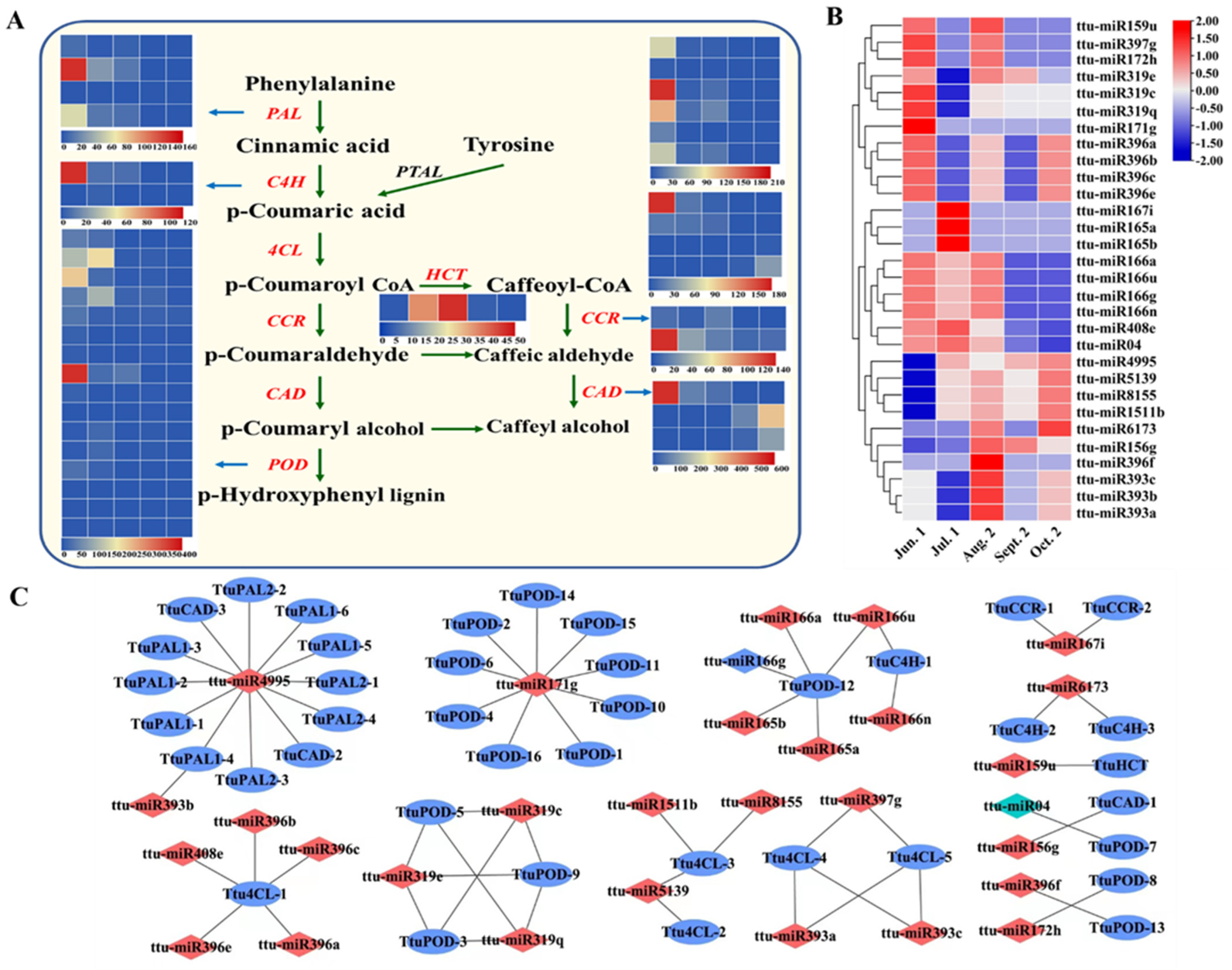
3.5. Target Prediction of miRNAs and Function Enrichment Analysis
3.6. Transcriptional Regulatory miRNA-mRNA Networks
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tang, Y.; Ren, Z. Geographical distribution of tilia linn. J. Syst. Evol. 1996, 34, 254–264. [Google Scholar]
- Yao, W.F.; Shen, Y.B. Effects of gibberellic acid and magnetically treated water on physiological characteristics of Tilia miqueliana seeds. Can. J. Res. 2018, 48, 554–558. [Google Scholar] [CrossRef] [Green Version]
- Shi, F.H.; Shen, Y.B.; Shi, J.S. Protection, development and utilization of Tilia miqueliana seeds. J. For. Eng. 2012, 26, 11–14. [Google Scholar]
- Wu, Y.; Shen, Y.B. The structural and chemical characteristics of the pericarp are important in Tilia miqueliana seed dormancy. New Forests 2021, 52, 878–888. [Google Scholar] [CrossRef]
- Wu, Y.; Shen, Y.B. Dormancy in Tilia miqueliana is attributable to permeability barriers and mechanical constraints in the endosperm and seed coat. Braz. J. Bot. 2021, 44, 725–740. [Google Scholar] [CrossRef]
- Llave, C.; Kasschau, K.D.; Rector, M.A.; Carrington, J.C. Endogenous and silencing-associated small RNAs in plants. Plant Cell 2002, 14, 1605–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Jia, T.; Chen, X. The ‘how’ and ‘where’ of plant microRNAs. New Phytol. 2017, 216, 1002–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The, C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Park, W.; Li, J.; Song, R.; Messing, J.; Chen, X. Carpel Factory, a Dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr. Biol. 2002, 12, 1484–1495. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Wang, Y.; Gruber, M.Y.; Hannoufa, A. miR156/SPL10 modulates lateral root development, branching and leaf morphology in Arabidopsis by silencingAGAMOUS-LIKE 79. Front. Plant Sci. 2017, 8, 2226. [Google Scholar] [CrossRef] [Green Version]
- Palatnik, J.F.; Allen, E.; Wu, X.; Schommer, C.; Schwab, R.; Carrington, J.C.; Weigel, D. Control of leaf morphogenesis by microRNAs. Nature 2003, 425, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Aukerman, M.J.; Sakai, H. Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell 2003, 15, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Czech, B.; Weigel, D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 2009, 138, 738–749. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Liu, Z.; Xing, L.; Wei, Y.; Mao, J.; Meng, Y.; Bao, L.; Han, M.; Zhao, C.; Zhang, D. miRNAs associated with auxin signaling, stress response, and cellular activities mediate adventitious root formation in apple rootstocks. Plant Physiol. Biochem. 2019, 139, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yang, Y.; Pan, H.; Zhu, J.; Zhu, M.; Xu, T.; Li, Z.; Dong, T. Molecular characterization and target prediction of candidate miRNAs related to abiotic stress responses and/or storage root development in Sweet Potato. Genes 2022, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.W.; Weigel, D.; Poethig, R.S. