Taxonomic and Functional Diversity of a Quercus pyrenaica Willd. Rhizospheric Microbiome in the Mediterranean Mountains

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Site Selection and Sample Collection
2.2. Sequencing of Environmental DNA
2.3. Phylogenetic Assignment and Functional Analysis of Metagenomic Sequences
2.4. Taxonomic Classification of 16S rRNA Metagenomic Sequences
3. Results
3.1. Physico-Chemical Properties of the Soils
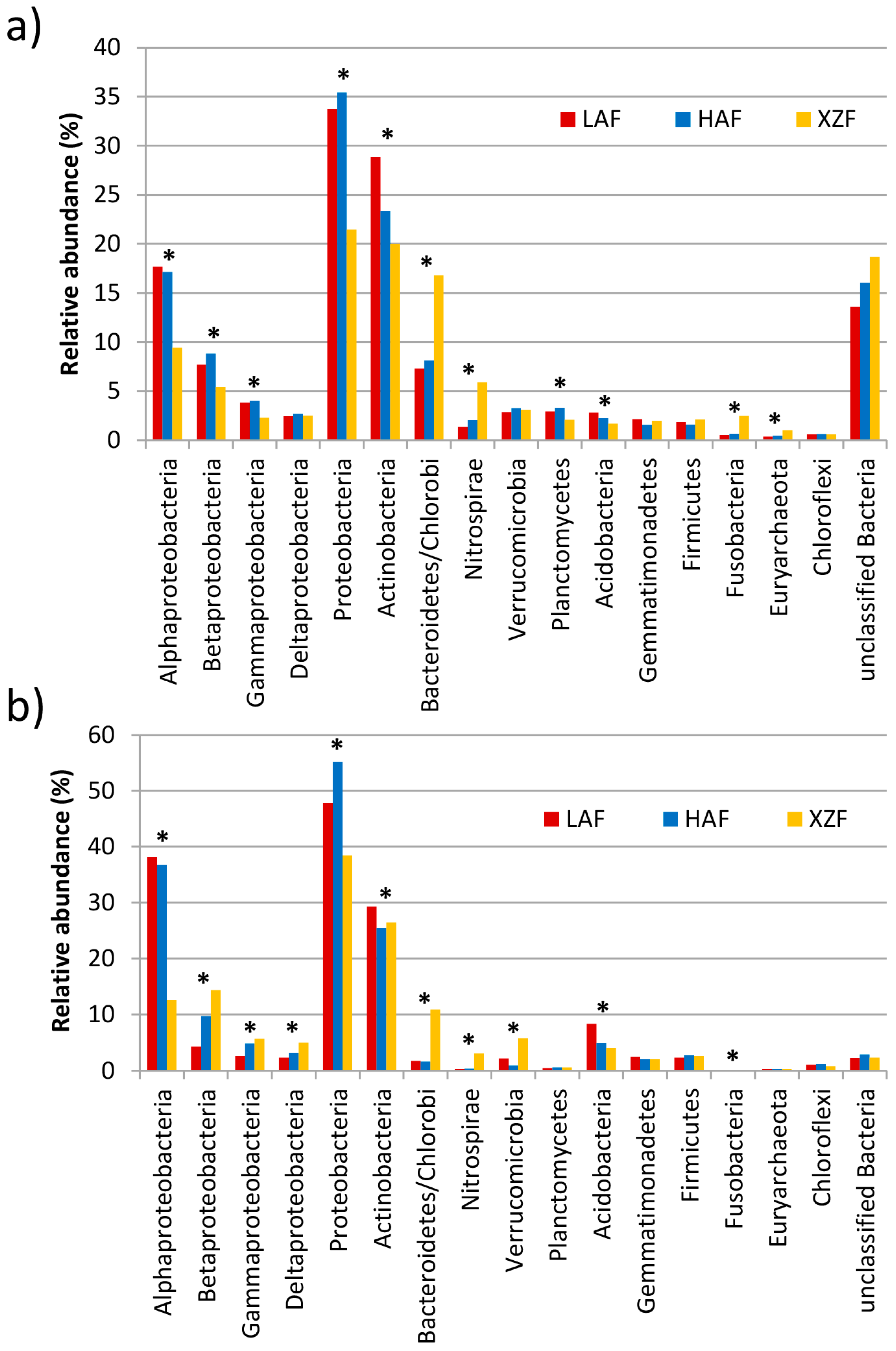
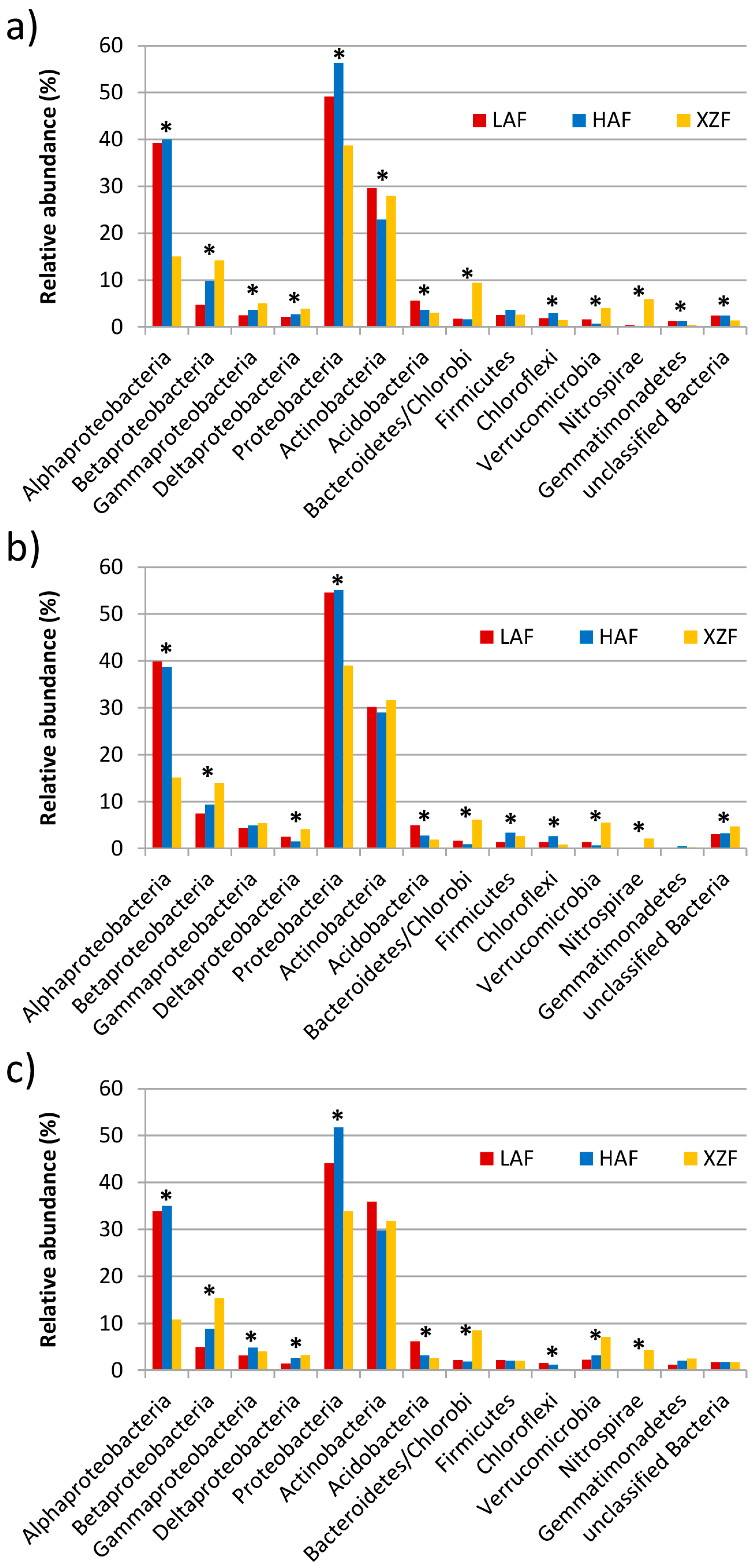
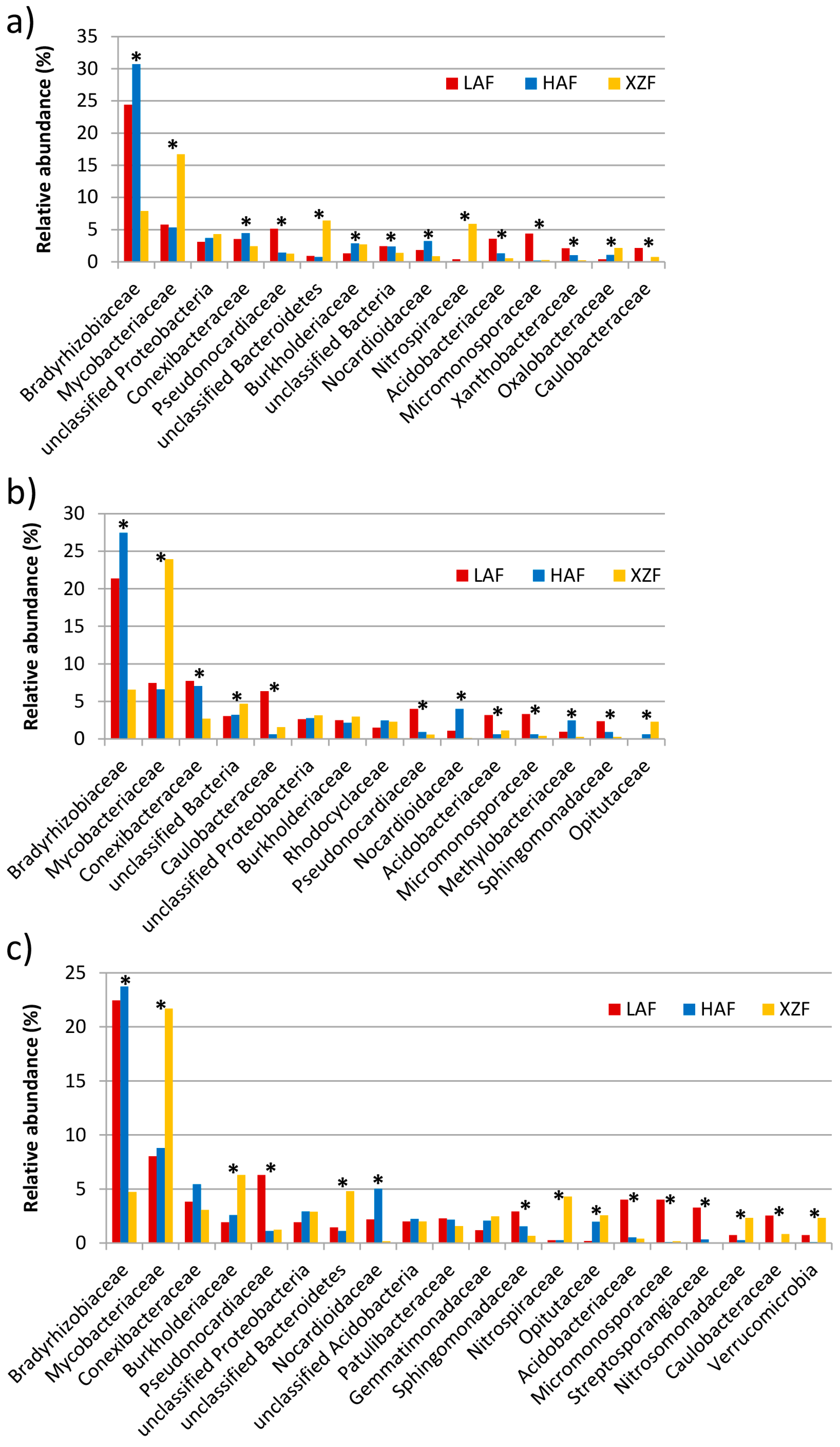
3.2. Taxonomic and Phylogenetic Analyses of the Metagenomes
3.3. Functional Analysis of the Metagenomes
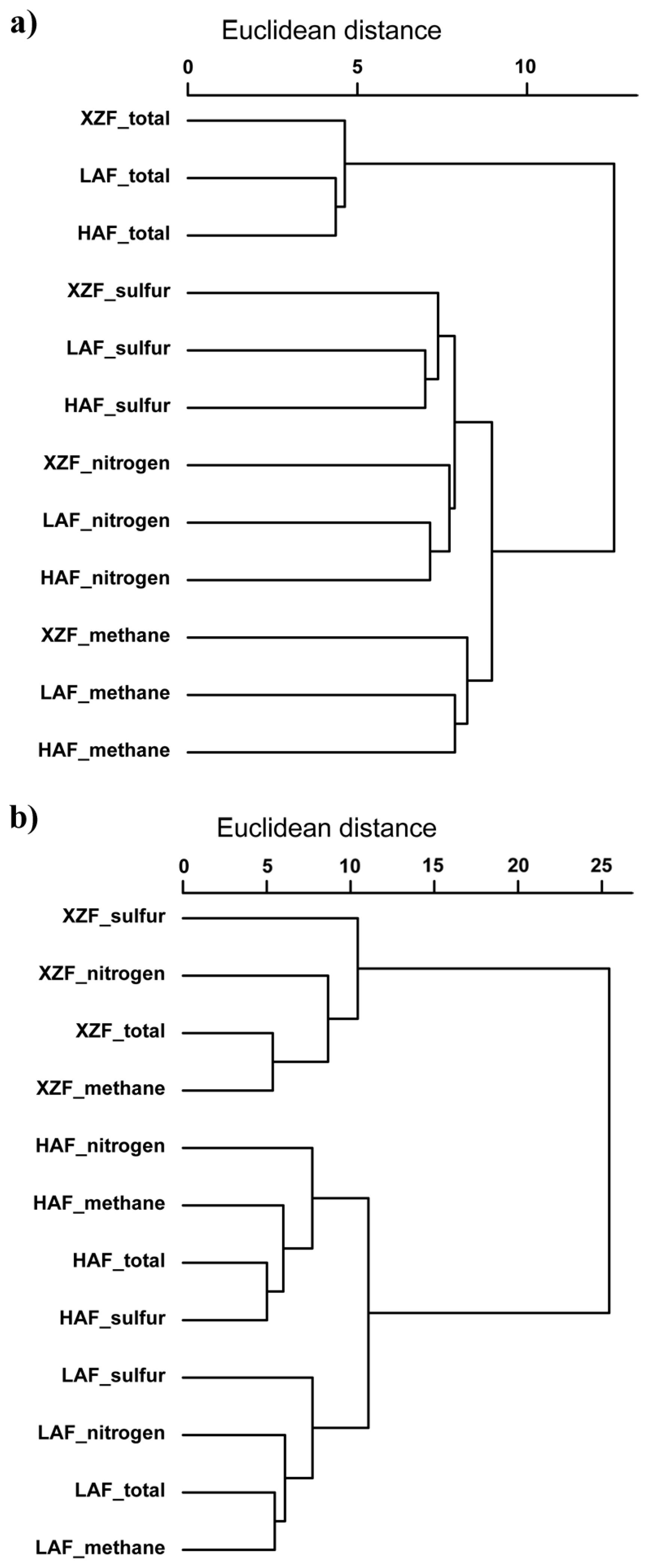
3.4. Functional Analysis of Methane, Sulfur and Nitrogen Metabolism Genes
3.5. Phylogenetic Analysis of Methane, Sulfur and Nitrogen Metabolism Sequences
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bardgett, R.D.; Mawdsley, J.L.; Edwards, S.; Hobbs, P.J.; Rodwell, J.S.; Davies, W.J. Plant species and nitrogen effects on soil biological properties of temperate upland grasslands. Funct. Ecol. 1999, 13, 650–660. [Google Scholar]
- Bragazza, L.; Bardgett, R.D.; Mitchell, E.A.D.; Buttler, A. Linking soil microbial communities to vascular plant abundance along a climate gradient. New Phytol. 2015, 205, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijden, M.G.A.; Bardgett, R.D.; van Straalen, N.M. The unseen majority: Soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol. Lett. 2008, 11, 296–310. [Google Scholar] [PubMed]
- Strickland, M.S.; Lauber, C.; Fierer, N.; Bradford, M.A. Testing the functional significance of microbial community composition. Ecology 2009, 90, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Nazaries, L.; Munro, S.; Anderson, I.; Campbell, C.D. Use of multiplex terminal restriction fragment length polymorphism for rapid and simultaneous analysis of different components of the soil microbial community. Appl. Environ. Microbiol. 