1. Introduction
WRKY transcription factors (TFs) are a unique class of proteins commonly present in higher plants and throughout the entire green lineage. These proteins are characterized by a distinct 60-amino acid WRKY domain, which encompasses a highly conserved WRKYGQK sequence and a zinc finger motif, enabling them to bind robustly to W-box
cis-elements [
1,
2,
3]. According to the number of WRKY domains and their zinc finger motif structure, WRKY proteins can be divided into three major categories [
4]. These proteins exhibit a wide range of functions, influencing various physiological processes. These include aging [
5], seed dormancy and germination [
6], embryogenesis [
7], trichome development [
8], regulation of flowering time [
9,
10], determination of fruit flavor [
11], and biosynthesis of secondary metabolites [
12,
13,
14].
Research has underscored the role of WRKY TFs in plant defense responses against both biotic and abiotic stresses. These TFs are responsive to dehydration stress and phosphate deficiency. For instance,
VvWRKY11 from grapevines is implicated in dehydration stress response [
15], while
AtWRKY75, induced during phosphate deficiency, has been linked to increased phosphate stress susceptibility when suppressed [
16]. In maize, salt tolerance is facilitated by the interaction between transcription factor ZmWRKY20 and ZmWRKY115, which represses
ZmbZIP111 expression [
17]. Furthermore, the
BnD11 gene expression from the WRKY transcription factor family augments salt tolerance and drought resistance in rapeseed [
18]. Moreover, WRKY TFs play a pivotal role in signal transduction processes, especially those regulated by plant hormones, such as abscisic acid (ABA) and salicylic acid (SA) [
6]. They work in conjunction with other elements to fine-tune responses to environmental stimuli. Studying the evolution of
WRKY gene family provides valuable insights into the development of biotic and abiotic stress responses and signal transduction mechanisms.
Salinity stress, caused by the accumulation of high levels of salt in the soil, poses a major threat to agricultural productivity worldwide. As the global climate changes and human activities lead to soil salinization, understanding the molecular mechanisms that govern plant responses to salinity stress becomes crucial for developing stress-tolerant crops and ensuring food security. Among the various transcription factors that play pivotal roles in mediating plant stress responses, the WRKY family has emerged as a key player in the regulatory network involved in salinity stress adaptation [
19]. In recent years, accumulating evidence has suggested that WRKY transcription factors play a central role in orchestrating plant responses to salinity stress [
19,
20,
21]. Several members of the WRKY family have been shown to exhibit differential expression under salinity stress, and their overexpression or suppression has been associated with enhanced or reduced tolerance to salt in different plant species [
22]. These findings highlight the potential of WRKY transcription factors as promising candidates for improving crop salinity tolerance through genetic engineering or breeding strategies.
Salinity stress responses in plants are complex and interconnected with various signaling pathways. Recently, gasotransmitters, like nitric oxide (NO), jasmonates (JA), and hydrogen sulfide (H
2S), have emerged as important players in these processes. NO, as a versatile signaling molecule, plays a crucial role in plants by acting as a highly reactive free radical gas [
23]. JA, on the other hand, are lipid-derived plant hormones that regulate diverse processes, including defense responses and developmental pathways [
24,
25]. H
2S, similar to NO, interacts with other hormone signaling pathways, such as ABA and ethylene, to modulate plant responses to environmental stresses [
26]. These small gaseous molecules serve as vital signaling mediators, facilitating cross-talk between different stress response pathways and regulating various physiological processes in plants [
27]. Combining salinity stress with the application of these stressors provides valuable insights into the molecular mechanisms underlying plant acclimation and adaptation to multiple environmental challenges.
Cyclocarya paliurus (
Batal.) Iljinskaja (
C. paliurus), also known as the sweet tea tree or the tea tree of immortality, is a deciduous tree native to China [
28,
29,
30]. It belongs to the
Juglandaceae family, which also includes walnuts and pecans [
30]. The tree is primarily found in the central and southern regions of China, particularly in the provinces of Hubei, Hunan, and Guizhou [
31].
Cyclocarya paliurus (
C. paliurus) has long been recognized for its medicinal properties, containing valuable biological components such as triterpenoids, polysaccharides, and flavonoids [
32,
33]. Its leaves are commonly used in traditional Chinese medicine to treat various ailments, and the tea made from
C. paliurus leaves has gained popularity due to its diverse health benefits [
32,
34]. However, the species is facing environmental challenges, especially salinity stress, which can harm plant growth [
35]. To protect
C. paliurus and understand its response to salinity stress, it is crucial to identify key genes involved in this process [
28,
36]. Among the gene families associated with salinity stress response, the
WRKY gene family has been extensively studied in other plant species but remains relatively unexplored in
C. paliurus [
37]. The recently completed
C. paliurus genome sequencing provides an opportunity to study the structure, expression, and evolutionary characteristics of the
WRKY gene family at the whole genome level [
38].
The study aims to bridge this knowledge gap by investigating the role of WRKY genes in the response to salt stress in C. paliurus. By identifying the WRKY family genes associated with salt tolerance and analyzing their expression profile, we hope to provide insights into the molecular mechanisms underlying salt tolerance in C. paliurus. This knowledge is crucial for enhancing the salt tolerance of C. paliurus and exploring the feasibility of cultivating it in coastal saline areas.
In this study, we focused on the WRKY gene family in C. paliurus and identified 80 WRKY genes categorized into three groups. Comprehensive analyses were conducted to understand their characteristics and evolution. We also examined their expression under different conditions, including salinity stress. Additionally, we explored the interplay between WRKY genes and gasotransmitters (NO, JA, H2S) during salinity stress. These findings provide insights into stress adaptation and offer potential strategies for enhancing crop resilience and global food security.
