
Structural Characterization of Trivalvaria costata Chloroplast Genome and Molecular Evolution of rps12 Gene in Magnoliids

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequencing and Sequence Preparation
2.2. SSR Analysis
2.3. Codon Usage Analysis
2.4. Phylogenetic Analysis of Magnoliids
2.5. Molecule Evolution Analysis of rps12
3. Results
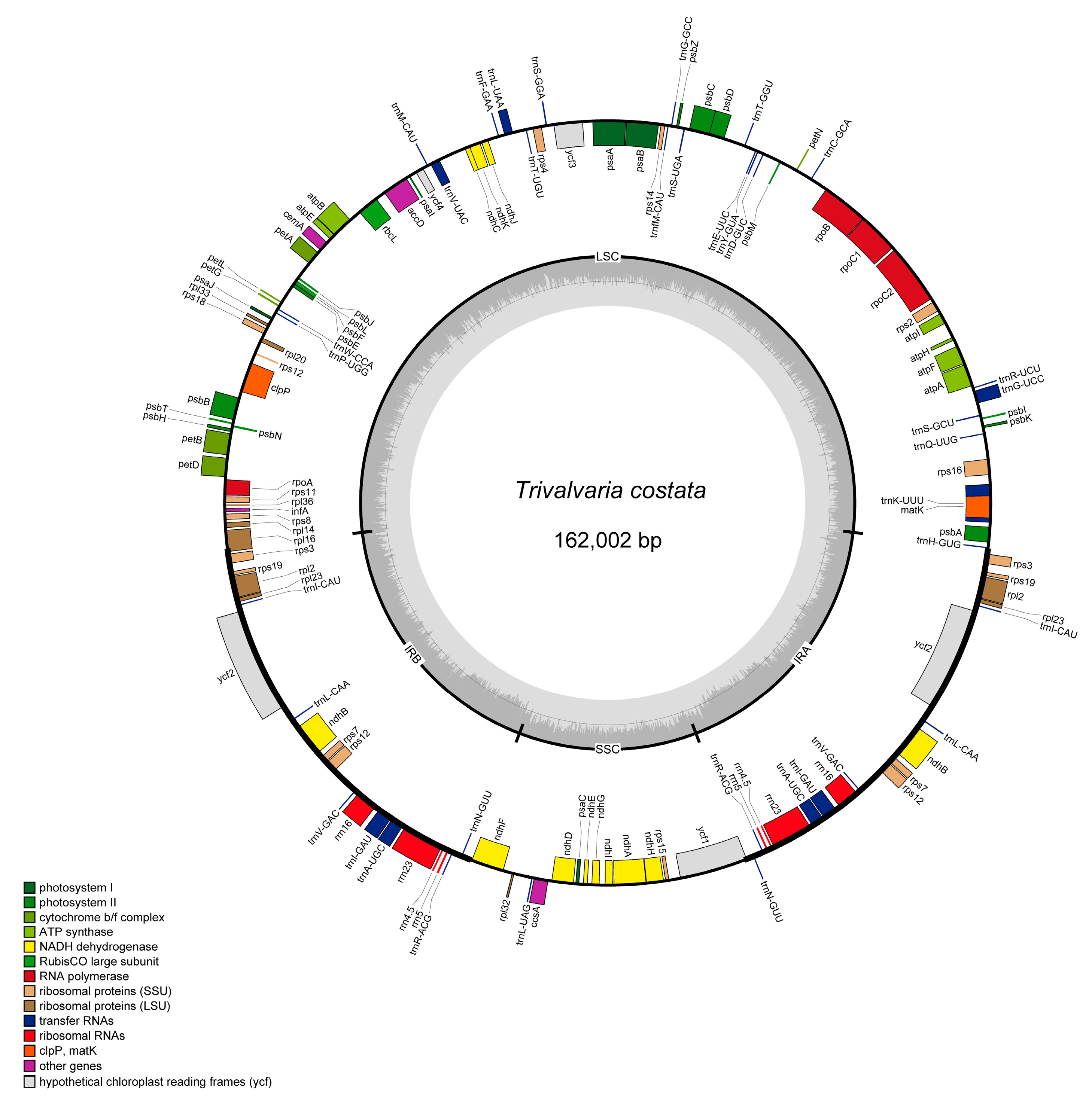
3.1. Structure Characteristics of T. costata cp Genome
3.2. Codon Usage Bias Analysis of T. costata
3.3. Information on cp Genome and rps12 in Sample Species
3.4. Evolutionary Rates and Selection Pressure of rps12
4. Discussion
4.1. Distribution Pattern of SSRs in T. costata
4.2. Codon Usage Bias in T. costata cp Genome
4.3. Expansion and Reduced Substitution Rates of IR Region
4.4. rps12 May Be Undergoing Adaptive Changes in Magnoliids
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.K.; Ruhlman, T.A. Plastid genomes of seed plants. In Genomics of Chloroplasts and Mitochondria; Bock, R., Knoop, V., Eds.; Springer: Dordrecht, Switzerland, 2012; Volume 35, pp. 103–126. [Google Scholar]
- Mower, J.P.; Vickrey, T.L. Structural diversity among plastid genomes of land plants. Adv. Bot. Res. 2018, 85, 263–292. [Google Scholar]
- Nguyen, H.Q.; Nguyen, T.N.L.; Doan, T.N.; Nguyen, T.T.N.; Phạm, M.H.; Le, T.L.; Sy, T.D.; Chu, H.H.; Chu, H.M. Complete chloroplast genome of novel Adrinandra megaphylla Hu species: Molecular structure, comparative and phylogenetic analysis. Sci. Rep. 2021, 11, 11731. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Miao, Y.-J.; Qian, J.; Zheng, Y.; Xia, C.-L.; Yang, Q.-S.; Liu, C.; Huang, L.-F.; Duan, B.-Z. Comparative analysis of complete chloroplast genome sequences of five endangered species and new insights into phylogenetic relationships of Paris. Gene 2022, 833, 146572. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhao, W.-J.; Xu, J.; Li, M.-F.; Zhang, Y.-J. Chloroplast genome evolution and species identification of Styrax (Styracaceae). BioMed Res. Int. 2022, 2022, 5364094. [Google Scholar] [CrossRef]
- Wu, L.-W.; Nie, L.-P.; Wang, Q.; Xu, Z.-C.; Wang, Y.; He, C.-N.; Song, J.-Y.; Yao, H. Comparative and phylogenetic analyses of the chloroplast genomes of species of Paeoniaceae. Sci. Rep. 2021, 11, 14643. [Google Scholar] [CrossRef]
- Xue, B.; Ding, H.-B.; Yao, G.; Shao, Y.-Y.; Fan, X.-J.; Tan, Y.-H. From Polyalthia to Polyalthiopsis (Annonaceae): Transfer of species enlarges a previously monotypic genus. PhytoKeys 2020, 148, 71. [Google Scholar] [CrossRef]