2006, 72, 7278–7285. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gao, Y.; Wang, S.; Xu, D.; Yu, H.; Wu, L.; Lin, Q.; Hu, Y.; Li, X.; He, Z.; et al. The microbial gene diversity along an elevation gradient of the Tibetan grassland. ISME J. 2014, 8, 430–440. [Google Scholar] [PubMed]
- Malhi, Y.; Silman, M.; Salinas, N.; Bush, M.; Meir, P.; Saatchi, S. Introduction, elevation gradients in the tropics, laboratories for ecosystem ecology and global change research. Glob. Chang. Biol. 2010, 16, 3171–3175. [Google Scholar] [CrossRef]
- Curiel-Yuste, J.; Fernandez-Gonzalez, A.J.; Fernandez-Lopez, M.; Ogaya, R.; Peñuelas, J.; Sardans, J.; Lloret, F. Strong functional stability of soil microbial communities under semiarid Mediterranean conditions is subjected to long-term shifts in baseline precipitation. Soil Biol. Biochem. 2014, 69, 223–233. [Google Scholar]
- Cong, J.; Yang, Y.; Liu, X.; Lu, H.; Zhou, J.; Li, D.; Yin, H.; Ding, J.; Zhang, Y. Analyses of soil microbial community compositions and functional genes reveal potential consequences of natural forest succession. Sci. Rep. 2015, 5, 10007. [Google Scholar] [PubMed]
- Bryant, J.A.; Lamanna, C.; Morlon, H.; Kerkhoff, A.J.; Enquist, B.J.; Green, J.L. Microbes on mountainsides: Contrasting elevational patterns of bacterial and plant diversity. Proc. Nat. Acad. Sci. USA 2008, 105, 11505–11511. [Google Scholar] [PubMed]
- Singh, D.; Shi, L.; Adams, J.M. Bacterial diversity in the mountains of South-West China: Climate dominates over soil parameters. J. Microbiol. 2013, 51, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Lee-Cruz, L.; Kim, W.S.; Kerfahi, D.; Chun, J.H.; Adams, J.M. Strong elevational trends in soil bacterial community composition on Mt. Halla, South Korea. Soil Biol. Biochem. 2014, 68, 140–149. [Google Scholar] [CrossRef]