2. Materials and Methods
2.1. Gene Identification
This study employed a systematic approach to identify and validate
WRKY genes in the
C. paliurus genome. The genome of
Cyclocarya paliurus (
Batal.) Iljinskaja
(C. paliurus) can be extracted at the website (
https://ngdc.cncb.ac.cn, accessed on 12 April 2023). This was achieved using hidden Markov models (HMMs) from the Pfam protein family database and by confirming the presence of core WRKY sequences. Further validation of the annotation of the predicted
WRKY gene model through RNA seq reading. These models were then curated, with some being validated through PCR amplification and sequencing, culminating in the final set of
WRKY gene models.
Additional information about the identified WRKY proteins was also gathered. This included data on sequence length, molecular weights, isoelectric points, and predictions of subcellular locations. These details were obtained using tools available on the ExPasy website (
https://www.expasy.org/, accessed on 15 April 2023).
2.2. Sequence Analysis
A series of bioinformatics tools and techniques were used to analyze the WRKY proteins in
C. paliurus. The WRKY domain sequence of protein characterizations were aligned using ClustalW 2.0 software (
https://www.ebi.ac.uk/Tools/msa/clustalw2/, accessed on 20 April 2023) and subsequently adjusted with the help of GeneDoc 2.7 software. Using Gene Structure Display Server (GSDS) (
http://gsds.cbi.pku.edu.cn/, accessed on 25 April 2023) to determine the Exon Intron Tissue of
C. paliurus WRKY gene [
39]. Furthermore, the MEME online program (
http://meme.nbcr.net/meme/intro.html, accessed on 13 May 2023) was employed to identify conserved motifs in the WRKY proteins we discovered [
40]. These steps provided valuable insights into the structure of these proteins and their potential functions within
C. paliurus. This tool was used with optimized parameters, including a maximum of 20 motifs and an optimum width between 5 and 100 residues.
2.3. Chromosomal Distribution and Gene Duplication
In this study, we used a variety of tools to analyze the
CpWRKY genes in
C. paliurus. First, we mapped these genes to the chromosomes of
C. paliurus using a tool called Circos [
41]. Next, we employed MCScanX to examine gene duplication events, using the software’s default parameters [
42]. Finally, we used Dual Synteny Plotter software (
https://github.com/CJ-Chen/Tbtools, accessed on 20 May 2023) to create syntenic analysis maps [
43]. These maps allowed us to visualize the synteny relationships between the orthologous
WRKY genes in
C. paliurus and those in other selected species.
2.4. Classification and Phylogenetic Analysis of C. paliurus WRKY Gene Family
The outgroup is a taxon closely related to, but not part of, the group of interest (ingroup). It helps root the phylogenetic tree, clarifying evolutionary relationships within the ingroup. By comparing the two, we can deduce the common ancestor and evolutionary path. In our study, we chose Molluginaceae as the outgroup based on its established taxonomic relationship and prior research [
44]. Using an outgroup like this enhances the precision of our phylogenetic analysis by offering a benchmark for evolutionary comparison. The
WRKY genes in
C. paliurus were classified into different groups using the At-WRKY classification scheme. This was achieved by aligning their WRKY domains with those of CpWRKY and AtWRKY proteins. The phylogenetic tree is constructed using the Poisson model and through the neighbor-joining (NJ) method in MEGA 5.0.
To compare the
C. paliurus WRKY genes with those of other species, we obtained WRKY protein sequences from
Arabidopsis and rice [
45,
46]. These sequences were retrieved from the Phytozome database, based on the descriptions provided in the corresponding literature. This comparative analysis allowed for a deeper understanding of the evolutionary relationships and potential functional similarities between the
WRKY genes in these different species.
2.5. RNA-Seq Data Analysis and GO Annotation
We sourced our transcriptome data from the National Center for Biotechnology Information (NCBI) BioProject database, specifically under the project number PRJNA799813. We then selected
CpWRKYs that had RPKM (reads per kilobase of transcript, per million mapped reads) and FPKM (fragments per kilobase of transcript per million mapped reads) values greater than 1 for further analysis. To visualize the expression patterns of these selected genes, we generated a heatmap using TBtools V1.098 [
47]. For gene ontology (GO) analysis, we refer to the NCBI database, using the Blast2GO program (
https://www.biobam.com/blast2go/, accessed on 30 May 2023) [
48]. The results of the study can be divided into the following three main categories: molecular function, biological processes, and cellular components.
2.6. Promoter Cis-Acting Element Prediction
To analyze promoter sequences, the sequence 1500 bp upstream of the transcription start site of the
CpWRKYs gene was extracted from transcriptome sequencing data using BEDtools (version 2.25.0) [
49]. The PlantCARE online tool (
https://bioinformatics.psb.ugent.be/webtools/plantcare/html/, accessed on 26 April 2023) was utilized to identify motifs present in promoter sequences.
2.7. Plant Materials and Treatments
High-salting pot experiment of C. paliurus: In order to better observe the effects of high-concentration salt stress on the growth of C. paliurus, in this study, two C. paliurus plants with good growth status were selected, one was treated with high concentration salt (4% NaCl), and the other was treated with distilled water as blank control. Pour 1L of the corresponding solution into the pot every 2 days. Observe and record the growth of the plants every day. Because of the high concentration of salt treatment for a long time, the growth of willow plants was seriously affected, and even led to the death of the plants. The plants treated with high salt can be used to view phenotypic changes but may cause low confidence in subsequent experimental data. Therefore, this study adopts the method of low salt treatment to carry out the following experiment.
