
Development and Validation of a 36K SNP Array for Radiata Pine (Pinus radiata D.Don)

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Development of the SNP Screening Array
Candidate SNPs
2.2. Plant Material for Testing the Screening Array
2.3. Genotyping Array Design and Testing
2.3.1. SNP Selection
2.3.2. Plant Material for Testing on the Genotyping Array
2.4. Genotyping Array Data Analysis
Population Structure Analysis
3. Results
3.1. Design and Performance of the Screening Array
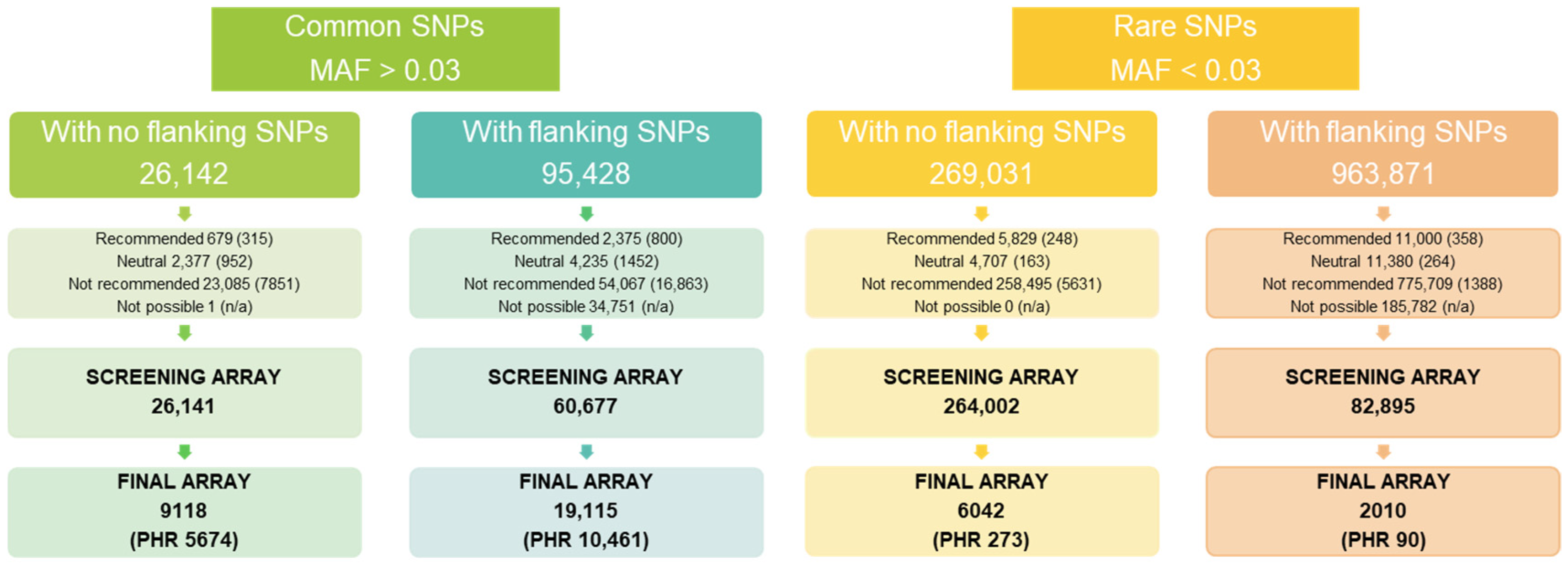
3.1.1. SNP Selection
3.1.2. Screening Array Genotyping
3.2. Design and Performance of the Genotyping 50K Array
3.2.1. Selection of SNPs for the Genotyping Array
3.2.2. Sample and SNP Performance
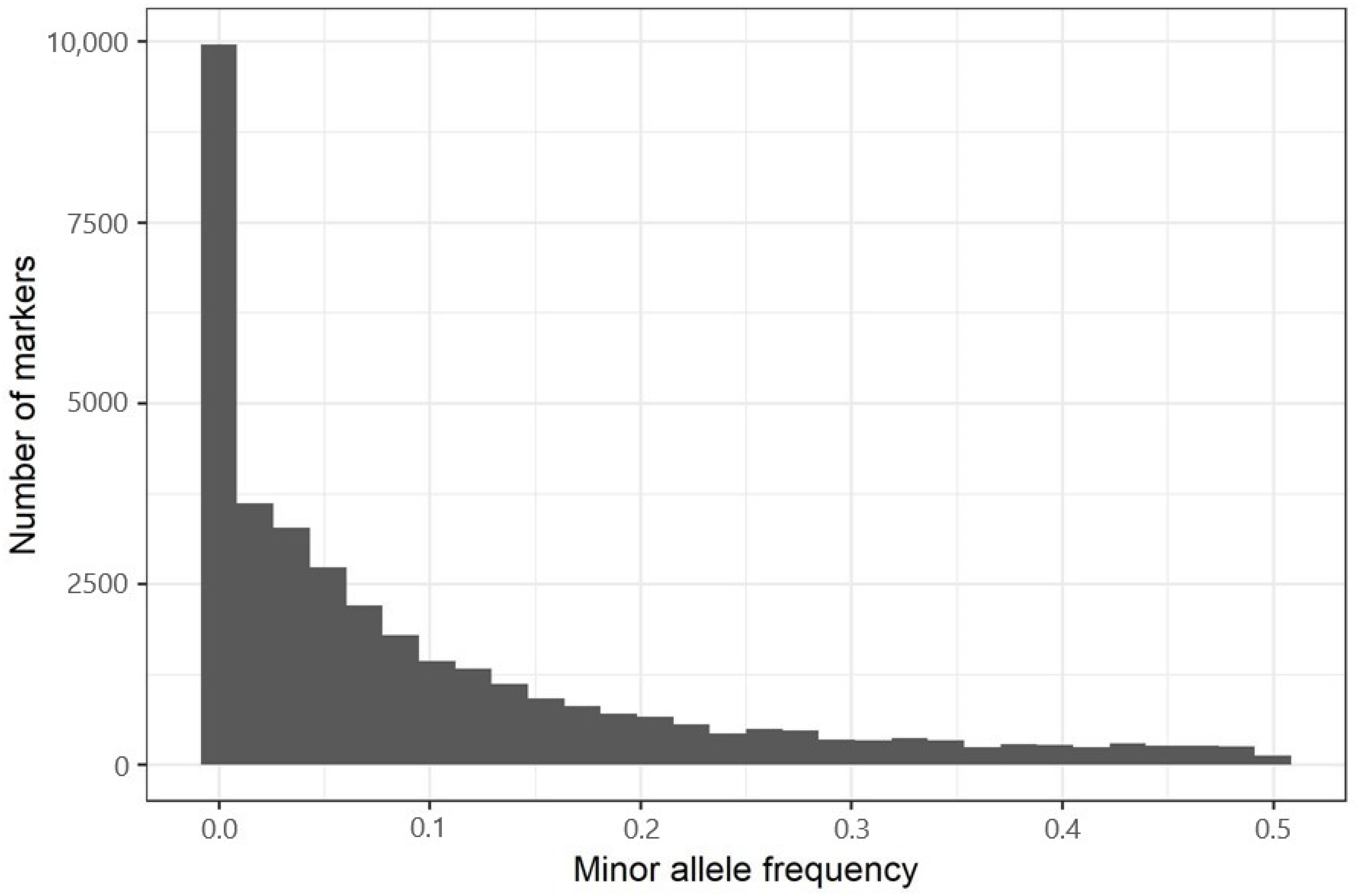
