
WGCNA Reveals Genes Associated with Lignification in the Secondary Stages of Wood Formation

 ,
,
Abstract
:1. Introduction
2. Results
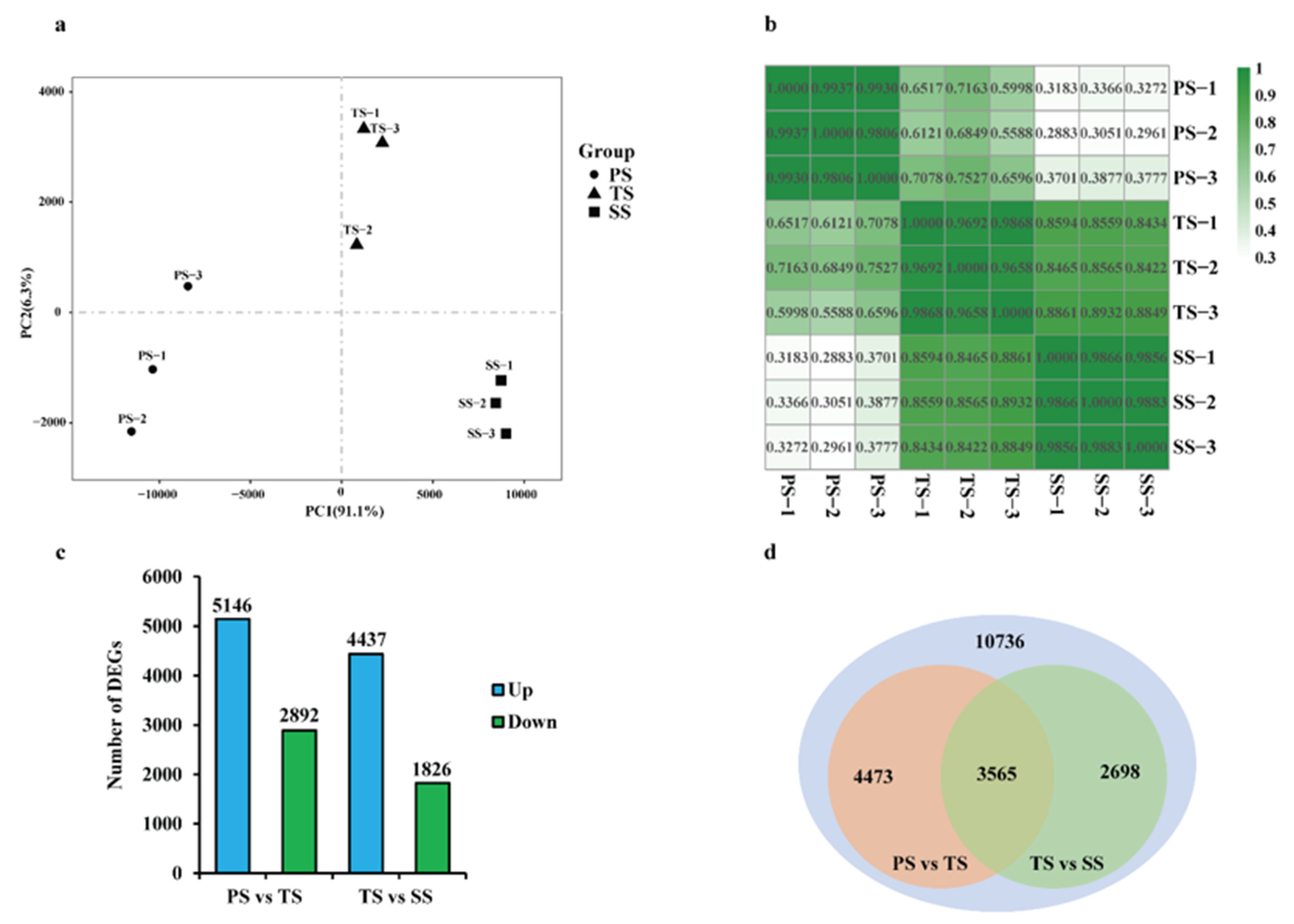
2.1. RNA-Seq Data Collection and Normalization of Poplar Stems during Various Stages of Wood Formation
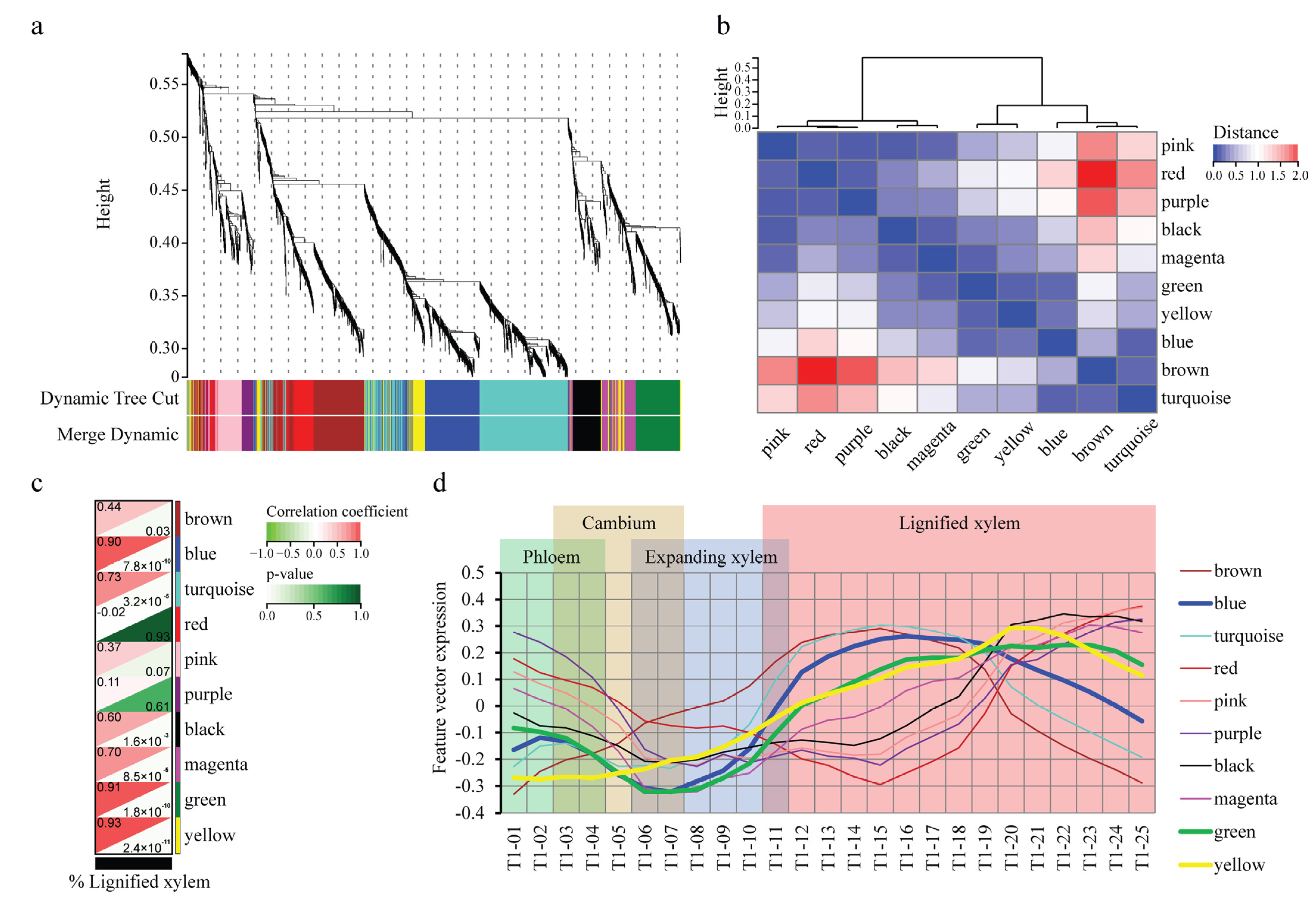
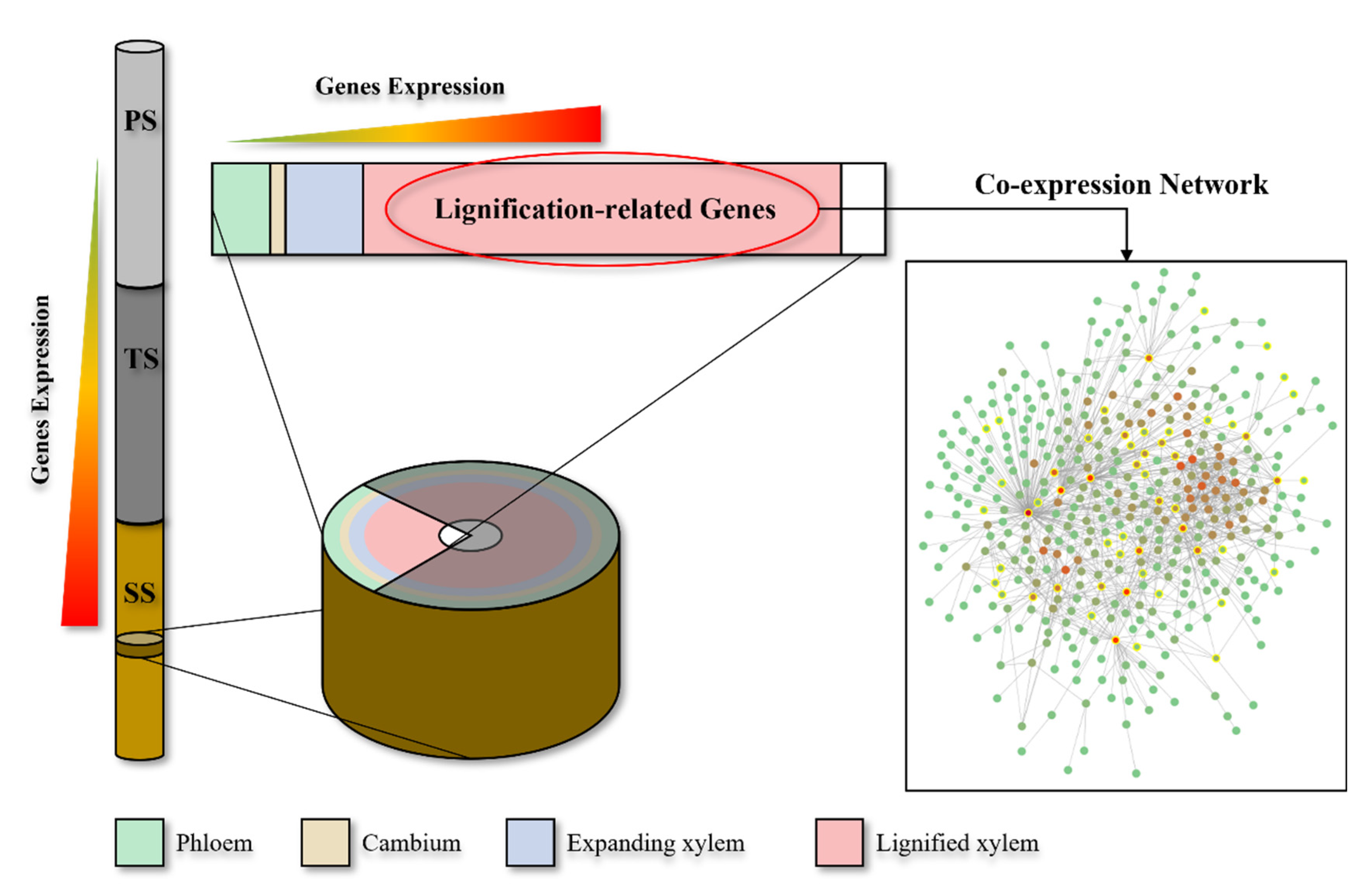
2.2. WGCNA Reveals Modules Related to Different Stages of Poplar Wood Formation
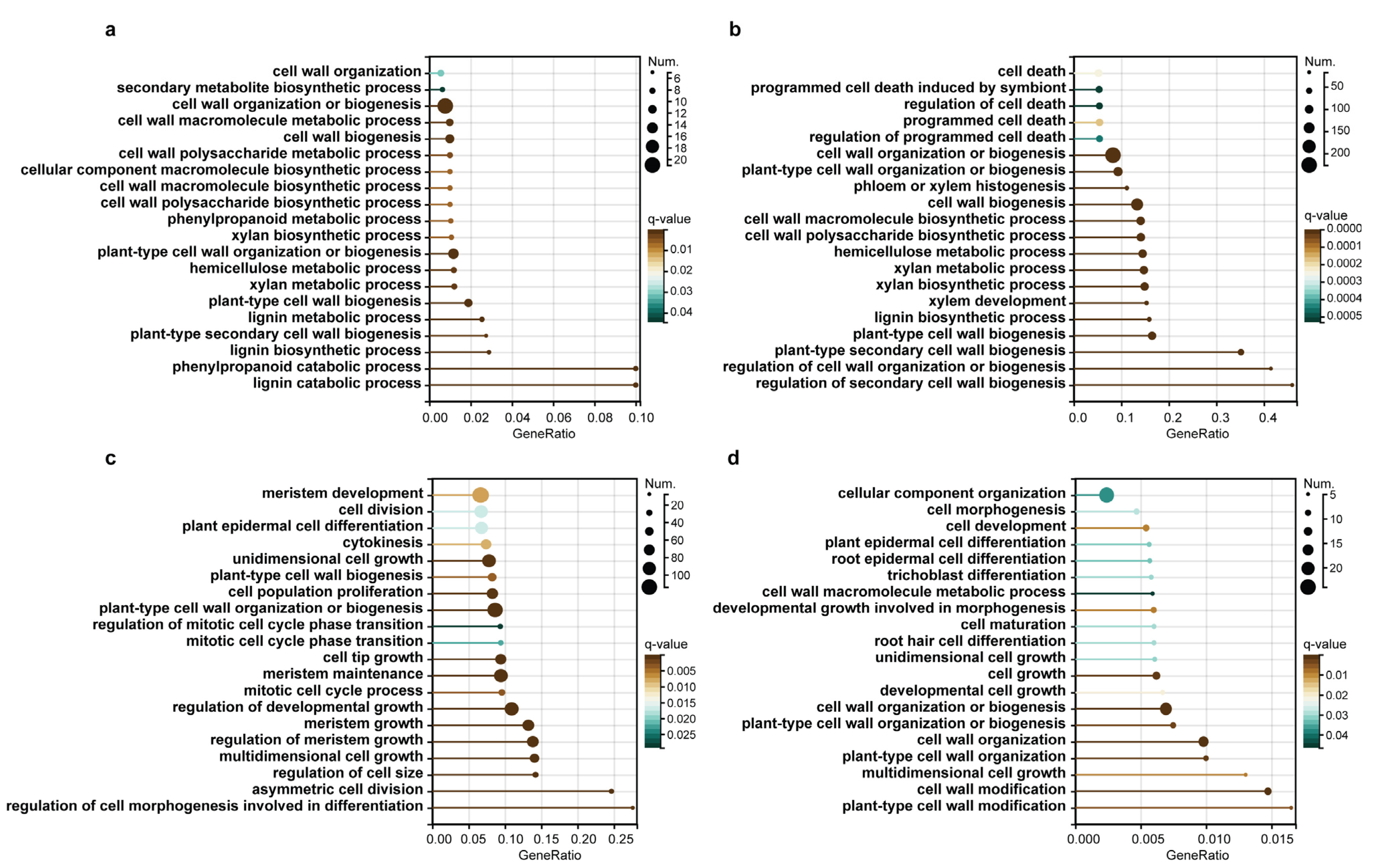
2.3. WGCNA Reveals Genes Associated with Lignification
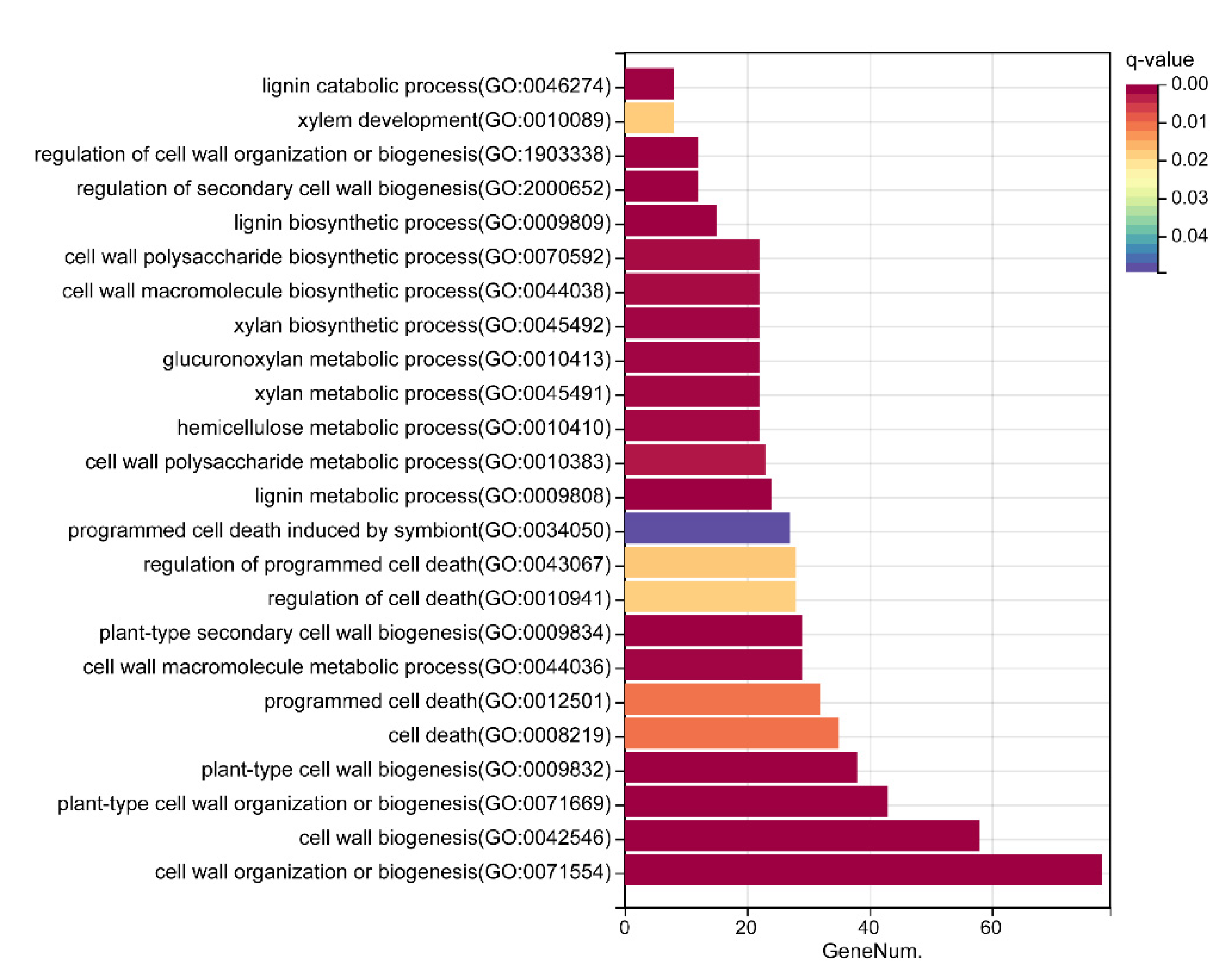
2.4. The Construction and Functional Analysis of the Co-Expression Network of the Modules Related to the Lignification of the Secondary Stem in Wood Formation
2.5. Confirmation of RNA-Seq Profiles by Quantitative PCR
3. Discussion
3.1. Candidate Genes Directly Related to Lignification of Secondary Stem
3.2. Co-Expression Network and Gene Function Related to Lignification of Secondary Stem in Poplar
4. Conclusions
5. Materials and Methods
5.1. RNA-Seq Data Collection
5.2. Gene Differential Expression Analysis
5.3. Weighted Gene Co-Expression Network Analysis
5.4. Gene Annotation and GO Enrichment Analysis
5.5. Downstream Target Gene Prediction and Protein Interaction Relationship Prediction
5.6. qRT-PCR Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhong, R.; McCarthy, R.L.; Lee, C.; Ye, Z.H. Dissection of the transcriptional program regulating secondary wall biosynthesis during wood formation in poplar. Plant Physiol. 2011, 157, 1452–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, N.; Awano, T.; Nakata, M.T.; Sano, Y.; Sakamoto, S.; Mitsuda, N.; Taniguchi, T. Populus NST/SND orthologs are key regulators of secondary cell wall formation in wood fibers, phloem fibers and xylem ray parenchyma cells. Tree Physiol. 2019, 39, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Li, C.; Lin, S.; Wang, J.; Zhou, B.; Jiang, T. Transcriptome analysis of salt-responsive and wood-associated NACs in Populus simonii × Populus nigra. BMC Plant Biol. 2020, 20, 317. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhao, X.; Ran, L.; Li, C.; Fan, D.; Luo, K. PtoMYB156 is involved in negative regulation of phenylpropanoid metabolism and secondary cell wall biosynthesis during wood formation in poplar. Sci. Rep. 2017, 7, 41209. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.S.; Wickneswari, R. Characterization of microRNAs expressed during secondary wall biosynthesis in Acacia mangium. PloS ONE 2012, 7, e49662. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.F.; Zhao, B.; Huang, C.C.; Chen, Z.W.; Zhao, T.; Liu, H.R.; Hu, G.J.; Shangguan, X.X.; Shan, C.M.; Wang, L.J.; et al. The miR319-Targeted GhTCP4 Promotes the Transition from Cell Elongation to Wall Thickening in Cotton Fiber. Mol. Plant 2020, 13, 1063–1077. [Google Scholar] [CrossRef]
- Du, Q.; Avci, U.; Li, S.; Gallego-Giraldo, L.; Pattathil, S.; Qi, L.; Hahn, M.G.; Wang, H. Activation of miR165b represses AtHB15 expression and induces pith secondary wall development in Arabidopsis. Plant J. 2015, 83, 388–400. [Google Scholar] [CrossRef]
- Xu, P.; Kong, Y.; Song, D.; Huang, C.; Li, X.; Li, L. Conservation and functional influence of alternative splicing in wood formation of Populus and Eucalyptus. BMC Genom. 2014, 15, 780. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.X.; Zhang, K.L.; Zhang, M.; Das, D.; Fang, Y.M.; Dai, L.; Zhang, J.; Zhu, F.Y. Alternative splicing and its regulatory role in woody plants. Tree Physiol. 2020, 40, 1475–1486. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, C.; Cheng, H.; Tian, S.; Liu, Y.; Wang, S.; Zhang, H.; Saqib, M.; Wei, H.; Wei, Z. DNA methylation and its effects on gene expression during primary to secondary growth in poplar stems. BMC Genom. 2020, 21, 498. [Google Scholar] [CrossRef]
- Ali, S.; Ehtram, A.; Arora, N.; Manjunath, P.; Roy, D.; Ehtesham, N.Z.; Hasnain, S.E. The M. tuberculosis Rv1523 Methyltransferase Promotes Drug Resistance Through Methylation-Mediated Cell Wall Remodeling and Modulates Macrophages Immune Responses. Front. Cell. Infect. Microbiol. 2021, 11, 622487. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, J.; Xu, P.; Zhang, R.; Li, L. Intron-mediated alternative splicing of WOOD-ASSOCIATED NAC TRANSCRIPTION FACTOR1B regulates cell wall thickening during fiber development in Populus species. Plant Physiol. 2014, 164, 765–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Ci, D.; Li, T.; Li, P.; Song, Y.; Chen, J.; Quan, M.; Zhou, D.; Zhang, D. The Role of DNA Methylation in Xylogenesis in Different Tissues of Poplar. Front. Plant Sci. 2016, 7, 1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prassinos, C.; Ko, J.H.; Yang, J.; Han, K.H. Transcriptome profiling of vertical stem segments provides insights into the genetic regulation of secondary growth in hybrid aspen trees. Plant Cell Physiol. 