The study conducted eight treatments. These treatments included a control group (CK) that was treated with distilled water, a group subjected to salt stress with 0.4% NaCl, a Methyl Jasmonate (MeJA) group treated with 0.2 mM MeJA and distilled water, and a combined salt + MeJA group. We also had a Sodium Nitroprusside (SNP) group treated with 0.25 mM SNP and distilled water, a combined salt + SNP group, a Sodium Hydrosulfide (NaHS) group treated with 0.5 mM NaHS and distilled water, and a combined salt + NaHS group. The NaCl solution was gradually introduced into the planting substrate to induce salt stress. The MeJA, SNP, and NaHS solutions were sprayed onto the plants to induce various physiological responses.
After a period of 30 days, leaf samples were collected from each plant. We ensured a representative sample by collecting two leaves from the upper, middle, and lower parts of each plant. These samples were immediately frozen in liquid nitrogen to halt any biological activity and then stored at −80 °C until further analysis. This rigorous process ensured the preservation of the samples and the reliability of the subsequent analyses.
2.8. Expression Profile under Salinity Stress and qRT-PCR Analysis
In the pot experiment, we quantified the transcript abundance using a metric known as fragments per kilobase of transcript per million mapped reads (FPKM) values. This measure provides a normalized count of the number of reads mapped to each gene, allowing for a comparison of gene expression levels across different samples.
We derived the expression profiles of C. paliurus from the transcriptome sequencing data under various treatments. These treatments included the control (CK), salt, salt + Sodium Nitroprusside (SNP), salt + Methyl Jasmonate (MeJA), and salt + Sodium Hydrosulfide (NaHS).
To visualize the expression patterns across these different treatments, we generated a heatmap using the TBtools software (
https://github.com/CJ-Chen/TBtools, accessed on 6 June 2023). This graphical representation allowed us to easily compare and contrast the gene expression levels under different conditions, providing valuable insights into the response of
C. paliurus to various stressors.
2.9. Sequence Analysis
For the statistical analysis of our data, we employed a one-way analysis of variance (ANOVA). This method allowed us to identify significant variations in gene expression across the different treatments. Following the ANOVA, we used the Duncan’s test for multiple comparisons to further investigate the differences between each treatment group.
All of these statistical analyses were performed using IBM SPSS Statistics Version 22 software (SPSS Inc., IBM Company Headquarters, Chicago, IL, USA). This software is a powerful tool for managing and analyzing data, and it provided us with robust and reliable results. The data obtained from these analyses were presented as means with their corresponding standard deviations (SD).
3. Results
3.1. Identification of WRKY Proteins in Cyclocarya paliurus (C. paliurus)
At the outset, we obtained a total of 95 candidate gene models corresponding to the Pfam WRKY family. To validate these gene models, we cross-checked their annotations using available
Cyclocarya paliurus (
C. paliurus) transcriptome data. Through a comparative analysis with the WRKY family in
Arabidopsis thaliana, HMM models, and Pfam results, we identified discrepancies and manually curated some incorrectly predicted
WRKY gene models. As a result, 12 redundant sequences were removed. Ultimately, we meticulously selected and annotated 80 gene models as
C. paliurus WRKY genes, ensuring they were included based on the presence of the intact WRKY domain. The validation sequences of
CpWRKY genes are provided in
Supplementary File S1. We successfully mapped these 80
WRKY genes onto the corresponding linkage groups and systematically renamed them as
CpWRKY1 to
CpWRKY80 based on their sequential arrangement on the linkage groups (
Figure S1).
We further analyzed gene characteristics, including the length of the coding sequence (CDS), length of the protein sequence, protein molecular weight (MW), isoelectric point (pI), and subcellular localization (
Supplementary Excel S1). Among the 80 CpWRKY proteins, CpWRKY42 was identified as the smallest protein, consisting of 152 amino acids (aa), while CpWRKY19 was the largest, with 818 aa. The MW of the proteins was from 17.7 to 89.6 kDa, and the pI was from 5.06 (CpWRKY50 and CpWRKY19) to 9.93 (CpWRKY3). In terms of subcellular localization, all 80 CpWRKY proteins were predicted to be located in the nucleus, as indicated by our predictions (
Supplementary Excel S1).