3.2.3. Minor Allele Frequencies and Heterozygosity
3.2.4. Detection of Contaminating DNA
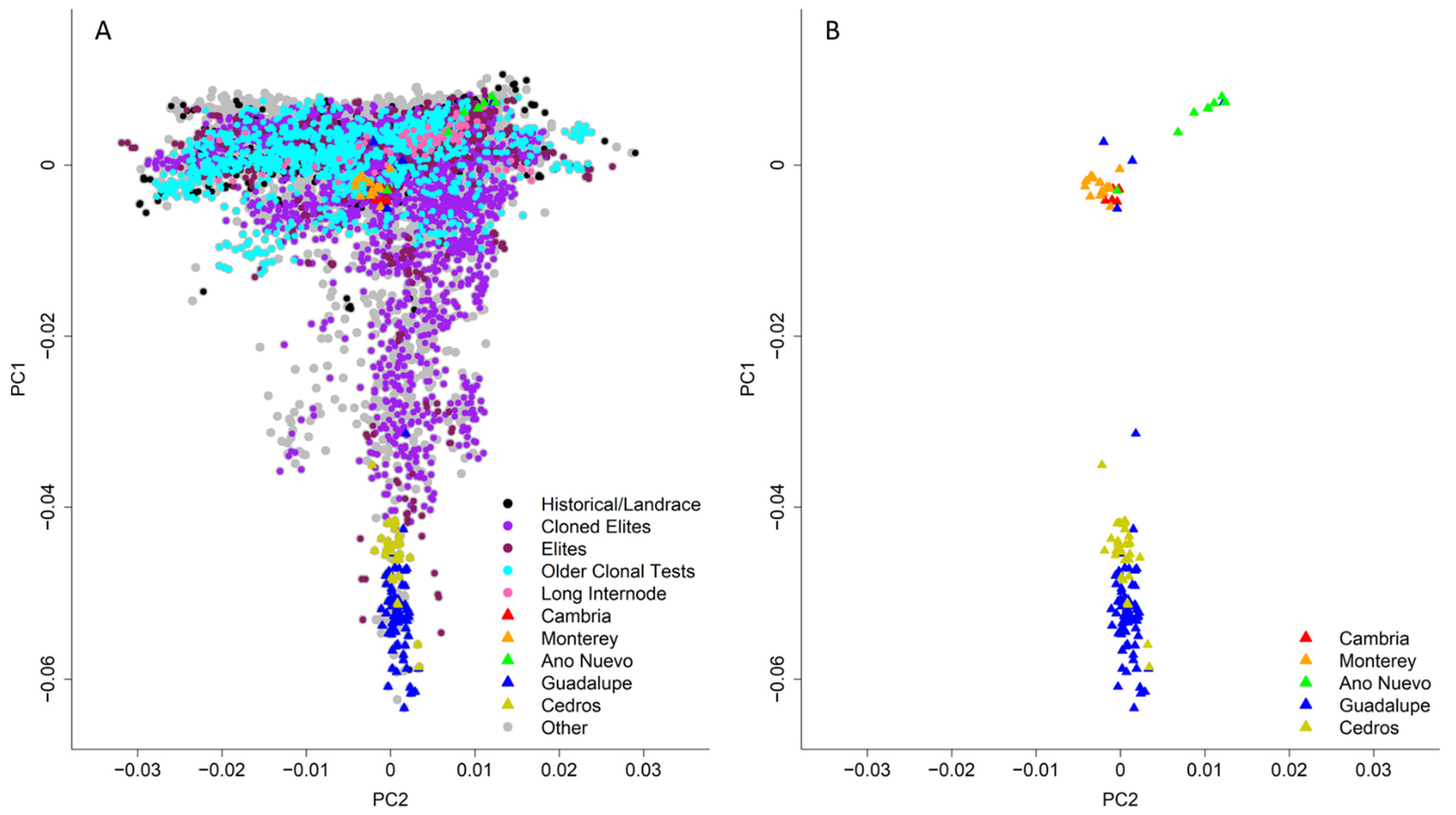
3.2.5. Native Provenance Performance
3.3. Population Structure
4. Discussion
4.1. Performance of the Genotyping Array
4.2. Native Provenances
4.3. Population Structure within the Breeding Program
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grattapaglia, D.; Silva-Junior, O.B.; Resende, R.T.; Cappa, E.P.; Müller, B.S.; Tan, B.; Isik, F.; Ratcliffe, B.; El-Kassaby, Y.A. Quantitative genetics and genomics converge to accelerate forest tree breeding. Front. Plant Sci. 2018, 9, 1693. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dungey, H.S. Expected benefit of genomic selection over forward selection in conifer breeding and deployment. PLoS ONE 2018, 13, e0208232. [Google Scholar] [CrossRef] [Green Version]
- Suontama, M.; Klápště, J.; Telfer, E.; Graham, N.; Stovold, T.; Low, C.; McKinley, R.; Dungey, H. Efficiency of genomic prediction across two Eucalyptus nitens seed orchards with different selection histories. Heredity 2019, 122, 370–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isik, F.; Bartholome, J.; Farjat, A.; Chancerel, E.; Raffin, A.; Sanchez, L.; Plomion, C.; Bouffier, L. Genomic selection in maritime pine. Plant Sci. 2016, 242, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Food and Agriculture Organization of the United Nations. Available online: http://www.fao.org/3/i0350e/i0350e02a.pdf (accessed on 15 December 2021).