- Chmielewski, M.; Meyza, K.; Chybicki, I.J.; Dzialuk, A.; Litkowiec, M.; Burczyk, J. Chloroplast microsatellites as a tool for phylogeographic studies: The case of white oaks in Poland. iForest 2015, 8, 765. [Google Scholar] [CrossRef] [Green Version]
- Fages-Lartaud, M.; Hundvin, K.; Hohmann-Marriott, M.F. Mechanisms governing codon usage bias and the implications for protein expression in the chloroplast of Chlamydomonas reinhardtii. Plant J. 2022, 112, 919–945. [Google Scholar] [CrossRef]
- Rao, Y.-S.; Wu, G.-Z.; Wang, Z.-F.; Chai, X.W.; Nie, Q.H.; Zhang, X.-Q. Mutation bias is the driving force of codon usage in the Gallus gallus genome. DNA Res. 2011, 18, 499–512. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.S.; Chaw, S.M. Evolutionary stasis in cycad plastomes and the first case of plastome GC-biased gene conversion. Genome Biol. Evol. 2015, 7, 2000–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.W.; Kuo, L.Y.; Pryer, K.M.; Rothfels, C.J. Genes translocated into the plastid inverted repeat show decelerated substitution rates and elevated GC content. Genome Biol. Evol. 2016, 8, 2452–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, A.-D.; Guo, W.-H.; Gupta, S.; Fan, W.S.; Mower, J.P. Evolutionary dynamics of the plastid inverted repeat: The effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016, 209, 1747–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norihiro, Z.; Keita, T.; Kazuo, S.; Masahiro, S. Trans splicing in vivo: Joining of transcripts from the ‘divided’ gene for ribosomal protein S12 in the chloroplasts. FEBS Lett. 1987, 210, 153–156. [Google Scholar]
- Ping, J.-Y.; Li, A.-M.; Feng, P.-P.; Zhu, M.; Su, Y.-J.; Wang, T. The highly conserved rps12 gene in ferns provides strong evidence for decreased substitution rates in the inverted repeat region. Plant Syst. Evol. 2021, 307, 26. [Google Scholar] [CrossRef]
- Ping, J.-Y.; Feng, P.-P.; Hao, J.; Li, J.-Y.; Su, Y.-J.; Wang, T. The molecular evolution pattern of rps12 gene in gymnosperms. Chin. Sci. Bull. 2021, 66, 3182–3193. (In Chinese) [Google Scholar] [CrossRef]
- Liu, S.-S.; Wang, Z.; Su, Y.-J.; Wang, T. Patterns and rates of plastid rps12 gene evolution inferred in a phylogenetic context using plastomic data of ferns. Sci. Rep. 2020, 10, 9394. [Google Scholar] [CrossRef]
- Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG II. Bot. J. Linn. Soc. 2003, 141, 399–436. [Google Scholar] [CrossRef] [Green Version]
- Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 2009, 161, 105–121. [Google Scholar] [CrossRef] [Green Version]