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, H.; Yu, S.; Kong, D.; Xiong, J.; Ma, X.; Chen, L.; Luo, L. Temporal responses of conserved miRNAs to drought and their associations with drought tolerance and productivity in rice. BMC Genom. 2020, 21, 232. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Xue, H.; Zhang, F.; Jiang, Q.; Yang, S.; Yue, P.; Wang, F.; Zhang, Y.; Li, L.; He, P.; et al. The miR156/SPL module regulates apple salt stress tolerance by activating MdWRKY100 expression. Plant Biotechnol. J. 2021, 19, 311–323. [Google Scholar] [CrossRef]
- Pegler, J.L.; Oultram, J.; Grof, C.; Eamens, A.L. Molecular manipulation of the miR399/PHO2 expression module alters the salt stress response of Arabidopsis thaliana. Plants 2020, 10, 73. [Google Scholar] [CrossRef]
- Niu, J.; Wang, J.; An, J.; Liu, L.; Lin, Z.; Wang, R.; Wang, L.; Ma, C.; Shi, L.; Lin, S. Integrated mRNA and miRNA transcriptome reveal a cross-talk between developing response and hormone signaling for the seed kernels of Siberian apricot. Sci. Rep. 2016, 6, 35675. [Google Scholar] [CrossRef] [Green Version]
- Mutum, R.D.; Kumar, S.; Balyan, S.; Kansal, S.; Mathur, S.; Raghuvanshi, S. Identification of novel miRNAs from drought tolerant rice variety Nagina 22. Sci. Rep. 2016, 6, 30786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Q.X.; Liu, Y.F.; Hu, X.Y.; Zhang, W.K.; Ma, B.; Chen, S.Y.; Zhang, J.S. Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biol. 2011, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Qiao, Y.; Zhang, J.; Shi, W.; Zhang, J. Genome wide identification of microRNAs involved in fatty acid and lipid metabolism of Brassica napus by small RNA and degradome sequencing. Gene 2017, 619, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Khemka, N.; Singh Rajkumar, M.; Garg, R.; Jain, M. Genome-wide profiling of miRNAs during seed development reveals their functional relevance in seed size/weight determination in chickpea. Plant Direct. 2021, 5, e00299. [Google Scholar] [CrossRef]
- Gupta, M.; Bhaskar, P.B.; Sriram, S.; Wang, P.H. Integration of omics approaches to understand oil/protein content during seed development in oilseed crops. Plant Cell Rep. 2017, 36, 637–652. [Google Scholar] [CrossRef]
- Guo, M.; Simmons, C.R. Cell number counts--the fw2.2 and CNR genes and implications for controlling plant fruit and organ size. Plant Sci. 2011, 181, 1–7. [Google Scholar] [CrossRef]
- Liu, P.P.; Montgomery, T.A.; Fahlgren, N.; Kasschau, K.D.; Nonogaki, H.; Carrington, J.C. Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J. 2007, 52, 133–146. [Google Scholar] [CrossRef]