- McEwan, A.; Marchi, E.; Spinelli, R.; Brink, M. Past, present and future of industrial plantation forestry and implication on future timber harvesting technology. J. For. Res. 2020, 31, 339–351. [Google Scholar] [CrossRef] [Green Version]
- Neagoe, M.; Taskhiri, M.; Turner, P. North and North-West Tasmania: Supply Chain and Infrastructure; Northern Tasmania Regional Forestry Hub: Hobart, Tasmania, 2020. [Google Scholar]
- Brooks, D.J. The outlook for demand and supply of wood: Implications for policy and sustainable management. Commonw. For. Rev. 1997, 76, 31–36. [Google Scholar]
- Bastin, J.-F.; Finegold, Y.; Garcia, C.; Mollicone, D.; Rezende, M.; Routh, D.; Zohner, C.M.; Crowther, T.W. The global tree restoration potential. Science 2019, 365, 76–79. [Google Scholar] [CrossRef]
- Bastin, J.-F.; Finegold, Y.; Garcia, C.; Mollicone, D.; Rezende, M.; Routh, D.; Zohner, C.M.; Crowther, T.W. Erratum for the Report: “The global tree restoration potential” by J.-F. Bastin, Y. Finegold, C. Garcia, D. Mollicone, M. Rezende, D. Routh, CM Zohner, TW Crowther and for the Technical Response “Response to Comments on ‘The global tree restoration potential’” by J.-F. Bastin, Y. Finegold, C. Garcia, N. Gellie, A. Lowe, D. Mollicone, M. Rezende, D. Routh, M. Sacande, B. Sparrow, C.M. Zohner, T.W. Crowther. Science 2020, 368, eabc8905. [Google Scholar]
- Cook-Patton, S.C.; Leavitt, S.M.; Gibbs, D.; Harris, N.L.; Lister, K.; Anderson-Teixeira, K.J.; Briggs, R.D.; Chazdon, R.L.; Crowther, T.W.; Ellis, P.W.; et al. Mapping carbon accumulation potential from global natural forest regrowth. Nature 2020, 585, 545–550. [Google Scholar] [CrossRef]
- Domke, G.M.; Oswalt, S.N.; Walters, B.F.; Morin, R.S. Tree planting has the potential to increase carbon sequestration capacity of forests in the United States. Proc. Natl. Acad. Sci. USA 2020, 117, 24649–24651. [Google Scholar] [CrossRef]
- New Zealand Forest Owners Association. New Zealand Plantation Forest Industry Facts and Figures 2019/2020. Available online: https://www.nzfoa.org.nz/images/Facts_Figures_2019_20_Web_FA3-updated.pdf (accessed on 15 December 2021).
- Burdon, R.; Libby, W.; Brown, A. Domestication of Radiata Pine; Springer: Berlin/Heidelberg, Germany, 2017; Volume 83. [Google Scholar]
- Telfer, E.; Graham, N.; Macdonald, L.; Li, Y.; Klápště, J.; Resende, M., Jr.; Neves, L.G.; Dungey, H.; Wilcox, P. A high-density exome capture genotype-by-sequencing panel for forestry breeding in Pinus radiata. PLoS ONE 2019, 14, e0222640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telfer, E.; Graham, N.; Macdonald, L.; Sturrock, S.; Wilcox, P.; Stanbra, L. Approaches to variant discovery for conifer transcriptome sequencing. PLoS ONE 2018, 13, e0205835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klápště, J.; Dungey, H.S.; Telfer, E.J.; Suontama, M.; Graham, N.J.; Li, Y.; McKinley, R. Marker Selection in Multivariate Genomic Prediction Improves Accuracy of Low Heritability Traits. Front. Genet. 2020, 11, 499094. [Google Scholar] [CrossRef]
- Klápště, J.; Dungey, H.S.; Graham, N.J.; Telfer, E.J. Effect of trait’s expression level on single-step genomic evaluation of resistance to Dothistroma needle blight. BMC Plant Biol. 2020, 20, 205. [Google Scholar] [CrossRef] [PubMed]