- Landesman, W.J.; Nelson, D.M.; Fitzpatrick, M.C. Soil properties and tree species drive β-diversity of soil bacterial communities. Soil Biol. Biochem. 2014, 76, 201–209. [Google Scholar] [CrossRef]
- Urich, T.; Lanzén, A.; Qi, J.; Huson, D.H.; Schleper, C.; Schuster, S.C. Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS ONE 2008, 3, e2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Cong, J.; Lu, H.; Yang, C.; Yang, Y.; Zhou, J.; Li, D. An integrated study to analyze soil microbial community structure and metabolic potential in two forest types. PLoS ONE 2014, 9, e93773. [Google Scholar] [CrossRef] [PubMed]
- Lionello, P.; Malanotte-Rizzoli, P.; Boscolo, R.; Alpert, P.; Artale, V.; Li, L.; Luterbacher, J.; May, W.; Trigo, R.; Tsimplis, M.; et al. The Mediterranean climate: An overview of the main characteristics and issues. In Mediterranean Climate Variability; Lionello, P., Malanotte-Rizzoli, P., Boscolo, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; pp. 1–26. [Google Scholar]
- Giorgi, F.; Lionello, P. Climate change projections for the Mediterranean region. Glob. Planet. Chang. 2008, 63, 90–104. [Google Scholar] [CrossRef]
- Felicísimo, A.M.; Muñoz, J.; Villalba, C.J.; Mateo, R.G. Impactos, vulnerabilidad y adaptación al cambio climático de la biodiversidad española. In Flora y Vegetación; PNACC; Ministerio de Medio Ambiente y Medio Rural y Marino: Madrid, Spain, 2011; Volume 1. [Google Scholar]
- Ruiz-Labourdette, D.; Schmitz, M.F.; Pineda, F.D. Changes in tree species composition in Mediterranean mountains under climate change: Indicators for conservation planning. Ecol. Indic. 2013, 24, 310–323. [Google Scholar] [CrossRef]
- Kohler, T.; Werhli, A.; Jurek, M. (Eds.) Mountains and climate change: A global concern. In Sustainable Mountain Development Series; Centre for Development and Environment (CDE), Swiss Agency for Development and Cooperation (SDC) and Geographica Bernensia: Berna, Switzerland, 2014. [Google Scholar]