- Punyasena, S.W.; Eshel, G.; McElwain, J.C. The influence of climate on the spatial patterning of Neotropical plant families. J. Biogeogr. 2008, 35, 117–130. [Google Scholar] [CrossRef]
- Blazier, J.C.; Ruhlman, T.A.; Weng, M.L.; Rehman, S.K.; Sabir, J.S.; Jansen, R.K. Divergence of RNA polymerase α subunits in angiosperm plastid genomes is mediated by genomic rearrangement. Sci. Rep. 2016, 6, 24595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.-T.; Gilbert, M.G. Annonaceae. In Flora of China; Wu, Z.-Y., Raven, P.H., Hong, D.-Y., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2011; Volume 30, pp. 672–713. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 25 January 2023).
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [PubMed] [Green Version]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; DePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.-J.; Moore, M.J.; Li, D.-Z.; Yi, T.-S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony (and other Methods), Version 4.0b10; Sinauer Associates: Sun-derland, MA, USA, 2002. [Google Scholar]
- Stamatakis, A. RaxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogeies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pond, S.L.; Frost, S.D.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [Green Version]
- IBM Corporation. SPSS Statistics (Version 22); IBM Corporation: New York, NY, USA, 2014. [Google Scholar]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Swanson, W.J.; Vacquier, V.D. Maximum-likelihood analysis of molecular adaptation in abalone sperm lysin reveals variable selective pressures among lineages and sites. Mol. Biol. Evol. 2000, 17, 1446–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.-H.; Wang, S.; Zhou, S.-L. Polymorphic chloroplast microsatellite loci in nelumbo (nelumbonaceae). Am J Bot. 2012, 99, 240–244. [Google Scholar] [CrossRef]
- Gao, R.; Wang, W.-Z.; Huang, Q.-Y.; Fan, R.-F.; Wang, X.; Feng, P.; Zhao, G.-M.; Bian, S.; Ren, H.-L.; Chang, Y. Complete chloroplast genome sequence of Dryopteris fragrans (L.) Schott and the repeat structures against the thermal environment. Sci. Rep. 2018, 8, 16635. [Google Scholar] [CrossRef] [Green Version]
- Gui, L.-J.; Jiang, S.-F.; Xie, D.-F.; Yu, L.-Y.; Huang, Y.; Zhang, Z.-J.; Liu, Y.-Y. Analysis of complete chloroplast genomes of Curcuma and the contribution to phylogeny and adaptive evolution. Gene 2020, 732, 144355. [Google Scholar] [CrossRef]
- Li, L.; Hu, Y.-F.; He, M.; Zhang, B.; Wu, W.; Cai, P.-M.; Huo, D.; Hong, Y.-C. Comparative chloroplast genomes: Insights into the evolution of the chloroplast genome of Camellia sinensis and the phylogeny of Camellia. BMC Genom. 2021, 22, 138. [Google Scholar] [CrossRef]