- Neves, L.G.; Davis, J.M.; Barbazuk, W.B.; Kirst, M. A high-density gene map of loblolly pine (Pinus taeda L.) based on exome sequence capture genotyping. G3 Genes Genomes Genet. 2014, 4, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.J.; Fahrenkrog, A.M.; Neves, L.G.; Resende, M.F.; Dervinis, C.; Davis, J.M.; Holliday, J.A.; Kirst, M. Exome resequencing reveals evolutionary history, genomic diversity, and targets of selection in the conifers Pinus taeda and Pinus Elliottii. Genome Biol. Evol. 2019, 11, 508–520. [Google Scholar] [CrossRef] [Green Version]
- Suren, H.; Hodgins, K.; Yeaman, S.; Nurkowski, K.; Smets, P.; Rieseberg, L.H.; Aitken, S.N.; Holliday, J.A. Exome capture from the spruce and pine giga-genomes. Mol. Ecol. Resour. 2016, 16, 1136–1146. [Google Scholar] [CrossRef]
- Azaiez, A.; Pavy, N.; Gérardi, S.; Laroche, J.; Boyle, B.; Gagnon, F.; Mottet, M.-J.; Beaulieu, J.; Bousquet, J. A catalog of annotated high-confidence SNPs from exome capture and sequencing reveals highly polymorphic genes in Norway spruce (Picea abies). BMC Genom. 2018, 19, 942. [Google Scholar] [CrossRef]
- Neves, L.G.; Davis, J.M.; Barbazuk, W.B.; Kirst, M. Whole-exome targeted sequencing of the uncharacterized pine genome. Plant J. 2013, 75, 146–156. [Google Scholar] [CrossRef]
- Liu, J.J.; Schoettle, A.W.; Sniezko, R.A.; Yao, F.; Zamany, A.; Williams, H.; Rancourt, B. Limber pine (Pinus flexilis James) genetic map constructed by exome-seq provides insight into the evolution of disease resistance and a genomic resource for genomics-based breeding. Plant J. 2019, 98, 745–758. [Google Scholar] [CrossRef]
- Rellstab, C.; Dauphin, B.; Zoller, S.; Brodbeck, S.; Gugerli, F. Using transcriptome sequencing and pooled exome capture to study local adaptation in the giga-genome of Pinus Cembra. Mol. Ecol. Resour. 2019, 19, 536–551. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Klápště, J.; Telfer, E.; Wilcox, P.; Graham, N.; Macdonald, L.; Dungey, H.S. Genomic selection for non-key traits in radiata pine when the documented pedigree is corrected using DNA marker information. BMC Genom. 2019, 20, 1026. [Google Scholar] [CrossRef] [PubMed]
- Bernhardsson, C.; Zan, Y.; Chen, Z.; Ingvarsson, P.K.; Wu, H.X. Development of a highly efficient 50K SNP genotyping array for the large and complex genome of Norway spruce (Picea abies L. Karst) by whole genome re-sequencing and its transferability to other spruce species. Mol. Ecol. Resour. 2020, 21, 880–896. [Google Scholar]
- Silva-Junior, O.B.; Faria, D.A.; Grattapaglia, D. A flexible multi-species genome-wide 60K SNP chip developed from pooled resequencing of 240 Eucalyptus tree genomes across 12 species. New Phytol. 2015, 206, 1527–1540. [Google Scholar] [CrossRef] [Green Version]
- Müller, B.S.; Neves, L.G.; de Almeida Filho, J.E.; Resende, M.F.; Muñoz, P.R.; dos Santos, P.E.; Paludzyszyn Filho, E.; Kirst, M.; Grattapaglia, D. Genomic prediction in contrast to a genome-wide association study in explaining heritable variation of complex growth traits in breeding populations of Eucalyptus. BMC Genom. 2017, 18, 524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ukrainetz, N.K.; Mansfield, S.D. Assessing the sensitivities of genomic selection for growth and wood quality traits in lodgepole pine using Bayesian models. Tree Genet. Genomes 2020, 16, 14. [Google Scholar] [CrossRef]
- Müller, B.S.; de Almeida Filho, J.E.; Lima, B.M.; Garcia, C.C.; Missiaggia, A.; Aguiar, A.M.; Takahashi, E.; Kirst, M.; Gezan, S.A.; Silva-Junior, O.B. Independent and Joint-GWAS for growth traits in Eucalyptus by assembling genome-wide data for 3373 individuals across four breeding populations. New Phytol. 2019, 221, 818–833. [Google Scholar] [CrossRef] [Green Version]