- Leal-García, J. Análisis del Crecimiento Vegetativo e Inversión Reproductiva de Quercus pyrenaica Willd. en Tres Localidades de Sierra Nevada. Master’s Thesis, University of de Granada, Granada, Spain, 2013. [Google Scholar]
- Camacho-Olmedo, M.T.; García-Martínez, P.; Jiménez-Olivencia, Y.; Menor-Toribio, J.; Paniza-Cabrera, A. Dinámica evolutiva del paisaje vegetal de la Alta Alpujarra granadina en la segunda mitad del siglo XX. Cuadernos Geográficos de la Universidad de Granada 2002, 32, 25–42. [Google Scholar]
- Ragazzi, A.; Moricca, S.; Dellavalle, I.; Turco, E. Italian expansion of oak decline. In Decline of Oak Species in Italy; Problems and Perspectives; Ragazzi, A., Dellavalle, I., Eds.; Universita di Firenze, Concilio Nazionale di Ricerca, Accademia Italiana di Scienze Forestali: Firenze, Italy, 2000; pp. 39–75. [Google Scholar]
- Benito, B.; Lorite, J.; Peñas, J. Simulating potential effects of climatic warming on altitudinal patterns of key species in Mediterranean-alpine ecosystems. Clim. Chang. 2011, 108, 471–483. [Google Scholar] [CrossRef]
- Leverkus, A.B.; Castro, J.; Delgado-Capel, M.J.; Molinas-González, C.; Pulgar, M.; Marañón-Jiménez, S.; Delgado-Huertas, A.; Querejeta, J.I. Restoring for the present or restoring for the future: Enhanced performance of two sympatric oaks (Quercus ilex and Quercus pyrenaica) above the current forest limit. Restor. Ecol. 2015, 23, 936–946. [Google Scholar] [CrossRef]
- Cobo-Díaz, J.F.; Fernández-González, A.J.; Villadas, P.J.; Robles, A.B.; Toro, N.; Fernández-López, M. Metagenomic assessment of the potential microbial nitrogen pathways in the rhizosphere of a Mediterranean forest after a wildfire. Microb. Ecol. 2015, 69, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Sheik, C.S.; Beasley, W.H.; Elshahed, M.S.; Zhou, X.; Luo, Y.; Krumholz, L.R. Effect of warming and drought on grassland microbial communities. ISME J. 2011, 5, 1692–1700. [Google Scholar] [CrossRef] [PubMed]