- Liu, S.-S.; Wang, Z.; Su, Y.-J.; Wang, T. Comparative genomic analysis of Polypodiaceae chloroplasts reveals fine structural features and dynamic insertion sequences. BMC Plant Biol. 2021, 21, 31. [Google Scholar] [CrossRef]
- Schneider, H.; Schuettpelz, E.; Pryer, K.M.; Cranfill, R.; Magallón, S.; Lupia, R. Ferns diversified in the shadow of angiosperms. Nature 2004, 428, 553–557. [Google Scholar] [CrossRef]
- Zhu, M.; Feng, P.-P.; Ping, J.-Y.; Li, J.-Y.; Su, Y.-J.; Wang, T. Phylogenetic significance of the characteristics of simple sequence repeats at the genus level based on the complete chloroplast genome sequences of Cyatheaceae. Ecol. Evol. 2022, 11, 14327–14340. [Google Scholar] [CrossRef] [PubMed]
- Ping, J.-Y.; Feng, P.-P.; Li, J.-Y.; Zhang, R.-J.; Su, Y.-J.; Wang, T. Molecular evolution and SSRs analysis based on the chloroplast genome of Callitropsis funebris. Ecol. Evol. 2021, 11, 4786–4802. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.R.; Nandhini, M.B.; Monalisha, E.; Murugan, K.; Sethuraman, T.; Nagarajan, S.; Rao, N.S.P.; Ganesh, D. Synonymous codon usage in chloroplast genome of Coffea arabica. Bioinformation 2012, 8, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.-J.; Shen, L.-W.; Chen, S.-Q.; Qu, S.; Hou, N. Codon usage profiling of chloroplast genome in Juglandaceae. Forests 2023, 14, 378. [Google Scholar] [CrossRef]
- Zhang, P.-P.; Xu, W.-B.; Lu, X.; Wang, L. Analysis of codon usage bias of chloroplast genomes in Gynostemma species. Physiol. Mol. Biol. Plants 2021, 27, 2727–2737. [Google Scholar] [CrossRef]
- Liu, H.-B.; Lu, Y.-Z.; Lan, B.-L.; Xu, J.-C. Codon usage by chloroplast gene is bias in Hemiptelea davidii. J. Genet. 2020, 99, 8. [Google Scholar] [CrossRef]
- Li, G.-L.; Pan, Z.-L.; Gao, S.-C.; He, Y.-Y.; Xia, Q.-Y.; Jin, Y.; Yao, H.-P. Analysis of synonymous codon usage of chloroplast genome in Porphyra umbilicalis. Genes Genom. 2019, 41, 1173–1181. [Google Scholar] [CrossRef]
- Wang, Z.-J.; Xu, B.-B.; Li, B.; Zhou, Q.-Q.; Wang, G.-Y.; Jiang, X.-Z.; Wang, C.-C.; Xu, Z.-D. Comparative analysis of codon usage patterns in chloroplast genomes of six Euphorbiaceae species. Peer J. 2020, 8, e8251. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Cai, X.-N.; Chen, Q.-Z.; Zhou, H.-X.; Cai, Y.; Ben, A.-L. Factors affecting synonymous codon usage bias in chloroplast genome of Oncidium Gower Ramsey. Evol. Bioinform. 2011, 7, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Wei, F.; Cai, Z.; Wei, Y.; Khan, A.; Miao, J.; Wei, K. Analysis of codon usage bias and evolution in the chloroplast genome of Mesona chinensis Benth. Dev. Genes Evol. 2021, 231, 1–9. [Google Scholar] [CrossRef]