- Dubos, C.; Plomion, C. Identification of water-deficit responsive genes in maritime pine (Pinus pinaster Ait.) roots. Plant Mol. Biol. 2003, 51, 249–262. [Google Scholar] [CrossRef]
- Baison, J.; Vidalis, A.; Zhou, L.; Chen, Z.Q.; Li, Z.; Sillanpää, M.J.; Bernhardsson, C.; Scofield, D.; Forsberg, N.; Grahn, T. Genome-wide association study identified novel candidate loci affecting wood formation in Norway spruce. Plant J. 2019, 100, 83–100. [Google Scholar] [CrossRef]
- Rellstab, C.; Gugerli, F.; Eckert, A.J.; Hancock, A.M.; Holderegger, R. A practical guide to environmental association analysis in landscape genomics. Mol. Ecol. 2015, 24, 4348–4370. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Wang, J.; Hu, Q.; Li, J.; Sun, Y.; Zhang, L.; Abbott, R.J.; Liu, J.; Mao, K. Ancient introgression drives adaptation to cooler and drier mountain habitats in a cypress species complex. Commun. Biol. 2019, 2, 213. [Google Scholar] [CrossRef] [PubMed]
- Doerksen, T.K.; Herbinger, C.M. Impact of reconstructed pedigrees on progeny-test breeding values in red spruce. Tree Genet. Genomes 2010, 6, 591–600. [Google Scholar] [CrossRef]
- Cros, D.; Sánchez, L.; Cochard, B.; Samper, P.; Denis, M.; Bouvet, J.-M.; Fernández, J. Estimation of genealogical coancestry in plant species using a pedigree reconstruction algorithm and application to an oil palm breeding population. Theor. Appl. Genet. 2014, 127, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Resende, M.; Munoz, P.; Acosta, J.; Peter, G.; Davis, J.; Grattapaglia, D.; Resende, M.; Kirst, M. Accelerating the domestication of trees using genomic selection: Accuracy of prediction models across ages and environments. New Phytol. 2012, 193, 617–624. [Google Scholar] [CrossRef] [Green Version]
- Bartholomé, J.; Van Heerwaarden, J.; Isik, F.; Boury, C.; Vidal, M.; Plomion, C.; Bouffier, L. Performance of genomic prediction within and across generations in maritime pine. BMC Genom. 2016, 17, 604. [Google Scholar] [CrossRef]
- Meuwissen, T.H.; Sonesson, A.K.; Gebregiwergis, G.; Woolliams, J.A. Management of Genetic Diversity in the Era of Genomics. Front. Genet. 2020, 11, 880. [Google Scholar] [CrossRef]
- Isik, F.; Acosta, J.J.; Eckert, A.J.; Sniezko, R.; Wegrzyn, J. Pine SNP Chip Consortium: Progress on Pine SNP Discovery and Array Design in Loblolly Pine. In Proceedings of the Plant and Animal Genome XXVI Conference (PAG 2018), San Diego, CA, USA, 13–17 January 2018. [Google Scholar]
- Caballero, M.; Lauer, E.; Bennett, J.; Zaman, S.; McEvoy, S.; Acosta, J.; Jackson, C.; Townsend, L.; Eckert, A.; Whetten, R.W. Toward genomic selection in Pinus taeda: Integrating resources to support array design in a complex conifer genome. Appl. Plant Sci. 2021, 9, e11439. [Google Scholar] [CrossRef]
- Jackson, C.; Christie, N.; Reynolds, S.M.; Marais, G.C.; Tiikuzu, Y.; Caballero, M.; Kampman, T.; Visser, E.A.; Naidoo, S.; Kain, D. A genome-wide SNP genotyping resource for tropical pine tree species. Mol. Ecol. Resour. 2021, 22, 695–710. [Google Scholar] [CrossRef]
- Telfer, E.J.; Graham, N.; Stanbra, L.K.; Manley, T.; Wilcox, P.L. Extraction of high purity genomic DNA from pine for use in a high-throughput Genotyping Platform. N. Z. J. For. Sci. 2013, 43, 3. [Google Scholar] [CrossRef] [Green Version]
