
Assessing the Genetic Diversity and Genealogical Reconstruction of Cypress (Cupressus funebris Endl.) Breeding Parents Using SSR Markers


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Isolation and PCR Amplification
2.3. Statistical Analysis of Genetic Diversity
2.4. Clustering Analysis
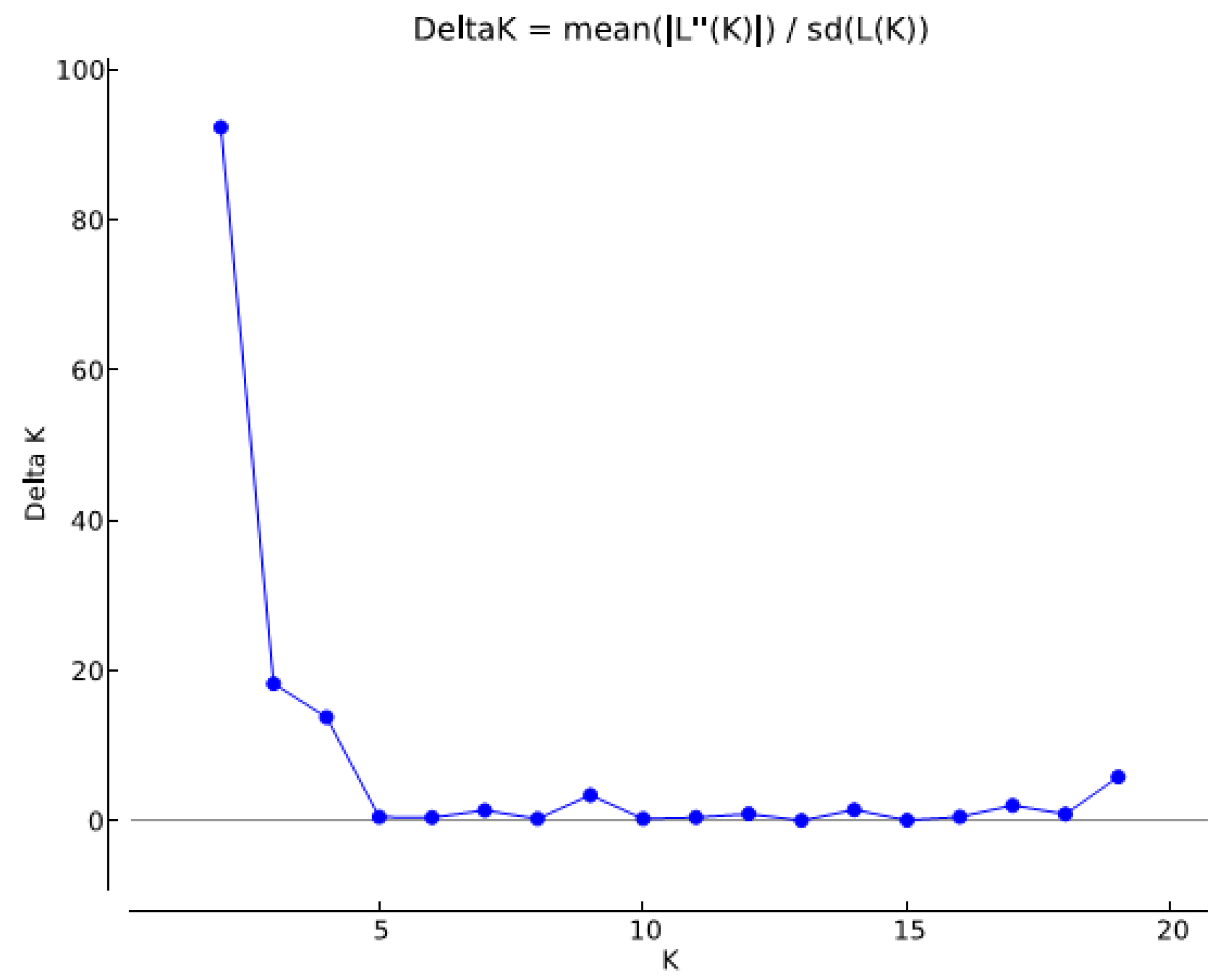
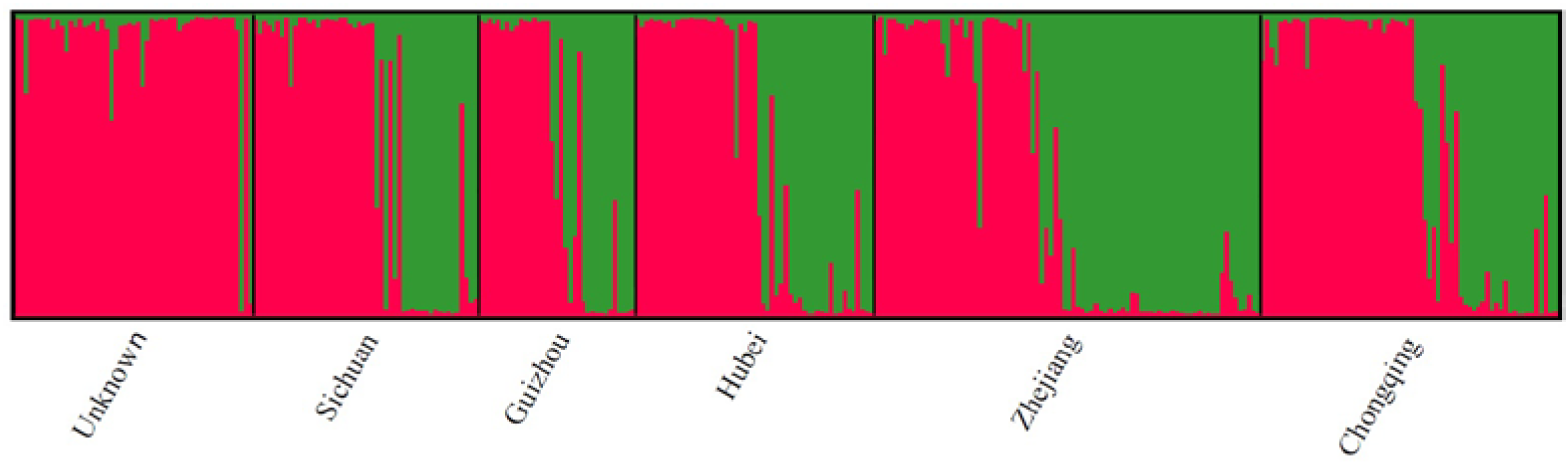
2.5. Population Structure
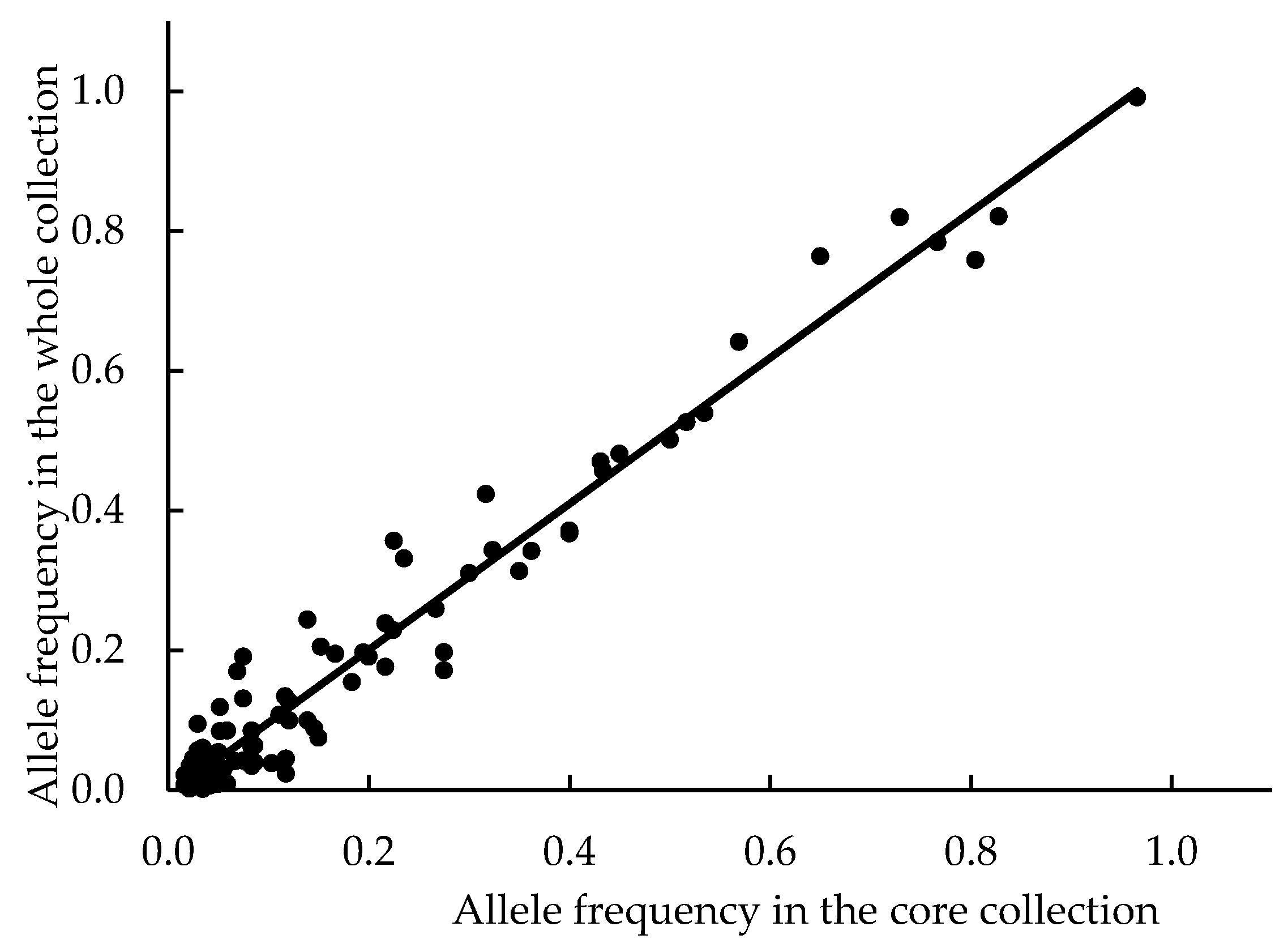
2.6. Construction of Genetic Core Collection
3. Results
3.1. Subsection
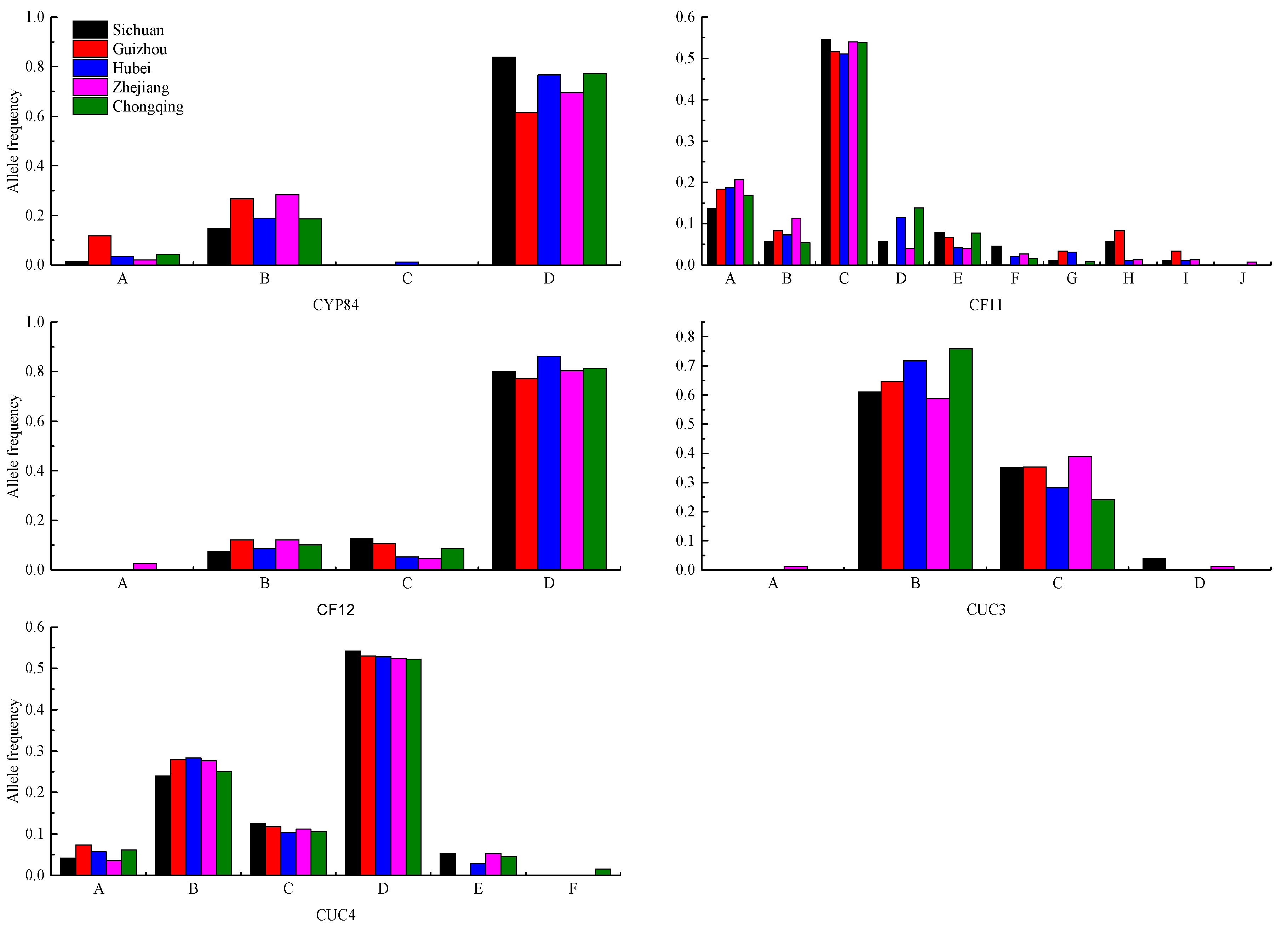
3.1.1. SSR Markers
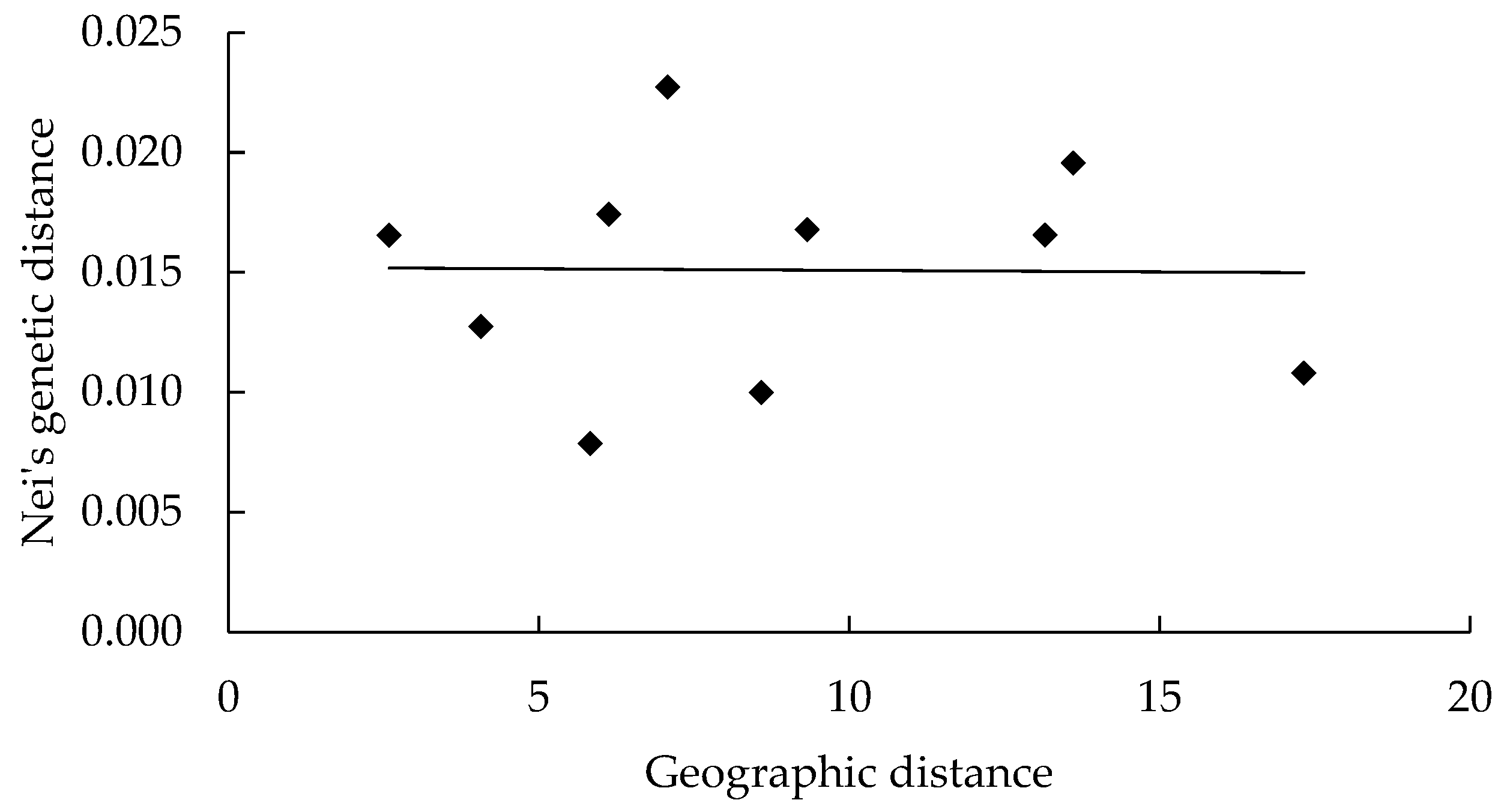
3.1.2. Genetic Relationship Analysis
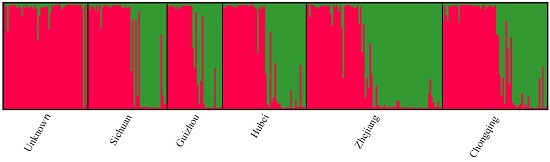
3.1.3. Population Diversity and Structure
3.1.4. Construction of Genetic Core Collection
4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Farjon, A. A Monograph of Cupressaceae and Sciadopitys; Royal Botanic Gardens, Kew: London, UK, 2005. [Google Scholar]
- Xu, L.; Xu, H.; Li, Z.; Shang, H.; Adams, R.P.; Mao, K. Genetic diversity and conservation implications of four cupressus species in china as revealed by microsatellite markers. Biochem. Genet. 2014, 52, 181–202. [Google Scholar]