- Li, C.-L.; Zhou, L.; Nie, J.-B.; Wu, S.-P.; Li, W.; Liu, Y.-H.; Liu, Y.-L. Codon usage bias and genetic diversity in chloroplast genomes of Elaeagnus species (Myrtiflorae: Elaeagnaceae). Physiol. Mol. Biol. Plants 2023, 29, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.-B.; Cao, L.; Zhou, M.; Xiu, L.-S. Analysis on codon usage of chloroplast genome of Eleutherococcus senticosus. China J. Chin. Mater. Med. 2013, 38, 661–665. (In Chinese) [Google Scholar]
- Hao, J.; Liang, Y.-Y.; Ping, J.-Y.; Li, J.-Y.; Shi, W.-X.; Su, Y.-J.; Wang, T. Chloroplast gene expression level is negatively correlated with evolutionary rates and selective pressure while positively with codon usage bias in Ophioglossum vulgatum L. BMC Plant Biol. 2022, 22, 580. [Google Scholar] [CrossRef]
- Hershberg, R.; Petrov, D.A. Selection on codon bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.-P.; Dang, Y.-K.; Zhou, M.; Li, L.; Yu, C.-H.; Fu, J.-J.; Chen, S.; Liu, Y. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. USA 2016, 26, e6117–e6125. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.-Y.; Xu, W.-J.; Xing, T.; Zhao, M.-M.; Li, N.-N.; Yan, L.; Xia, G.-M.; Wang, M.-C. Synonymous codon usage bias in the plastid genome is unrelated to gene structure and shows evolutionary heterogeneity. New Phytol. 2015, 209, 855–870. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Penaflor, C.; Kuehl, J.V.; Leebens-Mack, J.; Carlson, J.E.; de Pamphilis, C.W.; Boore, J.L.; Jansen, R.K. Complete plastid genome sequences of Drimys, Liriodendron, and Piper: Implications for the phylogenetic relationships of magnoliids. BMC Evol. Biol. 2006, 6, 77. [Google Scholar] [CrossRef] [Green Version]
- Hall, B.G. Comparison of the accuracies of several phylogenetic methods using protein and DNA sequences. Mol. Biol. Evol. 2005, 22, 792–802. [Google Scholar] [CrossRef] [Green Version]
- Liao, Q.; Ye, T.; Song, Y. Complete chloroplast genome sequence of a subtropical tree, Parasassafras confertiflorum (Lauranceae). Mitochondrial DNA Part B 2018, 3, 1216–1217. [Google Scholar] [CrossRef] [Green Version]
- Pal, L.; Dasgupta, B.; Chakrabarti, P. 310-Helix adjoining α-helix and β-strand: Sequence and structural features and their conservation. Biomolecules 2005, 78, 147–162. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order | Familly | Species Name | Genbank Accession No. | Cp Genome Size/bp | GC Content/% |
|---|---|---|---|---|---|
| Magnoliales | |||||