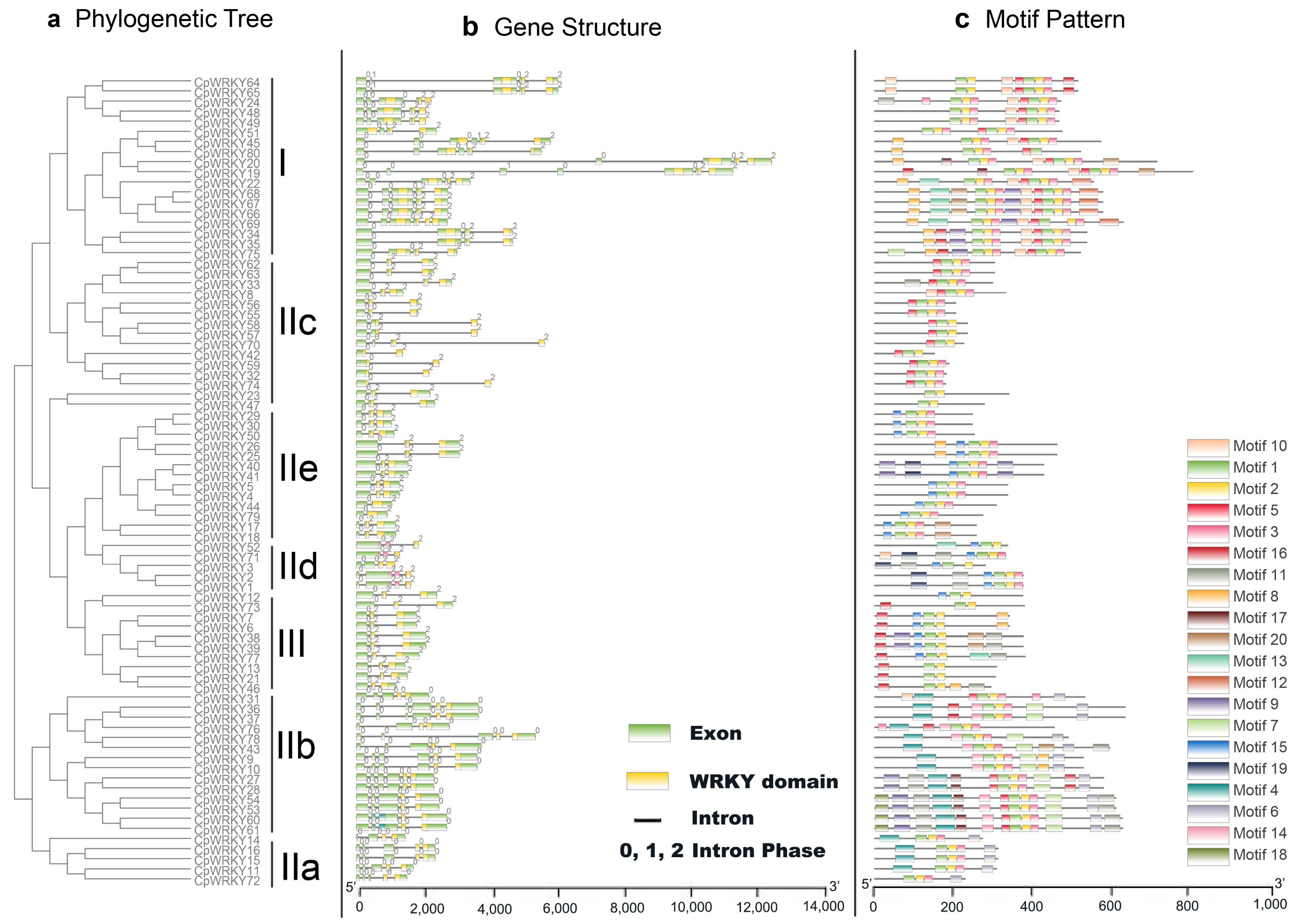
3.2. Multisequence Alignment, Phylogenetic Analysis, and Classification of CpWRKY Genes
The CpWRKY genes are classified based on phylogenetic analysis and multiple sequence alignment of its WRKY domain, which usually spans about 60 amino acids. As representatives for comparison, we randomly selected the WRKY domains of seven different Arabidopsis WRKY proteins (AtWRKY33, 18, 42, 48, 74, 69, 55) from each of the groups or subgroups.
Phylogenetic analysis (
Figure 1) identified three main groups in
C. paliurus, which corresponded to groups I, II, and III as identified in
Arabidopsis, a model plant species [
2]. Group I consisted of 18 CpWRKY proteins, all containing two WRKY domains and C2H2-type zinc finger motifs (C-X4-C-X22–23-H-X-H) (
Figure S2; Supplementary Excel S1). Through further analysis, we found that the WRKY domain sequences of 80 CpWRKY proteins had high conservatism. Specifically, 78 out of the 80 CpWRKY proteins displayed the highly conserved sequence WRKYGQK within their WRKY domain. This conservation suggests the functional importance of this sequence motif in WRKY proteins. However, it is worth noting that CpWRKY56 and CpWRKY55 exhibited a slight variation in this conserved sequence, differing by a single amino acid (
Figure S2). Notably, CpWRKY80, belonging to group I, possessed two WRKY domains and a C2H2-type zinc finger motif at its N-terminus, while lacking this motif at the
C-terminus. Group II contains 52 CpWRKY proteins, all of which possessed C2H2-type zinc finger (C-X4-5-C-X23-H-X-H), which can be further subdivided into 5 subgroups (IIa-IIe). Within group II, 5 WRKY proteins belonged to subgroup IIa, 14 to IIb, 15 to IIc, 5 to IId, and 13 to IIe. Lastly, group III included 10 CpWRKY members, all of which possessed C2HC-type zinc finger (C-X7-C-X23-H-X-C) (
Figure S2; Supplementary Excel S1).
3.3. Gene Structure and Motif Composition of C. paliurus WRKY Gene Family
To better understand the evolution of the
WRKY gene family in
C. paliurus, we examined the exon–intron organization of all the identified
CpWRKY genes.
Figure 2 illustrates that
CpWRKY genes exhibit diverse exon numbers, ranging from two to seven. Specifically, we observed that 6 genes have 2 exons, 33 genes have 3 exons, 10 genes have 4 exons, 21 genes have 5 exons, 9 genes have 6 exons, and 1 gene has 7 exons. Interestingly, we did not find any genes with only one exon in our analysis; moreover, we noticed a pattern where genes belonging to the same group tend to have similar exon–intron structures (
Figure 2a,b). For instance, all members of group IIe share a three-exon and two-intron configuration.