- Fu, L.G.; Yu, Y.F.; Farjon, A. Cupressaceae. In Flora of China; Raven, P.H., Wu, C.Y., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 1999; pp. 62–65. [Google Scholar]
- Bretting, P.; Wildrlechner, M. Genetic markers and plant genetic resource management. Plant Breed. Rev. 2010, 13, 11–86. [Google Scholar]
- Kearsey, M.J.; Farquhar, A.G.L. Qtl analysis in plants; where are we now? Heredity 1998, 80, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Vermigli, M. Genetic Varability in Italian Populations of Cupressus sempervirens L. Assessed by SSR and Rapd Markers. Ph.D. Thesis, University of Verona, Verona, Italy, 2005. [Google Scholar]
- Parker, P.G.; Snow, A.A.; Schug, M.D.; Booton, G.C.; Fuerst, P.A. What molecules can tell us about populations: Choosing andusing a molecular marker. Ecology 1998, 79, 361–382. [Google Scholar] [CrossRef]
- Hai-Fei, W.; Xu-Xiao, Z.; Jian-Ping, G.; Tao, Y.; Xue-Lian, S.; Yu, M.; Robert, R. Genetic diversity and relationship of global faba bean (Vicia faba L.) germplasm revealed by issr markers. Tag. Theor. Appl. Genet. Theor. Angew. Genet. 2012, 124, 789–797. [Google Scholar]
