

Grafting with Different Rootstocks Induced DNA Methylation Alterations in Pecan [Carya illinoinensis (Wangenh.) K. Koch]
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. WGBS Library Construction and Sequencing
2.3. Sequence Data Processing and Analysis
3. Results
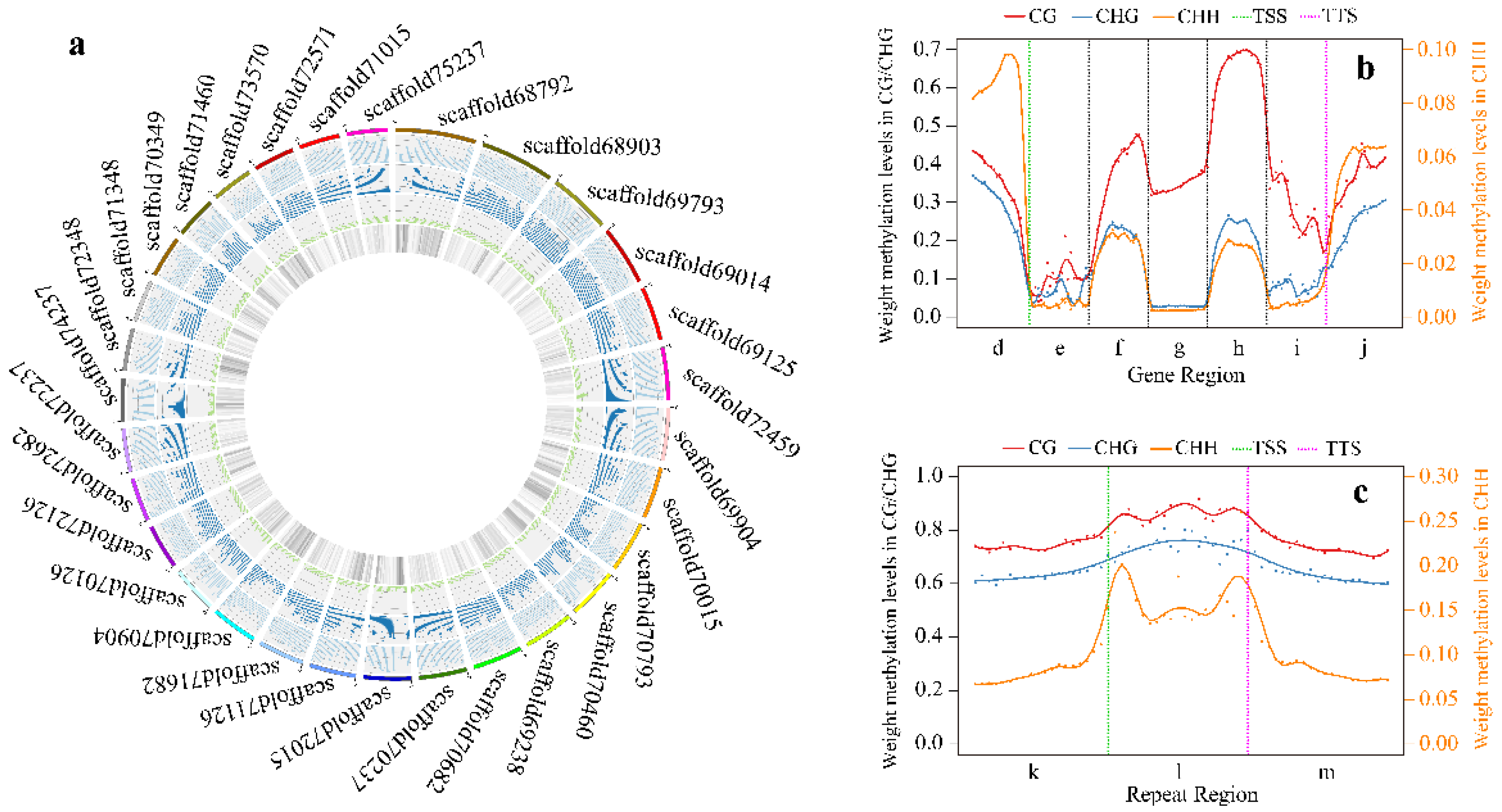
3.1. Pecan DNA Methylomes
3.2. Analysis of DMRs in Pecan Grafts with Different Growth Performances
3.3. GO and KEGG Analysis of DMGs
4. Discussion
4.1. Pecan Methylation Patterns
4.2. Methylation Alterations Induced by Grating with Different Rootstocks
4.3. Involvement of Methylation in Graft Growth Regulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Goldschmidt, E.E. Plant grafting: New mechanisms, evolutionary implications. Front. Plant Sci. 2014, 5, 727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seleznyova, A.; Thorp, T.; White, M.; Tustin, S.; Costes, E. Application of architectural analysis and AMAPmod methodology to study dwarfing phenomenon: The branch structure of ‘Royal Gala’apple grafted on dwarfing and non-dwarfing rootstock/interstock combinations. Ann. Bot. 2003, 91, 665–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallis, C.M.; Wallingford, A.K.; Chen, J. Grapevine rootstock effects on scion sap phenolic levels, resistance to Xylella fastidiosa infection, and progression of Pierce’s disease. Front. Plant Sci. 2013, 4, 502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, A.; Sharma, R. Effect of rootstocks on tree growth, yield, quality and leaf mineral composition of lemon (Citrus limon (L.) Burm.). Sci. Hortic. 2016, 200, 131–136. [Google Scholar] [CrossRef]
- Webster, A. Vigour mechanisms in dwarfing rootstocks for temperate fruit trees. In Proceedings of the Ist International Symposium on Rootstocks for Deciduous Fruit Tree Species, Zaragoza, Spain, 11–14 June 2002; Sanchez, M.A., Webster, A.D., Eds.; International Society Horticultural Science: Leuven, Belgium, 2004; pp. 29–41. [Google Scholar]
- Koepke, T.; Dhingra, A. Rootstock scion somatogenetic interactions in perennial composite plants. Plant Cell Rep. 2013, 32, 1321–1337. [Google Scholar] [CrossRef]
- Harada, T. Grafting and RNA transport via phloem tissue in horticultural plants. Sci. Hortic. 2010, 125, 545–550. [Google Scholar] [CrossRef]
- Movahedi, A.; Sun, W.; Zhang, J.; Wu, X.; Mousavi, M.; Mohammadi, K.; Yin, T.; Zhuge, Q. RNA-directed DNA methylation in plants. Plant Cell Rep. 2015, 34, 1857–1862. [Google Scholar] [CrossRef]
- Molnar, A.; Melnyk, C.W.; Bassett, A.; Hardcastle, T.J.; Dunn, R.; Baulcombe, D.C. Small silencing RNAs in plants are mobile and direct epigenetic modification in recipient cells. Science 2010, 328, 872–875. [Google Scholar] [CrossRef] [Green Version]