- Liu, X.; Huang, J.; Wang, Y.; Khanna, K.; Xie, Z.; Owen, H.A.; Zhao, D. The role of floral organs in carpels, an Arabidopsis loss-of-function mutation in MicroRNA160a, in organogenesis and the mechanism regulating its expression. Plant J. 2010, 62, 416–428. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, J.K.; Shin, J.S.; Suh, M.C. The SebHLH transcription factor mediates trans-activation of the SeFAD2 gene promoter through binding to E- and G-box elements. Plant Mol. Biol. 2007, 64, 453–466. [Google Scholar] [CrossRef]
- Li, D.; Jin, C.; Duan, S.; Zhu, Y.; Qi, S.; Liu, K.; Gao, C.; Ma, H.; Zhang, M.; Liao, Y.; et al. MYB89 transcription factor represses seed oil accumulation. Plant Physiol. 2017, 173, 1211–1225. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Guo, X.; Zhai, T.; Shu, A.; Zhao, L.; Liu, Z.; Zhang, S. Genome-wide identification and characterization of microRNAs responding to ABA and GA in maize embryos during seed germination. Plant Biol. 2020, 22, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, M.; Shen, Y.; Shi, S.; Tang, T. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform. 2012, 13, 140. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Chen, J.; Li, Z.; Li, X.; Hu, X.; Huang, Y.; Zhao, X.; Liang, C.; Wang, Y.; Sun, L.; et al. Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS ONE 2010, 5, e15224. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Storey, J.D. The positive false discovery rate: A Bayesian interpretation and the q-value. Ann. Stat. 2003, 31, 2013–2035. [Google Scholar] [CrossRef]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.J.; Ma, Y.K.; Tong, C.; Wang, M.; Wang, X.J. Psrobot: A web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012, 40, W22–W28. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Jones-Rhoades, M.W.; Bartel, D.P. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell 2004, 14, 787–799. [Google Scholar] [CrossRef]
- Harvey, J.J.; Lewsey, M.G.; Patel, K.; Westwood, J.; Heimstädt, S.; Carr, J.P.; Baulcombe, D.C. An antiviral defense role of AGO2 in plants. PLoS ONE 2011, 6, e14639. [Google Scholar] [CrossRef]
- Bao, D.; Ganbaatar, O.; Cui, X.; Yu, R.; Bao, W.; Falk, B.W.; Wuriyanghan, H. Down-regulation of genes coding for core RNAi components and disease resistance proteins via corresponding microRNAs might be correlated with successful Soybean mosaic virus infection in soybean. Mol. Plant Pathol. 2018, 19, 948–960. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi Khaksefidi, R.; Mirlohi, S.; Khalaji, F.; Fakhari, Z.; Shiran, B.; Fallahi, H.; Rafiei, F.; Budak, H.; Ebrahimie, E. Differential expression of seven conserved microRNAs in response to abiotic stress and their regulatory network in Helianthus annuus. Front. Plant Sci. 2015, 6, 741. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Xu, L.; Wang, Y.; Yu, R.; Zhu, X.; Luo, X.; Gong, Y.; Wang, R.; Limera, C.; Zhang, K.; et al. Identification of novel and salt-responsive miRNAs to explore miRNA-mediated regulatory network of salt stress response in radish (Raphanus sativus L.). BMC Genom. 2015, 16, 197. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Xian, Z.; Huang, W.; Li, Z. Evidence for the biological function of miR403 in tomato development. Sci. Hortic. 2015, 197, 619–626. [Google Scholar] [CrossRef]