- Khadivi-Khub, A.; Zamani, Z.; Fattahi, R.; Wünsch, A. Genetic variation in wild Prunus L. Subgen. Cerasus germplasm from iran characterized by nuclear and chloroplast ssr markers. Trees 2014, 28, 471–485. [Google Scholar] [CrossRef]
- Nimnual, A.; Romkaew, J.; Chukeatirote, E.; Nilthong, S. Evaluation of genetic relationship among some important Japanese and Thai soybean varieties using AFLP analysis. Aust. J. Crop Sci 2014, 8, 481–485. [Google Scholar]
- Escribano, P.; Viruel, M.A.; Hormaza, J.I. Comparison of different methods to construct a core germplasm collection in woody perennial species with simple sequence repeat markers. A case study in cherimoya (annona cherimola, annonaceae), an underutilised subtropical fruit tree species. Ann. Appl. Biol. 2008, 153, 25–32. [Google Scholar] [CrossRef]
- Wang, J.C.; Hu, J.; Xu, H.M.; Zhang, S. A strategy on constructing core collections by least distance stepwise sampling. Tag. Theo. Appl. Genet. Theor. Angew. Genet. 2007, 115, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, D.; Wang, M.; Sun, J.; Qi, Y.; Li, J.; Wei, X.; Han, L.; Qiu, Z.; Tang, S. A core collection and mini core collection of Oryza sativa L. In China. Tag. Theor. Appl. Genet. Theor. Angew. Genet. 2011, 122, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Belaj, A.; Atienza, S.G.; Urdíroz, N.M.; Rosa, R.D.L.; Satovic, Z.; Martín, A.; Kilian, A.; Trujillo, I.; Valpuesta, V. Developing a core collection of olive ( Olea europaea L.) based on molecular markers (DArTs, SSRs, SNPs) and agronomic traits. Tree Genet. Genomes 2012, 8, 365–378. [Google Scholar] [CrossRef]
- Guo, Y.; Li, Y.; Hong, H.; Qiu, L.J. Establishment of the integrated applied core collection and its comparison with mini core collection in soybean (Glycine max). Crop J. 2005, 44, 174–180. [Google Scholar] [CrossRef]
- Taniguchi, F.; Kimura, K.; Saba, T.; Ogino, A.; Yamaguchi, S.; Tanaka, J. Worldwide core collections of tea (Camellia Sinensis) based on genome-wide simple sequence repeat (SSR) markers. In Proceedings of the International Plant and Animal Genome Conference XXII, San Diego, CA, USA, 10–15 January 2014.