- Bai, S.; Kasai, A.; Yamada, K.; Li, T.; Harada, T. A mobile signal transported over a long distance induces systemic transcriptional gene silencing in a grafted partner. J. Exp. Bot. 2011, 62, 4561–4570. [Google Scholar] [CrossRef] [Green Version]
- Finnegan, E.J.; Genger, R.K.; Peacock, W.J.; Dennis, E.S. DNA methylation in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998, 49, 223–247. [Google Scholar] [CrossRef]
- Wu, R.; Wang, X.; Lin, Y.; Ma, Y.; Liu, G.; Yu, X.; Zhong, S.; Liu, B. Inter-species grafting caused extensive and heritable alterations of DNA methylation in Solanaceae plants. PLoS ONE 2013, 8, e61995. [Google Scholar] [CrossRef]
- Avramidou, E.; Kapazoglou, A.; Aravanopoulos, F.A.; Xanthopoulou, A.; Ganopoulos, I.; Tsaballa, A.; Madesis, P.; Doulis, A.G.; Tsaftaris, A. Global DNA methylation changes in Cucurbitaceae inter-species grafting. Crop. Breed. Appl. Biotechnol. 2015, 15, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Uthup, T.K.; Karumamkandathil, R.; Ravindran, M.; Saha, T. Heterografting induced DNA methylation polymorphisms in Hevea brasiliensis. Planta 2018, 248, 579–589. [Google Scholar] [CrossRef]
- Cerruti, E.; Gisbert, C.; Drost, H.; Valentino, D.; Portis, E.; Barchi, L.; Prohens, J.; Lanteri, S.; Comino, C.; Catoni, M. Epigenetic bases of grafting-induced vigour in eggplant. bioRxiv 2019, 831719. [Google Scholar]
- Zhang, R.; Peng, F.; Li, Y. Pecan production in China. Sci. Hortic. 2015, 197, 719–727. [Google Scholar] [CrossRef]
- Grauke, L.J.; Starr, J.L. Phenotypic screening of pecan seedling rootstocks in search of nematode resistance. Trees 2014, 28, 1333–1341. [Google Scholar] [CrossRef]
- Sanderlin, R.S. Susceptibility of some common pecan rootstocks to infection by Xylella fastidiosa. Hortscience 2015, 50, 1183–1186. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, F.; Peng, F.; Tan, P.; Zhu, K.; Feng, G.; Mo, Z.; Li, Y. Identification of grafting-responsive microRNAs associated with growth regulation in pecan [Carya illinoinensis (Wangenh.) K. Koch]. Forests 2020, 11, 196. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.Z.; Chen, T.; Peng, F.R.; Liang, Y.W.; Tan, P.P.; Mo, Z.H.; Cao, F.; Shang, Y.J.; Zhang, R.; Li, Y.R. Variation in cytosine methylation among pecan cultivars at different developmental stages. J. Am. Soc. Hortic. Sci. 2018, 143, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhou, F.; Shang, J.; Peng, F.; Mo, Z.; Li, Y. Changes of cytosine methylation in pecan tissues of different stages by quantitative methylation-sensitive amplified polymorphism. Biol. Plant. 2020, 64, 473–484. [Google Scholar] [CrossRef]
- Li, Q.; Hermanson, P.J.; Springer, N.M. Detection of DNA methylation by whole-genome bisulfite sequencing. Methods Mol. Biol. 2018, 185–196. [Google Scholar]