2005, 46, 1213–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dharmawardhana, P.; Brunner, A.M.; Strauss, S.H. Genome-wide transcriptome analysis of the transition from primary to secondary stem development in Populus trichocarpa. BMC Genom. 2010, 11, 150. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Reng, M.; Tian, S.; Liu, C.; Cheng, H.; Liu, Y.; Zhang, H.; Saqib, M.; Wei, H.; Wei, Z. Transcriptome-wide identification and characterization of microRNAs in diverse phases of wood formation in Populus trichocarpa. G3 Genes|Genomes|Genet. 2021, 11, jkab195. [Google Scholar] [CrossRef]
- Liu, J.; Hai, G.; Wang, C.; Cao, S.; Xu, W.; Jia, Z.; Yang, C.; Wang, J.P.; Dai, S.; Cheng, Y. Comparative proteomic analysis of Populus trichocarpa early stem from primary to secondary growth. J. Proteom. 2015, 126, 94–108. [Google Scholar] [CrossRef]
- Sundell, D.; Street, N.R.; Kumar, M.; Mellerowicz, E.J.; Kucukoglu, M.; Johnsson, C.; Kumar, V.; Mannapperuma, C.; Delhomme, N.; Nilsson, O.; et al. AspWood: High-Spatial-Resolution Transcriptome Profiles Reveal Uncharacterized Modularity of Wood Formation in Populus tremula. Plant Cell 2017, 29, 1585–1604. [Google Scholar] [CrossRef] [Green Version]
- Johnsson, C.; Fischer, U. Cambial stem cells and their niche. Plant Sci. 2016, 252, 239–245. [Google Scholar] [CrossRef]
- Mellerowicz, E.J.; Baucher, M.; Sundberg, B.; Boerjan, W. Unravelling cell wall formation in the woody dicot stem. Plant Mol. Biol. 2001, 47, 239–274. [Google Scholar] [CrossRef]
- Chen, J.; Pan, A.; He, S.; Su, P.; Yuan, X.; Zhu, S.; Liu, Z. Different MicroRNA Families Involved in Regulating High Temperature Stress Response during Cotton (Gossypium hirsutum L.) Anther Development. Int. J. Mol. Sci. 2020, 21, 1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Y.; Chen, G.; Jin, J.; Wang, Y.; Xu, M.; Peng, J.; Ding, Y. Small RNA and Transcriptome Sequencing Reveals miRNA Regulation of Floral Thermogenesis in Nelumbo nucifera. Int. J. Mol. Sci. 2020, 21, 3324. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Li, C.; Fu, H.; Yang, M.; Wu, H.; Ding, Y.; Li, L.; Lin, S. Genome-Wide Analysis of SQUAMOSA-Promoter-Binding Protein-like Family in Flowering Pleioblastus pygmaeus. Int. J. Mol. Sci. 2022, 23, 14035. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gao, C.; Wang, L.; Zheng, L.; Yang, C.; Wang, Y. Comprehensive transcriptional profiling of NaHCO3-stressed Tamarix hispida roots reveals networks of responsive genes. Plant Mol. Biol. 2014, 84, 145–157. [Google Scholar] [CrossRef]