| Annonaceae | Annona cherimola | KU563738 | 201,723 | 39.60 | |
| Annona reticulata | MT742547 | 196,038 | 39.90 | ||
| Annona muricata | MT742546 | 201,906 | 39.60 | ||
| Fissistigma oldhamii | MW136266 | 187,782 | 38.90 | ||
| Fissistigma polyanthum | MW829282 | 189,920 | 38.70 | ||
| Uvaria macrophylla | MH992130 | 192,782 | 38.70 | ||
| Artabotrys hexapetalus | MZ936420 | 178,457 | 38.80 | ||
| Artabotrys pilosus | OK216144 | 178,195 | 38.80 | ||
| Polyalthiopsis verrucipes | MW018366 | 159,965 | 39.00 | ||
| Miliusa glochidioides | OM047203 | 159,789 | 39.20 | ||
| Trivalvaria costata | OM914484 | 162,002 | 39.00 | ||
| Greenwayodendron suaveolens | MH924590 | 159,031 | 39.00 | ||
| Chieniodendron hainanense | MK035708 | 160,497 | 39.00 | ||
| Cananga odorata | MN016933 | 167,946 | 39.00 | ||
| Magnoliaceae | Liriodendron chinense | NC_030504 | 159,429 | 39.57 | |
| Magnolia grandiflora | NC_020318 | 159,623 | 39.30 | ||
| Michelia × alba | NC_037005 | 160,060 | 39.25 | ||
| Houpoea officinalis | NC_020317 | 160,050 | 39.25 | ||
| Yulania denudata | NC_056770 | 160,090 | 39.24 | ||
| Manglietia fordiana | NC_058549 | 160,074 | 39.27 | ||
| Parakmeria yunnanensis | NC_024545 | 160,085 | 39.27 | ||
| Pachylarnax sinica | NC_023241 | 160,044 | 39.26 | ||
| Woonyoungia septentrionalis | NC_015892 | 159,667 | 39.26 | ||
| Lirianthe delavayi | NC_053643 | 159,478 | 39.28 | ||
| Alcimandra cathcartii | NC_023234 | 159,950 | 39.22 | ||
| Oyama sieboldii | NC_041435 | 160,770 | 39.25 | ||
| Myristicaceae | Magnolia alba | NC_060714 | 155,775 | 39.21 | |
| Endocomia macrocoma subsp. prainii | NC_042225 | 155,695 | 39.21 | ||
| Horsfieldia amygdalina | NC_060835 | 155,682 | 39.24 | ||
| Laurales | |||||
| Lauraceae | Actinodaphne lecomtei | NC_058827 | 152,863 | 39.38 | |
| Lindera glauca | NC_035953 | 152,780 | 39.73 | ||
| Cinnamomum camphora | NC_035882 | 152,570 | 39.39 | ||
| Laurus nobilis | NC_034700 | 152,750 | 39.23 | ||
| Litsea pungens | NC_050368 | 152,655 | 39.58 | ||
| Iteadaphne caudata | NC_050361 | 152,863 | 39.40 | ||
| Machilus gamblei | NC_058716 | 152,589 | 39.63 | ||
| Neocinnamomum delavayi | NC_036003 | 150,850 | 38.84 | ||
| Neolitsea pallens | NC_050370 | 152,699 | 39.33 | ||
| Ocotea guianensis | NC_061545 | 152,656 | 39.74 | ||
| Phoebe zhennan | NC_036143 | 152,831 | 39.48 | ||
| Beilschmiedia purpurascens | NC_051917 | 158,416 | 38.99 | ||
| Alseodaphnopsis hainanensis | NC_057082 | 152,829 | 39.18 | ||
| Alseodaphne gracilis | NC_037489 | 153,099 | 39.05 | ||
| Caryodaphnopsis tonkinensis | NC_050345 | 149,016 | 39.05 | ||