- Huang, Y.; Xiao, L.; Zhang, Z.; Zhang, R.; Wang, Z.; Huang, C.; Huang, R.; Luan, Y.; Fan, T.; Wang, J. The genomes of pecan and Chinese hickory provide insights into Carya evolution and nut nutrition. GigaScience 2019, 8, giz036. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.D.; Schmitz, R.J.; Ecker, J.R. ‘Leveling’ the playing field for analyses of single-base resolution DNA methylomes. Trends Genet. 2012, 28, 583–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Xi, Y.; Rodriguez, B.; Park, H.J.; Tong, P.; Meong, M.; Goodell, M.A.; Li, W. MOABS: Model based analysis of bisulfite sequencing data. Genome Biol. 2014, 15, R38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lister, R.; Omalley, R.C.; Tontifilippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhu, J.; Hu, F.; Ge, S.; Ye, M.; Xiang, H.; Zhang, G.; Zheng, X.; Zhang, H.; Zhang, S. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genom. 2012, 13, 300. [Google Scholar] [CrossRef] [Green Version]
- Su, C.; Wang, C.; He, L.; Yang, C.; Wang, Y. Shotgun bisulfite sequencing of the Betula platyphylla genome reveals the tree’s DNA methylation patterning. Int. J. Mol. Sci. 2014, 15, 22874–22886. [Google Scholar] [CrossRef] [Green Version]
- An, Y.C.; Goettel, W.; Han, Q.; Bartels, A.; Liu, Z.; Xiao, W. Dynamic changes of genome-wide DNA methylation during soybean seed development. Sci. Rep. 2017, 7, 12263. [Google Scholar] [CrossRef] [Green Version]
- Jia, X.D.; Wang, T.; Zhai, M.; Li, Y.R.; Guo, Z.R. Genetic diversity and identification of Chinese-grown pecan using ISSR and SSR markers. Molecules 2011, 16, 10078–10092. [Google Scholar] [CrossRef] [Green Version]
- Conner, P.J.; Wood, B.W. Identification of pecan cultivars and their genetic relatedness as determined by randomly amplified polymorphic DNA analysis. J. Am. Soc. Hortic. Sci. 2001, 126, 474–480. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, Z.; Zhu, J.K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef]
- Chen, X.; Ge, X.; Wang, J.; Tan, C.; King, G.J.; Liu, K. Genome-wide DNA methylation profiling by modified reduced representation bisulfite sequencing in Brassica rapa suggests that epigenetic modifications play a key role in polyploid genome evolution. Front. Plant Sci. 2015, 6, 836. [Google Scholar] [CrossRef] [Green Version]
- Chodavarapu, R.K.; Feng, S.; Bernatavichute, Y.V.; Chen, P.Y.; Stroud, H.; Yu, Y.; Hetzel, J.A.; Kuo, F.; Kim, J.; Cokus, S.J.; et al. Relationship between nucleosome positioning and DNA methylation. Nature 2010, 466, 388–392. [Google Scholar] [CrossRef] [Green Version]
- Khankahdani, H.H.; Rastegar, S.; Golein, B.; Golmohammadi, M.; Jahromi, A.A. Effect of rootstock on vegetative growth and mineral elements in scion of different Persian lime (Citrus latifolia Tanaka) genotypes. Sci. Hortic. 2019, 246, 136–145. [Google Scholar] [CrossRef]