- Chen, X.; Xia, J.; Xia, Z.; Zhang, H.; Zeng, C.; Lu, C.; Zhang, W.; Wang, W. Potential functions of microRNAs in starch metabolism and development revealed by miRNA transcriptome profiling of cassava cultivars and their wild progenitor. BMC Plant Biol. 2015, 15, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Sharkawy, I.; Liang, D.; Xu, K. Transcriptome analysis of an apple (Malus × domestica) yellow fruit somatic mutation identifies a gene network module highly associated with anthocyanin and epigenetic regulation. J. Exp. Bot. 2015, 66, 7359–7376. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Zhang, R.; Li, D.; Wang, F. Transcriptomic and Coexpression Network Analyses Revealed Pine Chalcone Synthase Genes Associated with Pine Wood Nematode Infection. Int. J. Mol. Sci. 2021, 22, 11195. [Google Scholar] [CrossRef]
- Zhang, M.; Cheng, W.; Yuan, X.; Wang, J.; Cheng, T.; Zhang, Q. Integrated transcriptome and small RNA sequencing in revealing miRNA-mediated regulatory network of floral bud break in Prunus mume. Front. Plant Sci. 2022, 13, 931454. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, J.; Liu, Z.; Liu, X.; Ma, Y.; Zhang, B.; Chen, Y.; Li, X.; Feng, Z.; Yang, N.; et al. Identification of hub genes in colorectal cancer based on weighted gene co-expression network analysis and clinical data from The Cancer Genome Atlas. Biosci. Rep. 2021, 41, BSR20211280. [Google Scholar] [CrossRef]
- Tian, F.; Yang, D.C.; Meng, Y.Q.; Jin, J.; Gao, G. PlantRegMap: Charting functional regulatory maps in plants. Nucleic Acids Res. 2020, 48, D1104–D1113. [Google Scholar] [CrossRef]
- Hu, Q.; Xiao, S.; Wang, X.; Ao, C.; Zhang, X.; Zhu, L. GhWRKY1-like enhances cotton resistance to Verticillium dahliae via an increase in defense-induced lignification and S monolignol content. Plant Sci. 2021, 305, 110833. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Takada, R.; Tobimatsu, Y.; Suzuki, S.; Yamamura, M.; Osakabe, K.; Osakabe, Y.; Sakamoto, M.; Umezawa, T. Double knockout of OsWRKY36 and OsWRKY102 boosts lignification with altering culm morphology of rice. Plant Sci. 2020, 296, 110466. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Zhang, C.; Guo, X.; Li, H.; Lu, H. MYB Transcription Factors and Its Regulation in Secondary Cell Wall Formation and Lignin Biosynthesis during Xylem Development. Int. J. Mol. Sci. 2021, 22, 3560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, L.; Wang, Z.; Zhang, R.; Liu, P.; Liu, M.; Liu, Z.; Zhao, Z.; Wang, L.; Chen, X.; et al. The regulation of cell wall lignification and lignin biosynthesis during pigmentation of winter jujube. Hortic. Res. 2021, 8, 238. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.K.; Li, X.; Xu, Q.; Chen, J.Y.; Yin, X.R.; Ferguson, I.B.; Chen, K.S. EjAP2-1, an AP2/ERF gene, is a novel regulator of fruit lignification induced by chilling injury, via interaction with EjMYB transcription factors. Plant Biotechnol. J. 2015, 13, 1325–1334. [Google Scholar] [CrossRef]
- Bang, S.W.; Lee, D.K.; Jung, H.; Chung, P.J.; Kim, Y.S.; Choi, Y.D.; Suh, J.W.; Kim, J.K. Overexpression of OsTF1L, a rice HD-Zip transcription factor, promotes lignin biosynthesis and stomatal closure that improves drought tolerance. Plant Biotechnol. J. 2019, 17, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.-H.; Zhong, R. Molecular control of wood formation in trees. J. Exp. Bot. 2015, 66, 4119–4131. [Google Scholar] [CrossRef] [Green Version]
- Goujon, T.; Ferret, V.; Mila, I.; Pollet, B.; Ruel, K.; Burlat, V.; Joseleau, J.P.; Barrière, Y.; Lapierre, C.; Jouanin, L. Down-regulation of the AtCCR1 gene in Arabidopsis thaliana: Effects on phenotype, lignins and cell wall degradability. Planta 2003, 217, 218–228. [Google Scholar] [CrossRef]
- Leplé, J.C.; Dauwe, R.; Morreel, K.; Storme, V.; Lapierre, C.; Pollet, B.; Naumann, A.; Kang, K.Y.; Kim, H.; Ruel, K.; et al. Downregulation of cinnamoyl-coenzyme A reductase in poplar: Multiple-level phenotyping reveals effects on cell wall polymer metabolism and structure. Plant Cell 2007, 19, 3669–3691. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Stein, A.; Wittkop, B.; Sarvari, P.; Li, J.; Yan, X.; Dreyer, F.; Frauen, M.; Friedt, W.; Snowdon, R.J. A knockout mutation in the lignin biosynthesis gene CCR1 explains a major QTL for acid detergent lignin content in Brassica napus seeds. Theor. Appl. Genet. 2012, 124, 1573–1586. [Google Scholar] [CrossRef]