- Yergeau, E.; Bokhorst, S.; Kang, S.; Zhou, J.; Greer, C.W.; Aerts, R.; Kowalchuk, G.A. Shifts in soil microorganisms in response to warming are consistent across a range of Antarctic environments. ISME J. 2012, 6, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhu, H.; Ruan, J.; Qian, W.; Fang, X.; Shi, Z.; Li, Y.; Li, S.; Shan, G.; Kristiansen, K.; et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowitz, V.M.; Chen, I.A.; Chu, K.; Szeto, E.; Palaniappan, K.; Pillay, M.; Ratner, A.; Huang, J.; Pagani, I.; Tringe, S.G.; et al. IMG/M 4 version of the integrated metagenome comparative analysis system. Nucl. Acids Res. 2014, 42, D568–D573. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Paarmann, D.; D’souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinf. 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Beiko, R.G. Identifying biologically relevant differences between metagenomic communities. Bioinformatics 2010, 26, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucl. Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Pérez, C.A.; Carmona, M.R.; Fariña, J.M.; Armesto, J.J. Selective logging of lowland evergreen rainforests in Chiloé Island, Chile: Effects of changing tree species composition on soil nitrogen transformations. For. Ecol. Manag. 2009, 258, 1660–1668. [Google Scholar] [CrossRef]
- Shen, C.; Xiong, J.; Zhang, H.; Feng, Y.; Lin, X.; Li, X.; Liang, W.; Chu, H. Soil pH drives the spatial distribution of bacterial communities along elevation on Changbai Mountain. Soil Biol. Biochem. 2013, 57, 204–211. [Google Scholar] [CrossRef]
- Xu, M.; Li, X.; Cai, X.; Gai, J.; Li, X.; Christie, P.; Zhang, J. Soil microbial community structure and activity along a montane elevational gradient on the Tibetan Plateau. Eur. J. Soil Biol. 2014, 64, 6–14. [Google Scholar] [CrossRef]