- Lordan, J.; Fazio, G.; Francescatto, P.; Robinson, T. Effects of apple (Malus × domestica) rootstocks on scion performance and hormone concentration. Sci. Hortic. 2017, 225, 96–105. [Google Scholar] [CrossRef]
- Olien, W.C.; Lakso, A.N. Effect of rootstock on apple (Malus domestica) tree water relations. Physiol. Plant. 1986, 67, 421–430. [Google Scholar] [CrossRef]
- Kou, H.P.; Li, Y.; Song, X.X.; Ou, X.F.; Xing, S.C.; Ma, J.; Wettstein, D.V.; Liu, B. Heritable alteration in DNA methylation induced by nitrogen-deficiency stress accompanies enhaced tolerance by progenies to the stress in rice (Oryza sativa L.). J. Plant Physiol. 2011, 168, 1685–1693. [Google Scholar] [CrossRef]
- Peng, H.; Zhang, J. Plant genomic DNA methylation in response to stresses: Potential applications and challenges in plant breeding. Prog. Nat. Sci. 2009, 19, 1037–1045. [Google Scholar] [CrossRef]
- Wang, W.S.; Pan, Y.J.; Zhao, X.Q.; Dwivedi, D.; Zhu, L.H.; Ali, J.; Fu, B.Y.; Li, Z.K. Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J. Exp. Bot. 2011, 62, 1951–1960. [Google Scholar] [CrossRef]
- Domb, K.; Katz, A.; Harris, K.D.; Yaari, R.; Kaisler, E.; Nguyen, V.H.; Hong, U.V.T.; Griess, O.; Heskiau, K.G.; Ohad, N.; et al. DNA methylation mutants in Physcomitrella patens elucidate individual roles of CG and non-CG methylation in genome regulation. Proc. Natl. Acad. Sci. USA 2020, 117, 33700–33710. [Google Scholar] [CrossRef] [PubMed]
- Vining, K.J.; Pomraning, K.R.; Wilhelm, L.J.; Priest, H.D.; Pellegrini, M.; Mockler, T.C.; Freitag, M.; Strauss, S.H. Dynamic DNA cytosine methylation in the Populus trichocarpa genome: Tissue-level variation and relationship to gene expression. BMC Genom. 2012, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Liu, L.; Peng, Y.; Li, M.; Li, Y.; Liu, D.; Li, X.; Zhang, Z. UNBRANCHED3 expression and inflorescence development is mediated by UNBRANCHED2 and the distal enhancer, KRN4, in Maize. PLoS Genet. 2020, 16, e1008764. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.J.; Rytter, J.; Detwiler, E.A.; Travis, J.W.; McNellis, T.W. Rootstock effects on gene expression patterns in apple tree scions. Plant Mol. Biol. 2003, 53, 493–511. [Google Scholar] [CrossRef] [PubMed]
- Prassinos, C.; Ko, J.H.; Lang, G.; Iezzoni, A.F.; Han, K.H. Rootstock-induced dwarfing in cherries is caused by differential cessation of terminal meristem growth and is triggered by rootstock-specific gene regulation. Tree Physiol. 2009, 29, 927–936. [Google Scholar] [CrossRef] [Green Version]
- Cookson, S.J.; Ollat, N. Grafting with rootstocks induces extensive transcriptional re-programming in the shoot apical meristem of grapevine. BMC Plant Biol. 2013, 13, 147. [Google Scholar] [CrossRef]