| Cassytha filiformis | NC_036001 | 114,622 | 36.93 | ||
| Cryptocarya chinensis | NC_036002 | 157,718 | 39.07 | ||
| Endiandra microneura | NC_051910 | 158,598 | 39.05 | ||
| Nothaphoebe cavaleriei | NC_058724 | 152,728 | 39.43 | ||
| Parasassafras confertiflorum | NC_042696 | 152,555 | 39.48 | ||
| Persea americana | NC_031189 | 152,723 | 39.04 | ||
| Potameia microphylla | NC_051913 | 158,597 | 39.04 | ||
| Sassafras tzumu | NC_045268 | 151,797 | 39.93 | ||
| Sinopora hongkongensis | NC_051914 | 158,598 | 39.01 | ||
| Syndiclis anlungensis | NC_052917 | 158,573 | 39.01 | ||
| Calycanthaceae | Idiospermum australiense | NC_042743 | 154,767 | 39.23 | |
| Chimonanthus praecox | NC_042744 | 153,252 | 39.25 | ||
| Calycanthus floridus var. glaucus | NC_004993 | 153,337 | 39.27 | ||
| Piperales | |||||
| Aristolochiaceae | Aristolochia contorta | NC_036152 | 160,576 | 38.27 | |
| Asarum sieboldii | NC_037190 | 193,356 | 36.20 | ||
| Saururaceae | Gymnotheca chinensis | NC_056145 | 161,621 | 38.30 | |
| Saururus chinensis | NC_050853 | 161,489 | 38.47 | ||
| Houttuynia cordata | NC_047437 | 160,228 | 38.36 | ||
| Piperaceae | Piper auritum | NC_034697 | 159,909 | 38.31 | |
| Canellales | |||||
| Winteraceae | Drimys granadensis | NC_008456 | 160,604 | 38.79 | |
| Pseudowintera colorata | NC_050985 | 161,675 | 38.81 | ||
| Tasmannia lanceolata | NC_050986 | 160,424 | 38.86 | ||
| Chloranthales | |||||
| Chloranthaceae | Chloranthus spicatus | NC_009598 | 157,772 | 38.89 | |
| Sarcandra glabra | NC_039621 | 158,900 | 39.23 | ||
| Model | df | ℓ | Parameters | Positive Selection Site |
|---|---|---|---|---|
| Branch model | None | |||
| rps12-CDS | ||||
| M0 | 136 | −1138.323 | ω = 0.200 | |
| F | 269 | −1110.448 | ω1 = 2.263, ω2 =2.381, ω3....... | |
| Model2 | 137 | −1138.466 | ω1 = 0.198, ω2 = 0.314 | |
| Exon 1 | ||||
| M0 | 136 | −411.148 | ω = 0.083 | |
| Model2 | 137 | −413.675 | ω1 = 0.085, ω2 = 0 | |
| Exons 2–3 | ||||
| M0 | 136 | −663.025 | ω = 0.405 | |
| Model2 | 137 | −653.180 | ω1 = 0.413, ω2 = 999 | |
| Site model | ||||
| M1a | 137 | −1084.387 | P0 = 0.922, ω0 = 0.009 | Note allowed |
| (Nearly Neutral) | P1 = 0.078, ω1 = 1 | |||
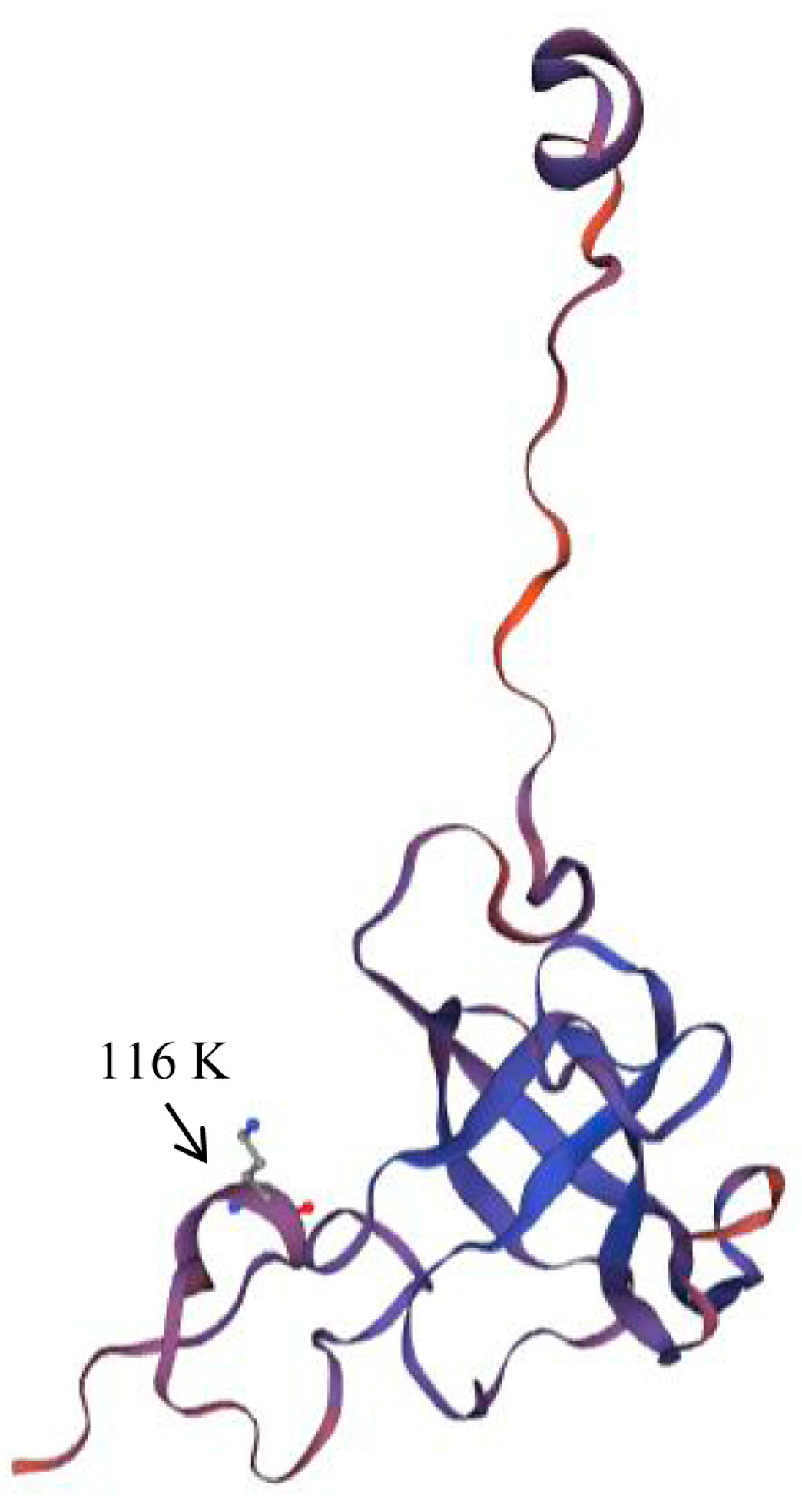
| M2a | 139 | −1058.347 | P0 = 0.910, ω0 = 0.009, | 116 K ** |
| (Positive Selection) | P1 = 0.082, ω1 = 1, | |||
| P2 = 0.008, ω2 = 12.760 | ||||
| M3 (Discrete) | 140 | −1059.785 | P0 = 0.929, ω0 = 0.015, | 16 R **, 18 V ** |
| P1 = 0.063, ω1 = 1.417, | 21 S **, 74 S **, | |||
| P2 = 0.008, ω2 = 12.571 | 109 D *, 116 K ** | |||
| M7 (β) | 137 | −1084.303 | P = 0.011, q = 0.077 | Note allowed |