- Zhang, B.; Liang, C.; He, H.; Zhang, X. Variations in soil microbial communities and residues along an altitude gradient on the Northern slope of Changbai Mountain, China. PLoS ONE 2013, 8, e66184. [Google Scholar] [CrossRef] [PubMed]
- Delmont, T.O.; Prestat, E.; Keegan, K.P.; Faubladier, M.; Robe, P.; Clark, I.M.; Pelletier, E.; Hirsch, P.R.; Meyer, F.; Gilbert, J.A.; et al. Structure, fluctuation and magnitude of a natural grassland soil metagenome. ISME J. 2012, 6, 1677–1687. [Google Scholar] [CrossRef] [PubMed]
- Uroz, S.; Ioannidis, P.; Lengelle, J.; Cébron, A.; Morin, E.; Buée, M.; Martin, F. Functional assays and metagenomic analyses reveals differences between the microbial communities inhabiting the soil horizons of a Norway spruce plantation. PLoS ONE 2013, 8, e55929. [Google Scholar] [CrossRef] [PubMed]
- Uroz, S.; Buée, M.; Murat, C.; Frey-Klett, P.; Martin, F. Pyrosequencing reveals a contrasted bacterial diversity between oak rhizosphere and surrounding soil. Environ. Microbiol. Rep. 2010, 2, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Andreote, F.D.; Jiménez, D.J.; Chaves, D.; Días, A.C.F.; Luvizotto, D.M.; Dini-Andreote, F.; Fasanella, C.C.; Lopez, M.V.; Baena, S.; Taketani, R.G.; et al. The Microbiome of Brazilian mangrove sediments as revealed by metagenomics. PLoS ONE 2012, 7, e38600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, C.; Beys-Da-Silva, W.; Santi, L.; Berger, M.; Vainstein, M.; Guimarães, J.; Vasconcelos, A.T. A potential source for cellulolytic enzyme discovery and environmental aspects revealed through metagenomics of Brazilian mangroves. AMB Express 2013, 3, 65. [Google Scholar] [CrossRef] [PubMed]
- Nazaries, L.; Tate, K.R.; Ross, D.J.; Singh, J.; Dando, J.; Saggar, S.; Baggs, E.M.; Millard, P.; Murrell, J.C.; Singh, B.K. Response of methanotrophic communities to afforestation and reforestation in New Zealand. ISME J. 2011, 5, 1832–1836. [Google Scholar] [CrossRef] [PubMed]