- Sunkar, R.; Zhou, X.; Zheng, Y.; Zhang, W.; Zhu, J.K. Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol. 2008, 8, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, M.; Wang, Y.; Yao, Y.; Xie, C.; Peng, H.; Ni, Z.; Sun, Q. Diverse set of microRNAs are responsive to powdery mildew infection and heat stress in wheat (Triticum aestivum L.). BMC Plant Biol. 2010, 10, 123. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.W.; Shi, B.J.; Huang, C.Y.; Langridge, P.; Baumann, U. Discovery of barley miRNAs through deep sequencing of short reads. BMC Genom. 2011, 12, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, C.; Wang, Z.; Zhang, L.; Yao, J.; Hua, K.; Liu, X.; Shi, H.; Zhu, J.K. The grain yield modulator miR156 regulates seed dormancy through the gibberellin pathway in rice. Nat. Commun. 2019, 10, 3822. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Koh, C.; Feurtado, J.A.; Tsang, E.W.; Cutler, A.J. MicroRNAs and their putative targets in Brassica napus seed maturation. BMC Genom. 2013, 14, 140. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.T.; Su, C.L.; Jean, W.H.; Chen, W.C.; Chang, Y.C.; Shih, M.C. Identification and characterization of the microRNA transcriptome of a moth orchid Phalaenopsis aphrodite. Plant Mol. Biol. 2014, 84, 529–548. [Google Scholar] [CrossRef] [Green Version]
- Nodine, M.D.; Bartel, D.P. MicroRNAs prevent precocious gene expression and enable pattern formation during plant embryogenesis. Genes Dev. 2010, 24, 2678–2692. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wu, K.; Yuan, Q.; Liu, X.; Liu, Z.; Lin, X.; Zeng, R.; Zhu, H.; Dong, G.; Qian, Q.; et al. Control of grain size, shape and quality by OsSPL16 in rice. Nat. Genet. 2012, 44, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, Z.; Gao, L.; Wang, L.; Gao, M.; Jiao, Z.; Qiao, H.; Yang, J.; Chen, M.; Yao, L.; et al. Genome-wide identification and characterization of microRNAs in developing grains of Zea mays L. PLoS ONE 2016, 11, e0153168. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Liu, H.; Wang, K.; Liu, L.; Wang, S.; Zhao, Y.; Yin, J.; Li, Y. Development-associated microRNAs in grains of wheat (Triticum aestivum L.). BMC Plant Biol. 2013, 13, 140. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Jian, H.; Wang, T.; Wei, L.; Li, J.; Li, C.; Liu, L. Identification of microRNAs actively involved in fatty acid biosynthesis in developing Brassica napus Seeds Using High-Throughput Sequencing. Front. Plant Sci. 2016, 7, 1570. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Bian, S.; Tang, M.; Lu, Q.; Li, S.; Liu, X.; Tian, G.; Nguyen, V.; Tsang, E.W.; Wang, A.; et al. MicroRNA-mediated repression of the seed maturation program during vegetative development in Arabidopsis. PLoS Genet. 2012, 8, e1003091. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Wei, K.; Wang, M.; Wang, L.; Cui, J.; Zhang, D.; Guo, J.; Zhao, M.; Zheng, Y. Identification and temporal expression analysis of conserved and novel MicroRNAs in the leaves of winter wheat grown in the field. Front Genet. 2019, 10, 779. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.D.; Ohlrogge, J.B. Compartmentation of triacylglycerol accumulation in plants. J. Biol. Chem. 2012, 287, 2288–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, P.D.; Stymne, S.; Ohlrogge, J. Biochemical pathways in seed oil synthesis. Curr. Opin. Plant Biol. 2013, 16, 358–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlrogge, J.B.; Jaworski, J.G. Regulation of fatty acid synthesis. Annu. Rev. Plant Physiol. Plant Mol. Biol 1997, 48, 109–136. [Google Scholar] [CrossRef] [Green Version]
- Li-Beisson, Y.; Shorrosh, B.; Beisson, F.; Andersson, M.X.; Arondel, V.; Bates, P.D.; Baud, S.; Bird, D.; Debono, A.; Durrett, T.P.; et al. Acyl-lipid metabolism. Arab. Book 2013, 11, e0161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, D.D.; Li, S.S.; Shu, Q.Y.; Gu, Z.Y.; Wu, Q.; Feng, C.Y.; Xu, W.Z.; Wang, L.S. Identification of microRNAs and long non-coding RNAs involved in fatty acid biosynthesis in tree peony seeds. Gene 2018, 666, 72–82. [Google Scholar] [CrossRef]
- Yang, Q.; Liu, S.; Han, X.; Ma, J.; Deng, W.; Wang, X.; Guo, H.; Xia, X. Integrated transcriptome and miRNA analysis uncovers molecular regulators of aerial stem-to-rhizome transition in the medical herb Gynostemma pentaphyllum. BMC Genom. 2019, 20, 865. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Jung, J.H.; Reyes, J.L.; Kim, Y.S.; Kim, S.Y.; Chung, K.S.; Kim, J.A.; Lee, M.; Lee, Y.; Narry Kim, V.; et al. microRNA-directed cleavage of ATHB15 mRNA regulates vascular development in Arabidopsis inflorescence stems. Plant J. 2005, 42, 84–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.H.; Park, C.M. MIR166/165 genes exhibit dynamic expression patterns in regulating shoot apical meristem and floral development in Arabidopsis. Planta 2007, 225, 1327–1338. [Google Scholar] [CrossRef]
- Han, S.Y.; Li, S.G.; Wan, F.; Yang, W.H.; Li, Y.L.; Xu, H.Y. Over-expression of miR166a inhibits cotyledon formation in somatic embryos and promotes lateral root development in seedlings of Larix leptolepis. Plant Cell Tiss. Org. 2016, 127, 461–473. [Google Scholar] [CrossRef]
- Llave, C.; Xie, Z.; Kasschau, K.D.; Carrington, J.C. Cleavage of scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science 2002, 297, 2053–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolle, C. The role of GRAS proteins in plant signal transduction and development. Planta 2004, 218, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Kim, B.; Song, S.K.; Heo, J.O.; Yu, N.I.; Lee, S.A.; Kim, M.; Kim, D.G.; Sohn, S.O.; Lim, C.E.; et al. Large-scale analysis of the GRAS gene family in Arabidopsis thaliana. Plant Mol. Biol. 2008, 67, 659–670. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Mai, Y.X.; Zhang, Y.C.; Luo, Q.; Yang, H.Q. MicroRNA171c-targeted SCL6-II, SCL6-III, and SCL6-IV genes regulate shoot branching in Arabidopsis. Mol. Plant 2010, 3, 794–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, A.; Godin, B.; Bonnet, M.; Sotta, B.; Marion-Poll, A. Maternal synthesis of abscisic acid controls seed development and yield in Nicotiana plumbaginifolia. Planta 2004, 218, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, L.; Liu, X.; Cui, D.; Chen, T.; Zhang, H.; Jiang, C.; Xu, C.; Li, P.; Li, S.; et al. Deep sequencing of maize small RNAs reveals a diverse set of MicroRNA in dry and imbibed seeds. PLoS ONE 2013, 8, e55107. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, R.; Reeves, W.; Ariizumi, T.; Steber, C. Molecular aspects of seed dormancy. Annu. Rev. Plant Biol. 2008, 59, 387–415. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, V.; North, H.; Frey, A.; Sotta, B.; Seo, M.; Okamoto, M.; Nambara, E.; Marion-Poll, A. Functional analysis of Arabidopsis NCED6 and NCED9 genes indicates that ABA synthesized in the endosperm is involved in the induction of seed dormancy. Plant J. 2006, 45, 309–319. [Google Scholar] [CrossRef]
- Turner, M.; Nizampatnam, N.R.; Baron, M.; Coppin, S.; Damodaran, S.; Adhikari, S.; Arunachalam, S.P.; Yu, O.; Subramanian, S. Ectopic expression of miR160 results in auxin hypersensitivity, cytokinin hyposensitivity, and inhibition of symbiotic nodule development in soybean. Plant Physiol. 2013, 162, 2042–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinweha, N.; Asvarak, T.; Viboonjun, U.; Narangajavana, J. Involvement of miR160/miR393 and their targets in cassava responses to anthracnose disease. J. Plant Physiol. 2015, 174, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.C.; Bartel, D.P.; Bartel, B. MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell 2005, 17, 1360–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, C.; Zuo, Z.; Qiu, J.L. Identification and characterization of ABA-responsive MicroRNAs in rice. J. Genet. Genom. 2015, 42, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Zhang, L.; Wang, H.; Liu, Z.; Zhang, Z.; Zheng, Y. Differential expression of miRNAs in response to salt stress in maize roots. Ann. Bot. 2009, 103, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, G.; Stewart, C.N., Jr.; Xiao, P.; Zhang, B. MicroRNA expression analysis in the cellulosic biofuel crop switchgrass (Panicum virgatum) under abiotic stress. PLoS ONE 2012, 7, e32017. [Google Scholar] [CrossRef] [Green Version]
- Salih, H.; Gong, W.; Mkulama, M.; Du, X. Genome-wide characterization, identification, and expression analysis of the WD40 protein family in cotton. Genome 2018, 61, 539–547. [Google Scholar] [CrossRef]