- Ruel, K.; Berrio-Sierra, J.; Derikvand, M.M.; Pollet, B.; Thévenin, J.; Lapierre, C.; Jouanin, L.; Joseleau, J.P. Impact of CCR1 silencing on the assembly of lignified secondary walls in Arabidopsis thaliana. New Phytol. 2009, 184, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Van Acker, R.; Leplé, J.C.; Aerts, D.; Storme, V.; Goeminne, G.; Ivens, B.; Légée, F.; Lapierre, C.; Piens, K.; Van Montagu, M.C.; et al. Improved saccharification and ethanol yield from field-grown transgenic poplar deficient in cinnamoyl-CoA reductase. Proc. Natl. Acad. Sci. USA 2014, 111, 845–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Rest, B.; Danoun, S.; Boudet, A.M.; Rochange, S.F. Down-regulation of cinnamoyl-CoA reductase in tomato (Solanum lycopersicum L.) induces dramatic changes in soluble phenolic pools. J. Exp. Bot. 2006, 57, 1399–1411. [Google Scholar] [CrossRef] [Green Version]
- Barakat, A.; Yassin, N.B.; Park, J.S.; Choi, A.; Herr, J.; Carlson, J.E. Comparative and phylogenomic analyses of cinnamoyl-CoA reductase and cinnamoyl-CoA-reductase-like gene family in land plants. Plant Sci. 2011, 181, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; Lee, C.; Zhou, J.; McCarthy, R.L.; Ye, Z.H. A battery of transcription factors involved in the regulation of secondary cell wall biosynthesis in Arabidopsis. Plant Cell 2008, 20, 2763–2782. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Luo, F.; Zhong, Y.; He, J.; Li, L. Modulation of NAC transcription factor NST1 activity by XYLEM NAC DOMAIN1 regulates secondary cell wall formation in Arabidopsis. J. Exp. Bot. 2020, 71, 1449–1458. [Google Scholar] [CrossRef]
- Wang, H.H.; Tang, R.J.; Liu, H.; Chen, H.Y.; Liu, J.Y.; Jiang, X.N.; Zhang, H.X. Chimeric repressor of PtSND2 severely affects wood formation in transgenic Populus. Tree Physiol. 2013, 33, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Jeon, S.J.; Yanders, S.; Park, S.C.; Kim, H.S.; Kim, S. MYB3 plays an important role in lignin and anthocyanin biosynthesis under salt stress condition in Arabidopsis. Plant Cell Rep. 2022, 41, 1549–1560. [Google Scholar] [CrossRef]
- Du, J.; Miura, E.; Robischon, M.; Martinez, C.; Groover, A. The Populus Class III HD ZIP transcription factor POPCORONA affects cell differentiation during secondary growth of woody stems. PloS ONE 2011, 6, e17458. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Song, D.; Sun, J.; Wang, X.; Li, L. PtrHB7, a class III HD-Zip gene, plays a critical role in regulation of vascular cambium differentiation in Populus. Mol. Plant 2013, 6, 1331–1343. [Google Scholar] [CrossRef]