| M8 (β and ω >1) | 139 | −1058.470 | P0 = 0.992, p = 0.012, | 116 K ** |
| q = 0.089 | ||||
| P1 = 0.008, ω = 13.023 | ||||
| M8a (β and ω = 1) | 138 | −1084.391 | P0 = 0.922, p = 1.007, | None |
| q = 99 | ||||
| P1 = 0.078, ω = 1 |
| Model | Comparison of Model | 2Δℓ | df | p-Value | |
|---|---|---|---|---|---|
| Branch model | |||||
| rps12-CDS | M0-F | 55.751 | 133 | 1 | |
| M0-Model2 | 0.286 | 1 | 0.593 | ||
| Exon 1 | M0-Model2 | 0.996 | 1 | 0.318 | |
| Exons 2–3 | M0-Model2 | 18.973 | 1 | 0 | |
| Site model | |||||
| M0-M3 | 157.077 | 4 | 0 | ||
| M1a-M2a | 52.079 | 2 | 0 | ||
| M7-M8 | 51.667 | 2 | 0 | ||
| M8-M8a | 51.842 | 1 | 0 | ||
| trsv | trst | ratio | dN | dS | ω | ||
|---|---|---|---|---|---|---|---|
| rps12 | SC-63 | 0.003 | 0.004 | 0.031 | 0.002 | 0.011 | 0.027 |
| IR-3 | 0.002 | 0.004 | 0 | 0.003 | 0.007 | 0 | |
| P1 | 0.931 | 0.794 | 0.839 | 0.727 | 1.000 | 0.794 | |
| Exon 1 | SC-63 | 0.002 | 0.009 | 0.019 | 0.003 | 0.029 | 0.006 |
| IR-3 | 00 | 0.012 | 0 | 0 | 0.032 | 0 | |
| P1 | 0.839 | 0.794 | 0.908 | 0.862 | 0.794 | 0.931 | |
| Exons 2–3 | SC-63 | 0.003 | 0.002 | 0.020 | 0.002 | 0.004 | 0.035 |
| IR-3 | 0.003 | 0 | 0 | 0.002 | 0 | 0 | |
| P1 | 0.839 | 0.839 | 0.908 | 1.000 | 0.662 | 0.727 | |
| SC-63 | Exon 1 | 0.002 | 0.009 | 0.019 | 0.003 | 0.029 | 0.006 |
| Exons 2–3 | 0.003 | 0.002 | 0.020 | 0.002 | 0.004 | 0.035 | |
| P2 | 0.390 | 0.003 | 0.715 | 0.190 | 0.006 | 0.021 | |
| IR-3 | Exon 1 | 0 | 0.012 | 0 | 0 | 0.032 | 0 |
| Exons 2–3 | 0.003 | 0 | 0 | 0.002 | 0 | 0 | |
| P2 | 0.317 | 0.317 | 1.000 | 0.317 | 0.317 | 1.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ping, J.; Zhong, X.; Wang, T.; Su, Y. Structural Characterization of Trivalvaria costata Chloroplast Genome and Molecular Evolution of rps12 Gene in Magnoliids. Forests 2023, 14, 1101. https://doi.org/10.3390/f14061101
Ping J, Zhong X, Wang T, Su Y. Structural Characterization of Trivalvaria costata Chloroplast Genome and Molecular Evolution of rps12 Gene in Magnoliids. Forests. 2023; 14(6):1101. https://doi.org/10.3390/f14061101
Chicago/Turabian StylePing, Jingyao, Xiaona Zhong, Ting Wang, and Yingjuan Su. 2023. "Structural Characterization of Trivalvaria costata Chloroplast Genome and Molecular Evolution of rps12 Gene in Magnoliids" Forests 14, no. 6: 1101. https://doi.org/10.3390/f14061101
APA StylePing, J., Zhong, X., Wang, T., & Su, Y. (2023). Structural Characterization of Trivalvaria costata Chloroplast Genome and Molecular Evolution of rps12 Gene in Magnoliids. Forests, 14(6), 1101. https://doi.org/10.3390/f14061101