- Noda, K.; Okuda, H.; Iwagaki, I. Indole acetic acid and abscisic acid levels in new shoots and fibrous roots of citrus scion-rootstock combinations. Sci. Hortic. 2000, 84, 245–254. [Google Scholar] [CrossRef]
- Davies, P.J. Plant Hormones: Physiology, Biochemistry and Molecular Biology, 2nd ed.; Kluwer Academic Publishers: London, UK, 1995. [Google Scholar]
- Hoth, S.; Morgante, M.; Sanchez, J.; Hanafey, M.K.; Tingey, S.V.; Chua, N. Genome-wide gene expression profiling in Arabidopsis thaliana reveals new targets of abscisic acid and largely impaired gene regulation in the abi1-1 mutant. J. Cell Sci. 2002, 115, 4891–4900. [Google Scholar] [CrossRef] [Green Version]
- Teale, W.D.; Paponov, I.A.; Palme, K. Auxin in action: Signalling, transport and the control of plant growth and development. Nat. Rev. Mol. Cell Biol. 2006, 7, 847–859. [Google Scholar] [CrossRef]
- Ren, H.; Gray, W.M. SAUR proteins as effectors of hormonal and environmental signals in plant growth. Mol. Plant 2015, 8, 1153–1164. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Clean Bases | GC (%) | Clean Reads | Unique Reads | Mapped (%) | Conversion Rate (%) |
|---|---|---|---|---|---|---|
| SV1 | 25,202,430,600 | 22.42 | 84,008,102 | 65,482,398 | 77.95 | 99.32 |
| SV2 | 19,950,784,200 | 21.94 | 66,502,614 | 51,044,261 | 76.76 | 99.27 |
| SV3 | 22,336,860,600 | 21.57 | 74,456,202 | 56,215,238 | 75.50 | 99.21 |
| PV1 | 22,141,198,200 | 21.53 | 73,803,994 | 56,631,166 | 76.73 | 99.29 |
| PV2 | 21,764,759,400 | 21.79 | 72,549,198 | 55,861,160 | 77.00 | 99.18 |
| PV3 | 21,762,984,300 | 21.78 | 72,543,281 | 54,898,021 | 75.68 | 99.24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Tan, P.; Liang, Y.; Shang, Y.; Zhu, K.; Peng, F.; Li, Y. Grafting with Different Rootstocks Induced DNA Methylation Alterations in Pecan [Carya illinoinensis (Wangenh.) K. Koch]. Forests 2023, 14, 4. https://doi.org/10.3390/f14010004
Liu Z, Tan P, Liang Y, Shang Y, Zhu K, Peng F, Li Y. Grafting with Different Rootstocks Induced DNA Methylation Alterations in Pecan [Carya illinoinensis (Wangenh.) K. Koch]. Forests. 2023; 14(1):4. https://doi.org/10.3390/f14010004
Chicago/Turabian StyleLiu, Zhuangzhuang, Pengpeng Tan, Youwang Liang, Yangjuan Shang, Kaikai Zhu, Fangren Peng, and Yongrong Li. 2023. "Grafting with Different Rootstocks Induced DNA Methylation Alterations in Pecan [Carya illinoinensis (Wangenh.) K. Koch]" Forests 14, no. 1: 4. https://doi.org/10.3390/f14010004
APA StyleLiu, Z., Tan, P., Liang, Y., Shang, Y., Zhu, K., Peng, F., & Li, Y. (2023). Grafting with Different Rootstocks Induced DNA Methylation Alterations in Pecan [Carya illinoinensis (Wangenh.) K. Koch]. Forests, 14(1), 4. https://doi.org/10.3390/f14010004