- Kejun, L.; Major, G.; Spencer, M.; Smith, J.S.; Ed, B.; John, D. Genetic structure and diversity among maize inbred lines as inferred from DNA microsatellites. Genetics 2003, 165, 2117–2128. [Google Scholar]
- Bagnoli, F.; Vendramin, G.G.; Buonamici, A.; Doulis, A.G.; González-Martínez, S.C.; La Porta, N.; Magri, D.; Raddi, P.; Sebastiani, F.; Fineschi, S. Is cupressus sempervirens native in italy? An answer from genetic and palaeobotanical data. Mol. Ecol. 2009, 18, 2276–2286. [Google Scholar] [CrossRef] [PubMed]
- Shahroodian, S.H.; Azadfar, D.; Soltanloo, H.; Ramezanpour, S.S. Genetic variability in natural iranian populations of cupressus sempervirens var. Horizontalis in Caspian sea coastward assessed by SSR markers. Plant Omics J. 2011, 4, 19–24. [Google Scholar]
- Webb, D.M.; Knapp, S.J. DNA extraction from a previously recalcitrant plant genus. Plant Mol. Biol. Rep. 1990, 8, 180–185. [Google Scholar] [CrossRef]
- Li, Z.; Xu, H.; Zhao, G. Isolation and characterization of polymorphic microsatellite loci primers for Cupressus funebris (cupressaceae). Conserv. Genet. Resour. 2012, 5, 307–309. [Google Scholar] [CrossRef]
- Xu, H.; Shi, D.; Wang, J.; Xu, T.; Wu, Y. Isolation and characterization of polymorphic microsatellite markers in Cupressus chenggiana S. Y. Hu (cupressaceae). Conserv. Genet. 2008, 9, 1023–1026. [Google Scholar] [CrossRef]
- Sebastiani, F.; Buonamici, A.; Fineschi, S.; Racchi, M.L.; Raddi, P.; Vendramin, G.G. Novel polymorphic nuclear microsatellites in Cupressus sempervirens L. Mol. Ecol. Notes 2005, 5, 393–394. [Google Scholar] [CrossRef]
- Marshall, T.C.; Slate, J.; Kruuk, L.E.; Pemberton, J.M. Statistical confidence for likelihood-based paternity inference in natural populations. Mol. Ecol. 1998, 7, 639–655. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. Genalex 6.5. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Paetkau, D.; Calvert, W.; Stirling, I.; Strobeck, C. Microsatellite analysis of population structure in Canadian polar bears. Mol. Ecol. 1995, 4, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin Version 3.01. An Integrated Software Package for Population Genetics Data Analysis; Computational and Molecular Population Genetic Lab, Institute of Zoology, University of Berne: Bern, Swizerland, 2006. [Google Scholar]
- Dice, L.R. Measures of the amount of ecologic association between species. Ecology 1945, 26, 297–302. [Google Scholar] [CrossRef]
- Rohlf, F.J. NTSYS-pc Numerical Taxonomy and Multivariate Analysis System (Version 1.80); State University of New York: New York, NY, USA, 1994. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 7, 574–578. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioniformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Hill, W.G.; Robertson, A. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 1968, 38, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Schoen, D.; Brown, A. Maximizing genetic diversity in core collections of wild crop relatives of crop species. In Core Collections of Plant Genetic Resources; Hodgkin, T., Brown, A.H.D., van Hintum, T.J.L., Morales, E.A.V., Eds.; John Wiley and Sons Ltd.: Chicester, UK, 1995; pp. 253–259. [Google Scholar]
- Cipriani, G.; Spadotto, A.; Jurman, I.; Gaspero, G.D.; Crespan, M.; Meneghetti, S.; Frare, E.; Vignani, R.; Cresti, M.; Morgante, M. The SSR-based molecular profile of 1005 grapevine (Vitis vinifera L.) accessions uncovers new synonymy and parentages, and reveals a large admixture amongst varieties of different geographic origin. Theor. Appl. Genet. 2010, 121, 1569–1585. [Google Scholar] [CrossRef] [PubMed]
- Hamrick, J.L.; Godt, M.J.W.; Sherman-Broyles, S.L. Factors influencing levels of genetic diversity in woody plant species. New For. 1992, 6, 95–124. [Google Scholar] [CrossRef]