- Oda, Y.; Fukuda, H. Secondary cell wall patterning during xylem differentiation. Curr. Opin. Plant Biol. 2012, 15, 38–44. [Google Scholar] [CrossRef]
- Goodson, H.V.; Jonasson, E.M. Microtubules and Microtubule-Associated Proteins. Cold Spring Harb. Perspect. Biol. 2018, 10, a022608. [Google Scholar] [CrossRef]
- Vilela, B.; Pagès, M.; Lumbreras, V. Regulation of MAPK signaling and cell death by MAPK phosphatase MKP2. Plant Signal. Behav. 2010, 5, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Jackson, D. Detection of MAPK3/6 Phosphorylation During Hypersensitive Response (HR)-Associated Programmed Cell Death in Plants. Methods Mol. Biol. (Clifton NJ) 2018, 1743, 153–161. [Google Scholar] [CrossRef]
- Lu, S.; Li, Q.; Wei, H.; Chang, M.J.; Tunlaya-Anukit, S.; Kim, H.; Liu, J.; Song, J.; Sun, Y.H.; Yuan, L.; et al. Ptr-miR397a is a negative regulator of laccase genes affecting lignin content in Populus trichocarpa. Proc. Natl. Acad. Sci. USA 2013, 110, 10848–10853. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.; Sun, Y.H.; Li, Q.; Heber, S.; Sederoff, R.; Chiang, V.L. Towards a systems approach for lignin biosynthesis in Populus trichocarpa: Transcript abundance and specificity of the monolignol biosynthetic genes. Plant Cell Physiol. 2010, 51, 144–163. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Gunasekara, C.; Guo, Y.; Zhang, H.; Lei, L.; Tunlaya-Anukit, S.; Busov, V.; Chiang, V.; Wei, H. Pop’s Pipes: Poplar gene expression data analysis pipelines. Tree Genet. Genomes 2014, 10, 1093–1101. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Von Mering, C.; Huynen, M.; Jaeggi, D.; Schmidt, S.; Bork, P.; Snel, B. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 2003, 31, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label | ID of Populus Trichocarpa | Symbol | ID of Arabidopsis | Function and References |
|---|---|---|---|---|
| TF7 | Potri.001G112200 | IXR11/KNAT7 | AT1G62990 | Downstream transcription factors regulated by PtrWND [1,37,45] |
| TF9 | Potri.014G104800 | ANAC043 | AT2G46770 | Regulating the formation of secondary cell walls in Arabidopsis [46] |
| TF10 | Potri.004G049300 | ANAC073/SND2 | AT4G28500 | PtrWND-regulated downstream TFs; dominant repression of PtSND2/NAC073 causes a reduction in secondary wall thickening in transgenic poplar wood [1,37,45,47] |
| TF12 | Potri.019G081500 | ATMYB3 | AT1G22640 | Involved in lignin biosynthesis under salt stress in Arabidopsis [48] |