Notably, each
CpWRKY gene was found to contain an intron within its WRKY domain. The distribution and phases of these introns corresponded with those observed in the
CpWRKY gene alignment. The V-type intron is a phase 0 intron, which is only found in IIa and IIb groups. On the other hand, type r intron is a phase 1 intron, which has been identified in all other groups, including groups I, IIc, IId, IIe, and III. This pattern mirrors observations made in both rice and
Arabidopsis [
24]. In a surprising finding, we discovered that no introns were present in the N-terminal WRKY domains of the group I genes. This absence of introns in these specific domains could suggest unique regulatory mechanisms or evolutionary paths for these genes.
Moving on to the motif composition of CpWRKY proteins, we conducted MEME motif analysis (
Figure 2c). In addition to the well-known WRKY domains (motifs 1 and 2), CpWRKY members within the same groups often shared similar motif compositions (
Supplementary Excel S2). For instance, motif 10 was unique to group I, while motif 4 and motif 6 were specific to group IIa and IIb, respectively. We also observed closely clustered pairs of CpWRKY proteins, such as CpWRKY4/5, CpWRKY44/79, CpWRKY23/47, CpWRKY32/74, and others, which exhibited highly similar motif distributions. The presence of these similar motif arrangements within specific subgroups suggests conserved protein architecture; however, the roles of the majority of these preserved motifs are still unknown.
In summary, the conserved motif composition and similar gene structure in the same taxa, combined with the results of our phylogenetic analysis, provide strong support for the reliability of taxa classification.
3.4. Evolutionary Analysis of Group III WRKY Genes in C. paliurus and Several Different Species
The group III
WRKY gene is thought to play a crucial role in plant adaptation and evolution, emerging after the differentiation of monocots and dicots [
29]. In order to further investigate the duplication and diversity of
C. paliurus group III genes, we detected available
WRKY III genes. The phylogenetic tree of WRKY III was constructed using the whole protein sequences of WRKY III of three monocots (
pineapple, rice,
maize) and four dicots (
Arabidopsis,
grapes, and
poplar) (
Supplementary Excel S3) [
4,
50,
51]. The phylogenetic analysis, performed using MEGA 5.0, revealed two distinct clades (
Figure S3). Within these clades, WRKY members from closely related species clustered together, signifying their evolutionary relationships. For instance, Clade 1a and Clade 1b consisted of members from rice and maize, two non-grass monocots. Clade 1c encompassed
WRKY genes from six different species, suggesting they might be orthologous genes originating from a common ancestor. Intriguingly, a small branch in the phylogenetic tree grouped together three
C. paliurus WRKY members with three poplar WRKY members and two grape WRKY members, indicating their evolutionary connection during the evolutionary process.
Through the use of the MEME web server, we identified 20 distinct conserved motifs among the WRKY III proteins. Notably, motifs 1, 2, and 10 were found to encode the WRKY domain. We observed that the WRKY members from the same lineage often shared similar motif compositions, suggesting potential functional similarities. Interestingly, motif 6 was unique to members of Clade 2, suggesting that it might play a critical role in the biological activities of certain WRKY III proteins. Additionally, motif 20 was found exclusively in WRKY III proteins from monocotyledonous plants, indicating that it may have played a significant role in the functional divergence of WRKY genes during the evolution of these plants.
In conclusion, our phylogenetic analysis and examination of motif composition among group III WRKY genes in C. paliurus and various species provide valuable insights into their evolutionary patterns and potential functional similarities.
3.5. Chromosomal Distribution and Synteny Analysis of AcWRKY Genes
In our study, we observed an uneven distribution of the
CpWRKY genes across the 16
C. paliurus linkage groups (chromosomes, Chr), with the exception of Chr 14 (
Figure S4). Chr05 had the highest number of
CpWRKY genes, totaling 11. Some linkage groups, such as Chr05, Chr04, and Chr02, had a higher number of
CpWRKY genes, while others, like Chr10 and Chr12, had fewer.
We also identified tandem duplication events, which are a common mechanism for gene family expansion. Notably, Chr05 revealed four clusters of CpWRKY genes, indicating that it was a hotspot for WRKY gene distribution. In total, the 47 CpWRKY genes were clustered into 23 locations across various linkage groups, further highlighting the occurrence of tandem duplication events.
Interestingly, we found that the quantity of
WRKY genes did not positively correlate with the length of the chromosomes. This suggests that other factors, such as selective pressure or functional diversification, might have influenced the distribution and expansion of the
CpWRKY genes. In addition to tandem duplication events, the study also identified that 30
WRKY genes from segmental duplication events and 3
WRKY genes are dispersed duplication by using BLASTP and MCScanX methods (
Supplementary Excel S4). These results suggest that gene duplication, both tandem and segmental, played a significant role in the evolution of
CpWRKY genes.
To further investigate the phylogenetic mechanisms of the
CpWRKY gene family, comparative syntenic maps were constructed for
C. paliurus with the following two representative species:
Arabidopsis (a dicot) and rice (a monocot) (
Figure 3). A total of 24
CpWRKY genes showed syntenic relationships with genes in
Arabidopsis, followed by rice with 11 syntenic genes (
Supplementary File S2). The numbers of orthologous gene pairs between rice and
Arabidopsis were 22 and 31. Some
CpWRKY genes were discovered to be linked to at least three syntenic gene pairs, indicating their significance in the evolution of the
WRKY gene family.