- Chen, J.; Zheng, Y.; Qin, L.; Wang, Y.; Chen, L.; He, Y.; Fei, Z.; Lu, G. Identification of miRNAs and their targets through high-throughput sequencing and degradome analysis in male and female asparagus officinalis. BMC Plant Bio. 2016, 16, 80. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Hu, X.; Cai, W.; Huang, W.; Zhou, X.; Luo, Q.; Yang, H.; Wang, J.; Huang, J. Arabidopsis miR171-targeted scarecrow-like proteins bind to GT cis-elements and mediate gibberellin-regulated chlorophyll biosynthesis under light conditions. PLoS Genet. 2014, 10, e1004519. [Google Scholar] [CrossRef] [Green Version]
- Novaes, E.; Kirst, M.; Chiang, V.; Winter-Sederoff, H.; Sederoff, R. Lignin and biomass: A negative correlation for wood formation and lignin content in trees. Plant Physiol. 2010, 154, 555–561. [Google Scholar] [CrossRef] [Green Version]
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Li, Q.; Wei, H.; Chang, M.J.; Tunlaya-Anukit, S.; Kim, H.; Liu, J.; Song, J.; Sun, Y.H.; Yuan, L.; et al. Ptr-miR397a is a negative regulator of laccase genes affecting lignin content in Populus trichocarpa. Proc. Natl. Acad. Sci. USA 2013, 110, 10848–10853. [Google Scholar] [CrossRef]
- Wang, C.Y.; Zhang, S.; Yu, Y.; Luo, Y.C.; Liu, Q.; Ju, C.; Zhang, Y.C.; Qu, L.H.; Lucas, W.J.; Wang, X.; et al. MiR397b regulates both lignin content and seed number in Arabidopsis via modulating a laccase involved in lignin biosynthesis. Plant Biotechnol. J. 2014, 12, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, B.; Shen, W.H.; Huang, H.; Dong, A. TCP transcription factors interact with AS2 in the repression of class-I KNOX genes in Arabidopsis thaliana. Plant J. 2012, 71, 99–107. [Google Scholar] [CrossRef]
- Schommer, C.; Palatnik, J.F.; Aggarwal, P.; Chételat, A.; Cubas, P.; Farmer, E.E.; Nath, U.; Weigel, D. Control of jasmonate biosynthesis and senescence by miR319 targets. PLoS Biol. 2008, 6, e230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nag, A.; King, S.; Jack, T. miR319a targeting of TCP4 is critical for petal growth and development in Arabidopsis. Proc. Natl. Acad. Sci. USA 2009, 106, 22534–22539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.; Zhang, Z.M.; Gao, J.; Zeng, X.; Pan, G.T. The role of miR319 in plant development regulation. Hereditas 2011, 33, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Schommer, C.; Bresso, E.G.; Spinelli, S.V.; Palatnik, J.F. Role of microRNA miR319 in plant development. Signal. Commun. Plants 2012, 15, 29–47. [Google Scholar] [CrossRef]
- Ori, N.; Cohen, A.R.; Etzioni, A.; Brand, A.; Yanai, O.; Shleizer, S.; Menda, N.; Amsellem, Z.; Efroni, I.; Pekker, I.; et al. Regulation of LANCEOLATE by miR319 is required for compound-leaf development in tomato. Nat. Genet. 2007, 39, 787–791. [Google Scholar] [CrossRef]
- Sun, X.; Wang, C.; Xiang, N.; Li, X.; Yang, S.; Du, J.; Yang, Y.; Yang, Y. Activation of secondary cell wall biosynthesis by miR319-targeted TCP4 transcription factor. Plant Biotechnol. J. 2017, 15, 1284–1294. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Jun. 1 | Jul. 1 | Aug. 2 | Sept. 2 | Oct. 2 |
|---|---|---|---|---|---|
| Raw reads | 11,780,752 | 15,366,592 | 13,279,485 | 12,378,261 | 11,573,778 |