- Bruford, M.W.; Ciofi, C.; Funk, S.M. Characteristics of microsatellites. In Molecular Tools for Screening Biodiversity: Plants and Animals; Karp, A., Isaac, P.G., Ingram, D.S., Eds.; Chapman & Hall: London, UK, 1998; pp. 202–205. [Google Scholar]
- Wei, L.; Luca, D.; Paolo, D.F.; Roberta, P.; Silviero, S.; Stefano, T. Genetic diversity, population structure and construction of a core collection of apple cultivars from Italian germplasm. Plant Mol. Bio. Rep. 2014, 33, 458–473. [Google Scholar]
- Hamilton, M. Population Genetics; Wiley-Blackwell: New York, NY, USA, 2009. [Google Scholar]
- Freeland, J.; Kirk, H.; Petersen, S. Molecualr Ecology, 2nd ed.; John Wiley & Sons: Chichester, UK, 2011. [Google Scholar]
- Korori, A.; Azadfar, D.; Shirvany, A.; Valipour, K.; Matinizadeh, M. Assessment of genetic variability in long-lived Cupressus sempervirens var. Horizontalis using SSR markers. Plant Gene Trait 2012, 3, 43–49. [Google Scholar]
- Zheng, W.; Fu, L. Cupressus linn. In Editorial Committee of Flora Reipublicae Popularis Sinicae Flora Republicae Popularis Sinicae Tomus 7: Gymnospermae; Science Press: Beijing, China, 1978; pp. 328–336. [Google Scholar]
- Hamrick, J.L.; Godt, M.J.W. Allozyme diversity in plant species. In Plant Population Genetics, Breeding and Genetic Resources; Brown, A.H.D., Clegg, M.T., Kahler, A.L., Weir, B.S., Eds.; Sinauer: Sunderland, MA, USA, 1989; pp. 43–63. [Google Scholar]
- Weissinger, A. Technologies for germplasm conservation ex situ. In The Preservation and Valuation of Biological Resources; Orians, G.H., Brown, G.M., Kunin, W.E., Swierbinski, J.E., Eds.; University of Washington Press: Seattle, WA, USA, 1990; pp. 3–31. [Google Scholar]
- Lia, V.V.; Poggio, L.; Confalonieri, V.A. Microsatellite variation in maize landraces from northwestern argentina: Genetic diversity, population structure and racial affiliations. Tag. Theor. Appl. Genet. Theor. Angew. Genet. 2009, 119, 1053–1067. [Google Scholar] [CrossRef] [PubMed]
- Emanuelli, F.; Lorenzi, S.; Grzeskowiak, L.; Catalano, V.; Stefanini, M.; Troggio, M.; Myles, S.; Martinez-Zapater, J.M.; Zyprian, E.; Moreira, F.M. Genetic diversity and population structure assessed by SSR and SNP markers in a large germplasm collection of grape. BMC Plant Biol. 2013, 13, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Q.; Wang, B.; Wei, Z.; Zhang, D.; Li, B. Genetic diversity and population structure of Chinese white poplar (Populus tomentosa) revealed by SSR markers. J. Hered. 2012, 103, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Adugna, A. Analysis of in situ diversity and population structure in Ethiopian cultivated Sorghum bicolor (L.) landraces using phenotypic traits and SSR markers. Springerplus 2014, 3, 212. [Google Scholar] [CrossRef] [PubMed]
- Jena, K.K. The species of the genus oryza and transfer of useful genes from wild species into cultivated rice, O. sativa. Breed. Sci. 2010, 60, 518–523. [Google Scholar] [CrossRef]
- Tripp, E.A.; Siti, F.; Iain, D.; Mcdade, L.A. Origin of African Physacanthus (Acanthaceae) via wide hybridization. PLoS ONE 2013, 8, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.; Crossa, J.; Warburton, M.; Taba, S. Sampling strategies for conserving maize diversity when forming core subsets using genetic markers. Crop Sci 2006, 46, 854–864. [Google Scholar] [CrossRef]
- Richter, T.; Soltis, P.; Soltis, D. Genetic variation within and among populations of the narrow endemic, Delphinium viridescens (Ranunculaceae). Am. J. Bot. 1994, 81, 1070–1076. [Google Scholar] [CrossRef]