Interestingly, certain collinear gene pairs identified between C. paliurus and rice were not found between C. paliurus and Arabidopsis, such as CpWRKY9/OsWRKY9 and CpWRKY13/OsWRKY47. These orthologous pairs may have developed following the split between dicotyledonous and monocotyledonous plants, according to this evidence. Additionally, certain collinear pairs between C. paliurus and both rice and Arabidopsis (with eight CpWRKY genes) were discovered, suggesting that these orthologous pairs may have existed before the original separation.
3.6. GO Annotation and Cis-Element Analyses of the CpWRKYs
To understand the biological functions associated with
CpWRKY genes, GO annotation was performed on the 80 identified CpWRKY proteins (
Figure S5; Supplementary Excel S5). The GO annotation results categorized the CpWRKY proteins into the following three primary gene ontology (GO) terms: In the context of gene ontology (GO) analysis, CC refers to the cellular component, MF denotes molecular function, and BP stands for biological process. In the molecular function (MF) category, the majority of CpWRKY proteins (74 out of 80) were annotated for “molecular function”, with a significant emphasis on “nucleic acid binding” and “DNA binding”. These functions are closely related to the primary roles of transcription factors (TFs), which are to regulate gene expression by binding to specific DNA sequences. In the cellular component (CC) category, the majority of CpWRKY proteins (31 out of 80) were assigned to the nucleus, which is consistent with their role as transcription factors. However, only a few CpWRKY proteins were found to be located in the cytoplasm (3 out of 80), organelle parts (CpWRKY6/7), plastids (3 out of 80), and chloroplasts (3 out of 80) (
Figure S5; Supplementary Excel S5). This distribution reflects the primary function of these proteins in the nucleus, where they interact with DNA to regulate gene expression.
In terms of the BP aspect, CpWRKY proteins were found to participate in various biological processes. The largest number of CpWRKYs (74 out of 80) were annotated to be involved in multiple biosynthetic and metabolic processes (
Figure S5; Supplementary Excel S5). Additionally, CpWRKY proteins were implicated in the regulation of biological processes, such as the regulation of cellular processes (74 out of 80), transcription (74 out of 80), DNA-templated processes (74 out of 80), and gene expression (74 out of 80). Furthermore, the analysis of biological processes (BP) showed that a significant number of CpWRKYs (44 out of 80) are involved in responding to stimuli, encompassing various types of biotic and abiotic stressors.
In this study, we analyzed conserved motifs in the gene promoter regions of
CpWRKY genes, which act as recognition and binding sites for proteins. These
cis-regulatory elements (CREs) were categorized into the following three main groups based on their functions: plant growth and development, phytohormone responsiveness, and responses to abiotic and biotic stresses (
Figure 4).
This study identified multiple motifs within the CpWRKY genes, with CAT-box and O2-site being the most frequently found, associated with meristem expression and zein metabolism regulation, respectively. Other motifs related to palisade mesophyll cell differentiation (HD-Zip 1) and cell cycle regulation (MSA-like) were less prevalent. The study also discovered RY-element and GCN4-motif in the promoters of CpWRKY genes, known for seed-specific regulation and endosperm expression, respectively. Insights into the potential roles of these motifs in regulating gene expression in response to various biological processes and stress conditions were provided. Additionally, various phytohormone-responsive elements (ABRE, CGTCA-motif, TGACG-motif, and TCA element) and stress response elements (ARE, MBS, LTR, GC-motif, TC-rich, and ERE) were identified, shedding light on the potential regulatory mechanisms allowing CpWRKY genes to respond to different stimuli in plant growth, development, and stress responses.
3.7. Salinity Stress Elicits WRKY Genes’ Response in C. paliurus