- Tate, K.R. Soil methane oxidation and land-use change–from process to mitigation. Soil Biol. Biochem. 2015, 80, 260–272. [Google Scholar] [CrossRef]
- Ollivier, J.; Töwe, S.; Bannert, A.; Hai, B.; Kastl, E.M.; Meyer, A.; Su, M.X.; Kleineidam, K.; Schloter, M. Nitrogen turnover in soil and global change. FEMS Microbiol. Ecol. 2011, 78, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Uroz, S.; Tech, J.J.; Sawaya, N.A.; Frey-Klett, P.; Leveau, J.H.J. Structure and function of bacterial communities in ageing soils: Insights from the Mendocino ecological staircase. Soil Biol. Biochem. 2014, 69, 265–274. [Google Scholar] [CrossRef]
- Xu, Z.; Hansen, M.A.; Hansen, L.H.; Jacquiod, S.; Sørensen, S.J. Bioinformatic approaches reveal metagenomic characterization of soil microbial community. PLoS ONE 2014, 9, e93445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cong, J.; Lu, H.; Li, G.; Qu, Y.; Su, X.; Zhou, J.; Li, D. Community structure and elevational diversity patterns of soil Acidobacteria. J. Environ. Sci. 2014, 26, 1717–1724. [Google Scholar] [CrossRef] [PubMed]
- Kersters, K.; de Vos, P.; Gillis, M.; Swings, J.; Vandamme, P.; Stackebrandt, E. Introduction to the Proteobacteria. In The Prokaryotes; Dwarkin, M., Falkow, S., Rosenberg, E., Schleifer, K.H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; Volume 5, pp. 3–37. [Google Scholar]
- Salminen, J.P.; Roslin, T.; Karonen, M.; Sinkkonen, J.; Pihlaja, K.; Pulkkinen, P. Seasonal variation in the content of hydrolyzable tannins, flavonoid glycosides, and proanthocyanidins in oak leaves. J. Chem. Ecol. 2004, 30, 1693–1711. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Schimel, J.P.; Holden, P.A. Variations in microbial community composition through two soil depth profiles. Soil Biol. Biochem. 2003, 35, 167–176. [Google Scholar] [CrossRef]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Cederlund, H.; Wessén, E.; Enwall, K.; Jones, C.M.; Juhanson, J.; Pell, M.; Philippot, L.; Hallin, S. Soil carbon quality and nitrogen fertilization structure bacterial communities with predictable responses of major bacterial phyla. Appl. Soil Ecol. 2014, 84, 62–68. [Google Scholar] [CrossRef]