- Bengtsson, B.O.; Weibull, P.; Ghatnekar, L. The loss of alleles by sampling: A study of the common outbreeding grass Festuca Ovina over three geographic scales. Hereditas 1995, 122, 221–238. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Origin | Numbers | Latitude | Longitude | Collection Time |
|---|---|---|---|---|
| unknown | 53 | - | - | 1981 |
| Chongqing Province, China | 66 | 28°88′ N | 106°68′ E | 2012 |
| Zhejiang Province, China | 86 | 30°26′ N | 120°22′ E | 2011 |
| Hubei Province, China | 53 | 32°57′ N | 111°19′ E | 2014 |
| Guizhou Province, China | 35 | 26°47′ N | 107°62′ E | 2014 |
| Sichuan Province, China | 50 | 30°37′ N | 102°90′ E | 2010 |
| Total | 343 |
| Locus | Na | Ne | PIC | H | I | He | Ho | uHe | Fis | PI | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Breeding parents from the five provinces (n = 290) | CF01 a | 9 | 6.298 | 0.689 | 0.832 | 1.977 | 0.841 | 0.407 | 0.843 | 0.513 *** | 0.04 |
| CF02 a | 2 | 1.021 | 0.682 | 0.021 | 0.058 | 0.021 | 0.007 | 0.021 | 0.702 *** | 0.96 | |
| CF03 | 9 | 4.191 | 0.64 | 0.682 | 1.685 | 0.761 | 0.719 | 0.763 | 0.048 ns | 0.09 | |
| CF04 | 8 | 4.679 | 0.802 | 0.693 | 1.740 | 0.786 | 0.781 | 0.788 | −0.014 ns | 0.07 | |
| CF09 | 7 | 3.947 | 0.772 | 0.618 | 1.544 | 0.747 | 0.736 | 0.748 | 0.002 ns | 0.11 | |
| CF11 | 10 | 3.007 | 0.722 | 0.614 | 1.506 | 0.667 | 0.866 | 0.669 | −0.293 ** | 0.14 | |
| CF12 a | 4 | 1.479 | 0.59 | 0.308 | 0.639 | 0.324 | 0.194 | 0.324 | 0.369 *** | 0.48 | |
| CUC1 | 3 | 1.575 | 0.572 | 0.275 | 0.608 | 0.365 | 0.431 | 0.366 | −0.202 ** | 0.46 | |
| CUC2 | 4 | 1.285 | 0.393 | 0.210 | 0.478 | 0.222 | 0.236 | 0.222 | −0.073 ns | 0.62 | |
| CUC3 a | 4 | 1.843 | 0.424 | 0.246 | 0.707 | 0.457 | 0.664 | 0.458 | −0.474 *** | 0.39 | |
| CUC4 | 6 | 2.732 | 0.547 | 0.382 | 1.235 | 0.634 | 0.853 | 0.635 | −0.363 ** | 0.19 | |
| CUC6 a | 6 | 1.428 | 0.48 | 0.292 | 0.636 | 0.300 | 0.172 | 0.300 | 0.414 *** | 0.51 | |
| CUC7 a | 4 | 2.064 | 0.575 | 0.149 | 0.784 | 0.515 | 0.885 | 0.516 | −0.730 *** | 0.35 | |
| CUC8 | 5 | 2.995 | 0.61 | 0.609 | 1.207 | 0.666 | 0.591 | 0.667 | 0.118 ns | 0.18 | |
| CYP52 | 4 | 3.554 | 0.739 | 0.527 | 1.322 | 0.719 | 0.951 | 0.720 | −0.321 ** | 0.13 | |
| CYP84 a | 4 | 1.688 | 0.37 | 0.392 | 0.701 | 0.407 | 0.192 | 0.408 | 0.530 *** | 0.41 | |
| CYP101 a | 3 | 2.075 | 0.471 | 0.057 | 0.774 | 0.518 | 0.962 | 0.519 | −0.856 *** | 0.35 | |
| Mean | 5.412 | 2.698 | 0.593 | 0.406 | 1.035 | 0.527 | 0.568 | 0.528 | −0.037 | - | |
| SD | 0.582 | 0.350 | 0.133 | 0.046 | 0.129 | 0.056 | 0.076 | 0.056 | 0.108 | - | |
| Total | 92 | 45.859 | - | - | - | - | - | - | - | 2.37 × 10−11 | |
| Breeding parents with unknown origin (n = 53) | CF01 a | 7 | 4.886 | 0.674 | 0.788 | 1.731 | 0.795 | 0.286 | 0.805 | 0.641 *** | 0.07 |
| CF02 a | 1 | 1.000 | 0.65 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | - | 1.00 | |
| CF03 a | 5 | 2.136 | 0.594 | 0.501 | 0.955 | 0.532 | 0.368 | 0.546 | 0.307 *** | 0.29 | |
| CF04 | 8 | 4.723 | 0.786 | 0.699 | 1.770 | 0.788 | 0.913 | 0.806 | −0.158 ** | 0.07 | |
| CF09 | 3 | 2.504 | 0.733 | 0.382 | 0.982 | 0.601 | 0.778 | 0.609 | −0.295 ** | 0.24 | |
| CF11 | 6 | 2.658 | 0.667 | 0.583 | 1.292 | 0.624 | 0.642 | 0.630 | −0.029 ns | 0.17 | |
| CF12 a | 2 | 1.328 | 0.565 | 0.205 | 0.413 | 0.247 | 0.288 | 0.249 | −0.169 ** | 0.60 | |
| CUC1 | 3 | 1.286 | 0.496 | 0.214 | 0.457 | 0.223 | 0.132 | 0.225 | 0.407 *** | 0.62 | |
| CUC2 a | 4 | 2.509 | 0.304 | 0.391 | 1.075 | 0.601 | 0.943 | 0.607 | −0.569 *** | 0.23 | |
| CUC3 a | 3 | 2.091 | 0.514 | 0.168 | 0.797 | 0.522 | 0.925 | 0.527 | −0.772 *** | 0.34 | |
| CUC4 | 6 | 2.301 | 0.641 | 0.560 | 1.135 | 0.565 | 0.160 | 0.571 | 0.717 *** | 0.23 | |
| CUC6 a | 5 | 2.445 | 0.584 | 0.247 | 1.041 | 0.591 | 0.925 | 0.597 | −0.564 *** | 0.25 | |