Salinity stress can negatively affect the growth and development of plants. To cope with this stress and adapt to harsh conditions, plants regulate their gene expression. Previous studies have explored the role of the
WRKY gene family in salinity stress in other species, such as pineapple [
52]. However, there is a lack of research specifically focusing on the involvement of
WRKY genes in salinity stress in
C. paliurus.
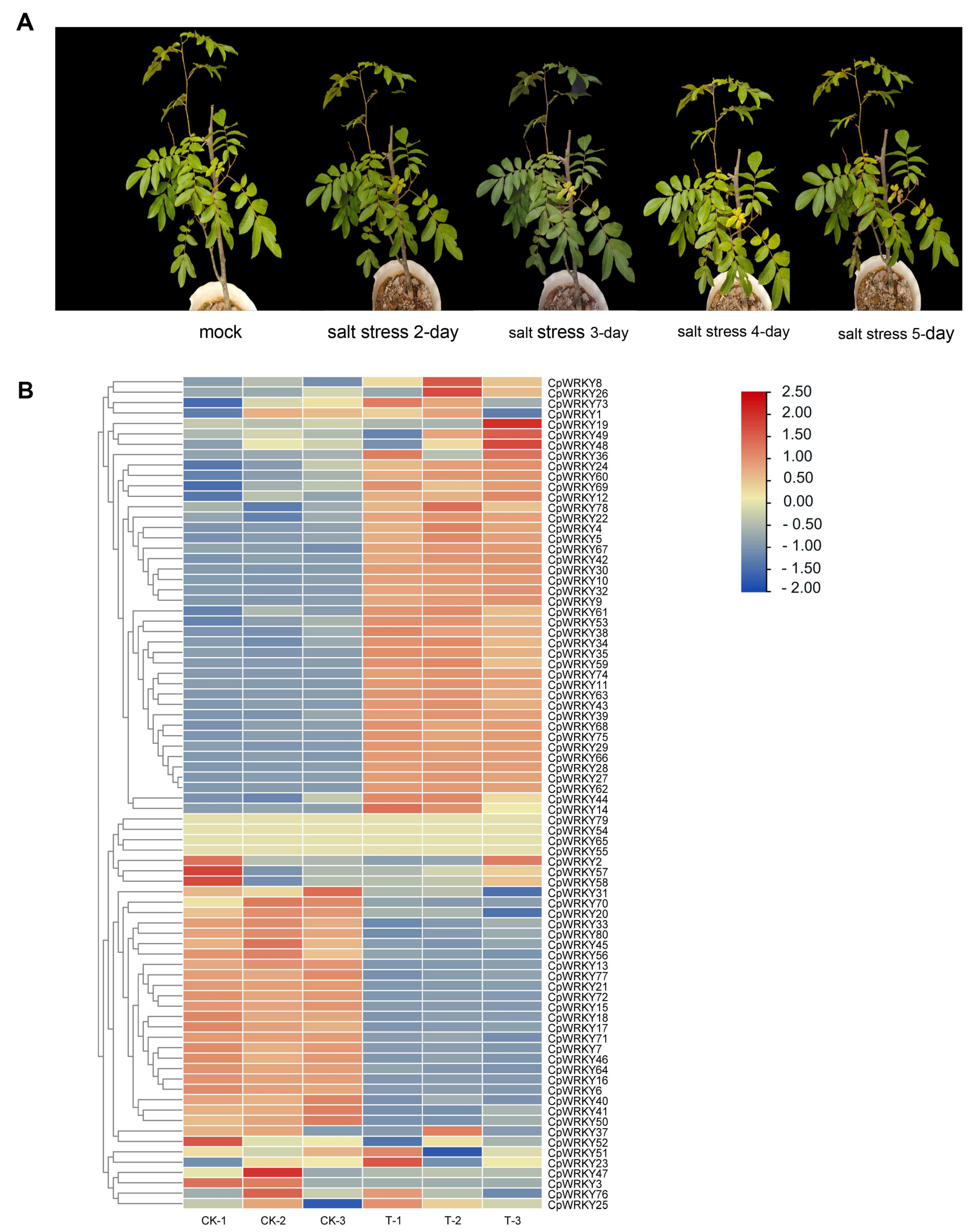
To address this gap, we conducted a growth experiment under salinity stress conditions to investigate how
C. paliurus responds to such stress. The results were presented in
Figure 5A, which showed that the plant indeed demonstrated a response to salinity stress.
Next, we analyzed the expression changes of the
WRKY gene family under salinity stress. The findings, depicted in
Figure 5B, revealed that 43.8% (38) of the
WRKY genes were significantly up-regulated, indicating that they play important roles in responding to salinity stress. On the other hand, 27.5% (22) of the genes were down-regulated, while 28.7% (23) showed no significant change. This suggests that salt stress alone is a potent inducer of
WRKY gene expression, indicating their involvement in the plant’s response to salinity stress.
3.8. Profiling of Expressed CpWRKY Genes under Combined Salinity Stress
Jasmonates (JA), hydrogen sulfide (H2S), and nitric oxide (NO) function as signaling molecules that modulate plant growth, development, and responses to environmental stresses. In this study, we aimed to investigate the response of the WRKY gene family to salinity stress in combination with the following specific stressors: Methyl jasmonate treatment (MeJA), sodium hydrosulfide treatment (NaHS), and sodium nitroprusside treatment (SNP). These treatments induce JA, H2S, and NO production, respectively. To further explore this phenomenon, we conducted a comprehensive analysis of CpWRKY gene expression patterns under various conditions using transcriptome data.
We observed that 80
CpWRKY genes were expressed under these treatments, and the combination treatment exhibited a superior capacity to induce the up-regulation of
CpWRKY gene expression compared to the individual treatments (
Figure S6A). For instance, when MeJA was administered alone, 26.3% (21) of
CpWRKY genes showed up-regulation; however, under the combined treatment of salt and MeJA, 48.8% (39) of
CpWRKY genes displayed up-regulation (
Figure S6B). Similar trends were observed for NaHS, SNP, NaCl + NaHS, and NaCl + SNP treatments. As depicted in
Figure S6C, only 5.0% (4) of
CpWRKY genes were up-regulated under NaHS alone; nevertheless, under the combined treatment of salt and NaHS, 41.3% (33) of
CpWRKY genes exhibited up-regulation. Under the SNP stress treatment condition alone, only 6.3% (5) of
CpWRKY genes demonstrated up-regulation; however, under the combined treatment with salt and SNP stress conditions applied together resulted in an increase in expression levels for 50.0% (40) of
CpWRKY genes (
Figure S6D).
Additionally, we verified the expression patterns of 11 genes selected at random that had exhibited up-regulation in response to salt-induced stress through qRT-PCR analysis. The results obtained from qRT-PCR were consistent with those from transcriptome data. Specifically, two genes showed up-regulation under MeJA treatment, six genes were down-regulated under NaHS treatment, and three genes were down-regulated under SNP treatment. However, most of these genes demonstrated up-regulation when subjected to combined salinity stress with MeJA or NaHS or SNP treatments (
Figure 6A–C).
In summary, the combined treatment exhibited an antagonistic effect on individual treatments, resulting in a significant up-regulation of CpWRKY genes. This suggests a synergistic interaction between salinity stress and other stressors in regulating the expression of these genes. These findings provide valuable insights into the intricate interplay among different stress signaling pathways and highlight the pivotal role of WRKY genes in plants’ response to salinity stress.