| TF13 | Potri.012G039400 | ATMYB52 | AT1G17950 | Involved in the regulation of secondary cell wall biosynthesis in Arabidopsis [45] |
| TF14 | Potri.001G099800 | AtMYB103 | AT1G63910 | Downstream transcription factors regulated by PtrWND [1,37,45] |
| TF15 | Potri.001G118800 | AtMYB42 | AT4G12350 | Downstream transcription factors regulated by PtrWND [1,37,45] |
| TF16 | Potri.003G114100 | AtMYB42 | AT4G12350 | Downstream transcription factors regulated by PtrWND [1,37,45] |
| TF17 | Potri.015G033600 | ATMYB52 | AT1G17950 | Involved in the regulation of secondary cell wall biosynthesis in Arabidopsis [45] |
| TF19 | Potri.005G186400 | ATMYB52 | AT1G17950 | Involved in the regulation of secondary cell wall biosynthesis in Arabidopsis [45] |
| TF20 | Potri.015G129100 | AtMYB85 | AT4G22680 | Downstream transcription factors regulated by PtrWND [1,37,45] |
| TF21 | Potri.013G156200 | ASL11/LBD15 | AT2G40470 | Downstream transcription factors regulated by PtrWND [1,37] |
| TF27 | Potri.001G188800 | ATHB-15 | AT1G52150 | Overexpression of a microRNA-resistant form causes delayed differentiation of secondary xylem and phloem fibres [49] |
| TF28 | Potri.003G050100 | ATHB-15 | AT1G52150 | Overexpression of a microRNA-resistant form causes delayed differentiation of secondary xylem and phloem fibres [49] |
| TF29 | Potri.018G045100 | ATHB-8 | AT4G32880 | Regulates differentiation of secondary xylem and phloem [50] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Xie, M.; Zhao, W.; Yan, P.; Wang, Y.; Gu, Y.; Jiang, T.; Qu, G. WGCNA Reveals Genes Associated with Lignification in the Secondary Stages of Wood Formation. Forests 2023, 14, 99. https://doi.org/10.3390/f14010099
Wang R, Xie M, Zhao W, Yan P, Wang Y, Gu Y, Jiang T, Qu G. WGCNA Reveals Genes Associated with Lignification in the Secondary Stages of Wood Formation. Forests. 2023; 14(1):99. https://doi.org/10.3390/f14010099
Chicago/Turabian StyleWang, Ruiqi, Miaomiao Xie, Wenna Zhao, Pingyu Yan, Yuting Wang, Yongmei Gu, Tingbo Jiang, and Guanzheng Qu. 2023. "WGCNA Reveals Genes Associated with Lignification in the Secondary Stages of Wood Formation" Forests 14, no. 1: 99. https://doi.org/10.3390/f14010099
APA StyleWang, R., Xie, M., Zhao, W., Yan, P., Wang, Y., Gu, Y., Jiang, T., & Qu, G. (2023). WGCNA Reveals Genes Associated with Lignification in the Secondary Stages of Wood Formation. Forests, 14(1), 99. https://doi.org/10.3390/f14010099