- Patterson, N.; Price, A.L.; Reich, D. Population Structure and Eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- Raj, A.; Stephens, M.; Pritchard, J.K. fastSTRUCTURE: Variational inference of population structure in large SNP data sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimin, A.; Stevens, K.A.; Crepeau, M.W.; Holtz-Morris, A.; Koriabine, M.; Marçais, G.; Puiu, D.; Roberts, M.; Wegrzyn, J.L.; de Jong, P.J.; et al. Sequencing and assembly of the 22-Gb loblolly pine genome. Genetics 2014, 196, 875–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdon, R.; Broekhuizen, P.; Zabkiewicz, J. Comparison of native-population and New Zealand land-race samples of Pinus radiata using cortical oleoresin monoterpenes. FRI Bull. 1997, 203, 50–56. [Google Scholar]
- Perry, A.; Wachowiak, W.; Downing, A.; Talbot, R.; Cavers, S. Development of a single nucleotide polymorphism array for population genomic studies in four European pine species. Mol. Ecol. Resour. 2020, 20, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Plomion, C.; Bartholomé, J.; Lesur, I.; Boury, C.; Rodríguez-Quilón, I.; Lagraulet, H.; Ehrenmann, F.; Bouffier, L.; Gion, J.M.; Grivet, D.; et al. High-density SNP assay development for genetic analysis in maritime pine (Pinus pinaster). Mol. Ecol. Resour. 2016, 16, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Howe, G.T.; Jayawickrama, K.; Kolpak, S.E.; Kling, J.; Trappe, M.; Hipkins, V.; Ye, T.; Guida, S.; Cronn, R.; Cushman, S.A.; et al. An Axiom SNP genotyping array for Douglas-fir. BMC Genom. 2020, 21, 9. [Google Scholar] [CrossRef] [Green Version]
- Mimura, M.; Aitken, S. Adaptive gradients and isolation-by-distance with postglacial migration in Picea Sitchensis. Heredity 2007, 99, 224–232. [Google Scholar] [CrossRef] [Green Version]
- Vitasse, Y.; Delzon, S.; Bresson, C.C.; Michalet, R.; Kremer, A. Altitudinal differentiation in growth and phenology among populations of temperate-zone tree species growing in a common garden. Can. J. For. Res. 2009, 39, 1259–1269. [Google Scholar] [CrossRef] [Green Version]
- Lopes, M.S.; El-Basyoni, I.; Baenziger, P.S.; Singh, S.; Royo, C.; Ozbek, K.; Aktas, H.; Ozer, E.; Ozdemir, F.; Manickavelu, A.; et al. Exploiting genetic diversity from landraces in wheat breeding for adaptation to climate change. J. Exp. Bot. 2015, 66, 3477–3486. [Google Scholar] [CrossRef]
- Funk, W.C.; Forester, B.R.; Converse, S.J.; Darst, C.; Morey, S. Improving conservation policy with genomics: A guide to integrating adaptive potential into U.S. Endangered Species Act decisions for conservation practitioners and geneticists. Conserv. Genet. 2019, 20, 115–134. [Google Scholar] [CrossRef]
- de Cara, M.Á.R.; Villanueva, B.; Toro, M.Á.; Fernández, J. Using genomic tools to maintain diversity and fitness in conservation programmes. Mol. Ecol. 2013, 22, 6091–6099. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| SNP Subset | MAF | Non-Target SNPs (MAF > 0.03) | No. of SNPs |
|---|---|---|---|
| Common_single | >0.03 | No | 26,142 |
| Common_multi | >0.03 | Yes | 95,428 |
| Rare_single | <0.03 | No | 269,031 |
| Rare_multi | <0.03 | Yes | 963,871 |
| Category * | No. of Samples (Total = 480) * | Purpose |
|---|---|---|
| Haploids | 50 | Identify multilocus (off-target) binding |
| Paired samples from needle and cambium | 34 (17 pairs) | Identify probes affected by tissue type, assess reproducibility |
| Triplicates | 72 (25 sets) | Assess reproducibility |
| Breeding program and client favourites | 334 | Assess general probe performance and allele frequencies, compare to exome capture data |