Overall, our study results suggest that salt stress combined with combined abiotic stressor treatment can lead to an up-regulated WRKY gene expression, emphasizing the complex and interactive nature of plant stress responses and highlighting the potential role of NASH in modulating these responses. Salt stress alone is a potent inducer of WRKY gene expression, indicating their involvement in plants’ response to salt stress.
4. Discussion
Cyclocarya paliurus (
C. paliurus) has been recognized for its medicinal properties and has been used for generations in traditional Chinese medicine to treat a variety of health conditions [
31,
32]. The plant contains a rich array of bioactive substances, including flavonoids, polysaccharides, triterpenic acids, and essential human trace elements [
29,
53,
54,
55]. These compounds contribute to the diverse biological and physiological activities exhibited by
C. paliurus, making it a valuable resource in the field of herbal medicine. One of the well-known medicinal virtues of
C. paliurus is its use in managing hyperlipidemia and coronary heart disease [
32,
56]. Studies have shown that
C. paliurus leaves possess cholesterol-lowering properties and can effectively reduce lipid levels in the blood, thereby promoting cardiovascular health [
57]. Additionally,
C. paliurus has been found to have anti-hyperglycemic effects, making it a potential natural remedy for managing high blood sugar levels in diabetes [
58]. The plant’s polysaccharides have been studied for their antioxidant properties, helping to combat oxidative stress in the liver and kidneys. Oxidative stress can cause damage to cells and tissues, leading to various diseases, but the polysaccharides from
C. paliurus have demonstrated protective effects against such stress-induced damage [
59]; moreover,
C. paliurus has been investigated for its potential to support the immune system. Some studies suggest that the plant’s bioactive compounds may enhance immunity, making it a promising candidate for developing immune-boosting supplements or functional foods [
34]. Environmental factors may cause a decline in leaf yield and quality of
C. paliurus. For example, seasonal changes affect leaf elongation and aging in
Cyclocarya paliurus, as well as their effects on the accumulation of flavonoids and phenolic compounds in the leaves [
60]. This paper mainly discusses the effect of salt stress on it.
Despite its remarkable medicinal properties,
C. paliurus faces challenges posed by environmental stresses, particularly salinity stress. Salinity stress can negatively impact the growth of
C. paliurus and, consequently, the yield and quality of its bioactive compounds. The
WRKY gene family plays a pivotal role in plants’ responses to various stresses, including salinity. Although salinity is also an abiotic stress source that seriously harms plant growth and crop yield, plants have evolved corresponding protection mechanisms to cope with external hazards. For example, WRKY transcription factor participates in JA regulation pathway, regulates H
2S signaling pathway and controls NO production in plants, thus enhancing plant resistance to stress. It has been reported that plants will produce NO after being harmed by salinity stress in the process of growth and development, in order to reduce the toxic effect of salting [
61]. The application of artificial exogenous JA increased the antioxidant reaction of plants and alleviated the harm of salinity stress [
62]. The H2S alleviates salt stress in plant by maintaining Na/K balance, regulating H2S metabolism and oxidative stress response. [
63]. Moreover, in response to drought stress, tomato SlWRKY81 transcription factor attenuates the effects of drought by regulating the production of intercellular NO [
64]. The transcription factor WRKY75 also plays an important role in plant defense against necrotic fungal pathogens by positively regulating the jasmonate-mediated mechanism [
65]. Understanding the genetic basis of its response to salinity stress, especially the role of the
WRKY gene family, can offer insights into potential strategies for protecting this valuable medicinal plant and harnessing its therapeutic potential.
This study delves into the WRKY gene family, a large group of transcription factors found in all plant species, with a focus on Cyclocarya paliurus (C. paliurus). We identified 80 WRKY genes in the C. paliurus genome, named CpWRKY1 to CpWRKY80 based on their chromosomal locations. Two of these genes, CpWRKY55 and CpWRKY56, showed sequence variations in their WRKY domains, suggesting potential differences in their interactions with downstream target genes.
The study also explored the evolution of the WRKY gene family. Unlike many monocotyledons, such as rice and maize, where a loss of WRKY domain is common, all group I WRKY proteins in C. paliurus have two domains, indicating unique evolutionary characteristics. The number of WRKY genes in C. paliurus is comparatively fewer than other sequenced monocot genomes, suggesting that duplication events have played a significant role in the expansion of the WRKY gene family.
The study also indicated that group III WRKY genes, which are thought to be the most advanced in terms of evolution, diversified prior to the divergence of monocots and dicots. This group’s size variation could be a potential cause for the diversity of the WRKY gene family size. Interestingly, C. paliurus has more group III WRKY genes than most other dicots, but without a lineage-specific radiation, possibly due to different duplication patterns.
We also investigated the expression patterns of the CpWRKY gene, revealing their potential role in physiological processes and stress responses. For instance, 95% of WRKY genes showed altered expression under NaCl stress, with eleven genes significantly up-regulated. These genes, mainly from group II, may play a key role in coping with salinity stress. In conclusion, this comprehensive analysis of the WRKY gene family in C. paliurus provides valuable insights into their potential functional roles and evolutionary characteristics. We explored the importance of the health benefits of C. paliurus and the regulatory mechanisms of the WRKY gene in a plant’s response to salt stress. It is a good way to know that regulating the WRKY gene can improve the salt tolerance of C. paliurus and increase the quality of its accessory products. Therefore, studying the WRKY gene aims to address the challenge of breeding a valuable resource, C. paliurus, under adverse conditions. These findings could guide further research into the genetic improvement of C. paliurus, particularly in terms of environmental resistance and agronomic traits.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