| clean reads | 10,918,208 | 14,491,495 | 12,750,811 | 10,816,291 | 10,274,213 |
| Total sRNA | 4,551,867 | 11,994,904 | 7,338,458 | 8,004,524 | 6,323,645 |
| Mapped sRNA | 1,576,482 | 7,945,688 | 4,785,383 | 6,154,905 | 5,191,238 |
| Types | Jun. 1 | Jun. 1 (Percent) | Jul. 1 | Jul. 1 (Percent) | Aug. 2 | Aug. 2 (Percent) | Sept. 2 | Sept. 2 (Percent) | Oct. 2 | Oct. 2 (Percent) |
|---|---|---|---|---|---|---|---|---|---|---|
| total | 1,576,482 | 100.00% | 7,945,688 | 100.00% | 4,785,383 | 100.00% | 6,154,905 | 100.00% | 5,191,238 | 100.00% |
| Known miRNA | 14,225 | 0.90% | 9212 | 0.12% | 6197 | 0.13% | 6381 | 0.10% | 4432 | 0.09% |
| rRNA | 551,669 | 34.99% | 2,946,068 | 37.08% | 1,417,839 | 29.63% | 2,168,153 | 35.23% | 1,116,058 | 21.50% |
| tRNA | 0 | 0.00% | 1 | 0.00% | 0 | 0.00% | 0 | 0.00% | 4 | 0.00% |
| snRNA | 2162 | 0.14% | 4574 | 0.06% | 2940 | 0.06% | 1507 | 0.02% | 2048 | 0.04% |
| snoRNA | 2248 | 0.14% | 2374 | 0.03% | 1524 | 0.03% | 1432 | 0.02% | 1271 | 0.02% |
| Novel miRNA | 24,604 | 1.56% | 28,171 | 0.35% | 23,649 | 0.49% | 9276 | 0.15% | 10,268 | 0.20% |
| other | 981,574 | 62.26% | 4,955,288 | 62.36% | 3,333,234 | 69.65% | 3,968,156 | 64.47% | 4,057,157 | 78.15% |
| Types | Total | Jun. 1 | Jul. 1 | Aug. 2 | Sept. 2 | Oct. 2 |
|---|---|---|---|---|---|---|
| Mapped mature | 189 | 153 | 127 | 127 | 85 | 105 |
| Mapped hairpin | 487 | 405 | 372 | 378 | 256 | 304 |
| Mapped uniq sRNA | 1935 | 329 | 409 | 418 | 366 | 413 |
| Mapped total sRNA | 40,447 | 14,225 | 9212 | 6197 | 6381 | 4432 |
| Types | Total | Jun. 1 | Jul. 1 | Aug. 2 | Sept. 2 | Oct. 2 |
|---|---|---|---|---|---|---|
| Mapped mature | 8 | 8 | 8 | 8 | 6 | 6 |
| Mapped star | 6 | 5 | 4 | 3 | 1 | 2 |
| Mapped hairpin | 8 | 8 | 8 | 8 | 6 | 7 |
| Mapped uniq sRNA | 2458 | 307 | 476 | 637 | 411 | 627 |
| Mapped total sRNA | 95,968 | 24,604 | 28,171 | 23,649 | 9276 | 10,268 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, X.; Liu, L.; Liu, P.; Wang, M.; Song, Y. Genome-Wide Identification of miRNAs and Its Downstream Transcriptional Regulatory Network during Seed Maturation in Tilia tuan. Forests 2022, 13, 1750. https://doi.org/10.3390/f13111750
Hao X, Liu L, Liu P, Wang M, Song Y. Genome-Wide Identification of miRNAs and Its Downstream Transcriptional Regulatory Network during Seed Maturation in Tilia tuan. Forests. 2022; 13(11):1750. https://doi.org/10.3390/f13111750
Chicago/Turabian StyleHao, Xuri, Lei Liu, Peng Liu, Menglei Wang, and Yuepeng Song. 2022. "Genome-Wide Identification of miRNAs and Its Downstream Transcriptional Regulatory Network during Seed Maturation in Tilia tuan" Forests 13, no. 11: 1750. https://doi.org/10.3390/f13111750
APA StyleHao, X., Liu, L., Liu, P., Wang, M., & Song, Y. (2022). Genome-Wide Identification of miRNAs and Its Downstream Transcriptional Regulatory Network during Seed Maturation in Tilia tuan. Forests, 13(11), 1750. https://doi.org/10.3390/f13111750