| Trios/duos | 79 | Assess inheritance pattern of SNPs |
| SNP Category | MAF | Probe Sets | No. of SNPs |
|---|---|---|---|
| Common | >0.03 | 1 | 16,608 |
| Common | >0.03 | 2 | 11,625 |
| Rare | <0.03 | 1 | 6748 |
| Rare | <0.03 | 2 | 1304 |
| TOTAL | 36,285 |
| Sample Category | No. of Samples |
|---|---|
| NZ progenitors (archive) | 784 |
| NZ progeny (clonal trials) | 2863 |
| NZ progeny (control pollinated trials) | 1309 |
| NZ progeny (open-pollinated trials) | 1182 |
| Australian progeny (clonal and open-pollinated trials) | 2110 |
| Quality control samples (miscellaneous) | 200 |
| TOTAL | 8448 |
| SNP Category * | Total | Percentage |
|---|---|---|
| PolyHighResolution | 21,078 | 4.8% |
| NoMinorHom | 29,245 | 6.7% |
| MonoHighResolution | 169,582 | 38.7% |
| CallRateBelowThreshold | 16,532 | 3.8% |
| OffTargetVariant | 5118 | 1.2% |
| Other | 195,885 | 44.7% |
| AAvarianceX | 91 | 0.02% |
| AAvarianceY | 148 | 0.03% |
| ABvarianceX | 213 | 0.1% |
| ABvarianceY | 180 | 0.04% |
| BBvarianceX | 149 | 0.03% |
| BBvarianceY | 186 | 0.04% |
| HomHomResolution | 337 | 0.1% |
| TOTAL | 438,744 | 100.0% |
| Statistic | Cambium and Needles | Cambium | Needles |
|---|---|---|---|
| Total sample number | 8448 | 366 | 8082 |
| Passed samples | 8397 (99.4%) | 365 (99.7%) | 8032 (99.4%) |
| Failed samples | 51 | 1 | 50 |
| Average Cluster Call Rate | 98.5% | 98.8% | 98.5% |
| Sample Reproducibility | 99.9% | 99.7% | 99.9% |
| SNP Category | No. of Markers | % of Markers |
|---|---|---|
| PolyHighResolution | 16,498 | 45.5 |
| NoMinorHom | 8802 | 24.3 |
| MonoHighResolution | 4044 | 11.1 |
| CallRateBelowThreshold | 5 | 0.0 |
| OffTargetVariant | 346 | 1.0 |
| Other | 6590 | 18.2 |
| TOTAL | 36,285 | 100.0 |
| Unrelated Samples | Related Samples | |||||
|---|---|---|---|---|---|---|
| (1) | (2) | (3) | (1) | (2) | (3) | |
| DNA Mixture (O:C) | O = AA C = BB | O = AA C = AB | O = AB C = AA | O = AA C = BB | O = AA C = AB | O = AB C = AA |
| 1:1 | 99.7% | 49.3% | 52.9% | 98.2% | 48.9% | 39.1% |
| 2:1 | 91.3% | 9.1% | 87.9% | 88.3% | 11.8% | 84.6% |
| 5:1 | 4.9% | 0.3% | 99.4% | 5.6% | 0.5% | 99.0% |
| 8:1 | 1.0% | 0.2% | 99.5% | 1.9% | 0.3% | 99.6% |
| 10:1 | 0.0% | 0.3% | 99.5% | 0.4% | 0.2% | 99.8% |
| 12:1 | 0.0% | 0.1% | 99.7% | 0.3% | 0.2% | 100.0% |
| 15:1 | 0.0% | 0.2% | 99.8% | 0.1% | 0.2% | 99.8% |
| 20:1 | 0.0% | 0.1% | 99.8% | 0.1% | 0.3% | 100.0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graham, N.; Telfer, E.; Frickey, T.; Slavov, G.; Ismael, A.; Klápště, J.; Dungey, H. Development and Validation of a 36K SNP Array for Radiata Pine (Pinus radiata D.Don). Forests 2022, 13, 176. https://doi.org/10.3390/f13020176
Graham N, Telfer E, Frickey T, Slavov G, Ismael A, Klápště J, Dungey H. Development and Validation of a 36K SNP Array for Radiata Pine (Pinus radiata D.Don). Forests. 2022; 13(2):176. https://doi.org/10.3390/f13020176
Chicago/Turabian StyleGraham, Natalie, Emily Telfer, Tancred Frickey, Gancho Slavov, Ahmed Ismael, Jaroslav Klápště, and Heidi Dungey. 2022. "Development and Validation of a 36K SNP Array for Radiata Pine (Pinus radiata D.Don)" Forests 13, no. 2: 176. https://doi.org/10.3390/f13020176
APA StyleGraham, N., Telfer, E., Frickey, T., Slavov, G., Ismael, A., Klápště, J., & Dungey, H. (2022). Development and Validation of a 36K SNP Array for Radiata Pine (Pinus radiata D.Don). Forests, 13(2), 176. https://doi.org/10.3390/f13020176