| CUC7 a | 2 | 2.000 | 0.428 | 0.000 | 0.693 | 0.500 | 1.000 | 0.505 | −1.000 *** | 0.38 | |
| CUC8 | 5 | 2.351 | 0.516 | 0.499 | 0.981 | 0.575 | 0.528 | 0.580 | 0.081 ns | 0.27 | |
| CYP52 a | 4 | 3.756 | 0.731 | 0.577 | 1.352 | 0.734 | 0.962 | 0.741 | −0.310 *** | 0.12 | |
| CYP84 a | 2 | 1.168 | 0.336 | 0.144 | 0.274 | 0.144 | 0.031 | 0.146 | 0.783 *** | 0.74 | |
| CYP101 a | 3 | 2.038 | 0.419 | 0.019 | 0.740 | 0.509 | 1.000 | 0.514 | −0.964 *** | 0.36 | |
| Mean | 4.059 | 2.422 | 0.567 | 0.352 | 0.923 | 0.503 | 0.581 | 0.509 | −0.118 | - | |
| SD | 0.473 | 0.270 | 0.14 | 0.045 | 0.116 | 0.054 | 0.091 | 0.055 | 0.140 | - | |
| Total | 69 | 41.180 | - | - | - | - | - | - | - | 3.29 × 10−10 |
| Population | Na | Ne | H | I | Ho | He | uHe | Fis | Na Freq. ≥ 5% | No. Private Alleles |
|---|---|---|---|---|---|---|---|---|---|---|
| Sichuan | 4.765 | 2.674 | 0.415 | 1.035 | 0.562 | 0.531 | 0.538 | −0.025 | 3.412 | 0.000 |
| (0.546) | (0.323) | (0.045) | (0.126) | (0.070) | (0.054) | (0.054) | (0.097) | (0.438) | (0.000) | |
| Guizhou | 4.294 | 2.628 | 0.400 | 1.004 | 0.553 | 0.525 | 0.534 | −0.067 | 3.353 | 0.000 |
| (0.567) | (0.317) | (0.047) | (0.129) | (0.079) | (0.056) | (0.057) | (0.115) | (0.420) | (0.000) | |
| Hubei | 4.882 | 2.552 | 0.385 | 0.998 | 0.588 | 0.511 | 0.517 | −0.070 | 3.235 | 0.059 |
| (0.562) | (0.302) | (0.045) | (0.126) | (0.079) | (0.056) | (0.056) | (0.112) | (0.369) | (0.059) | |
| Zhejiang | 5.176 | 2.817 | 0.418 | 1.055 | 0.570 | 0.538 | 0.542 | −0.058 | 3.176 | 0.176 |
| (0.577) | (0.391) | (0.046) | (0.131) | (0.076) | (0.055) | (0.056) | (0.103) | (0.413) | (0.095) | |
| Chongqing | 4.529 | 2.488 | 0.365 | 0.943 | 0.557 | 0.495 | 0.500 | −0.095 | 2.941 | 0.059 |
| (0.595) | (0.307) | (0.046) | (0.127) | (0.084) | (0.059) | (0.059) | (0.110) | (0.337) | (0.059) |
| Population | Sichuan | Guizhou | Hubei | Zhejiang | Chongqing |
|---|---|---|---|---|---|
| Sichuan | 0.008 | 0.005 | 0.005 | 0.008 | |
| Guizhou | 0.017 | 0.009 | 0.006 | 0.008 | |
| Hubei | 0.010 | 0.023 | 0.007 | 0.004 | |
| Zhejiang | 0.011 | 0.017 | 0.017 | 0.009 | |
| Chongqing | 0.013 | 0.017 | 0.008 | 0.020 |
| Source of Variation | d.f. | Sum of Squares | Variance Components | Percentage Variation (%) |
|---|---|---|---|---|
| Among populations | 4 | 22.589 | 0.015 | 0.332 |
| Within populations | 575 | 2226.637 | 4.473 | 99.668 |
| Total | 579 | 2249.226 | 4.488 |
| Population | Size | Na | Ne | I | Ho | He | Correlation of Allele Frequency |
|---|---|---|---|---|---|---|---|
| Overall breeding parents | 343 | 93 | 2.7 | 1.06 | 0.57 | 0.54 | 0.96 |
| Core collection | 30 | 93 | 3.0 | 1.16 | 0.57 | 0.57 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Zhang, R.; Jin, G.; Feng, Z.; Zhou, Z. Assessing the Genetic Diversity and Genealogical Reconstruction of Cypress (Cupressus funebris Endl.) Breeding Parents Using SSR Markers. Forests 2016, 7, 160. https://doi.org/10.3390/f7080160
Yang H, Zhang R, Jin G, Feng Z, Zhou Z. Assessing the Genetic Diversity and Genealogical Reconstruction of Cypress (Cupressus funebris Endl.) Breeding Parents Using SSR Markers. Forests. 2016; 7(8):160. https://doi.org/10.3390/f7080160
Chicago/Turabian StyleYang, Hanbo, Rui Zhang, Guoqing Jin, Zhongping Feng, and Zhichun Zhou. 2016. "Assessing the Genetic Diversity and Genealogical Reconstruction of Cypress (Cupressus funebris Endl.) Breeding Parents Using SSR Markers" Forests 7, no. 8: 160. https://doi.org/10.3390/f7080160
APA StyleYang, H., Zhang, R., Jin, G., Feng, Z., & Zhou, Z. (2016). Assessing the Genetic Diversity and Genealogical Reconstruction of Cypress (Cupressus funebris Endl.) Breeding Parents Using SSR Markers. Forests, 7(8), 160. https://doi.org/10.3390/f7080160