- Hortal, S.; Bastida, F.; Armas, C.; Lozano, Y.M.; Moreno, J.L.; García, C.; Pugnaire, F.I. Soil microbial community under a nurse-plant species changes in composition, biomass and activity as the nurse grows. Soil Biol. Biochem. 2013, 64, 139–146. [Google Scholar] [CrossRef]
- Wang, J.; Soininen, J.; Zhang, Y.; Wang, B.; Yang, X.; Shen, J. Contrasting patterns in elevational diversity between microorganisms and macroorganisms. J. Biogeogr. 2011, 38, 595–603. [Google Scholar] [CrossRef]
- Fierer, N.; Mccain, C.M.; Meir, P.; Zimmermann, M.; Rapp, J.M.; Silman, M.R.; Knight, R. Microbes do not follow the elevational diversity patterns of plants and animals. Ecology 2011, 92, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Defossez, E.; Courbaud, B.; Marcais, B.; Thuiller, W.; Granda, E.; Kunstler, G. Do interactions between plant and soil biota change with elevation? A study on Fagus sylvatica. Biol. Lett. 2011, 7, 699–701. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Franklin, J.F.; Spies, T.A. Contrasting microclimates among clearcut, edge and interior of old-growth Douglas-fir forest. Agric. For. Meteorol. 1993, 63, 219–237. [Google Scholar] [CrossRef]
- Pennanen, T. Microbial communities in boreal coniferous forest humus exposed to heavy metals and changes in soil Ph—A summary of the use of phospholipid fatty acids, Biolog® and 3H-thymidine incorporation methods in field studies. Geoderma 2001, 100, 91–126. [Google Scholar] [CrossRef]
- Villadas, P.J.; Fernández-López, M.; Ramírez-Saad, H.; Toro, N. Rhizosphere-bacterial community in Eperua falcata (Caesalpiniaceae) a putative Nitrogen-fixing tree from French Guiana rainforest. Microb. Ecol. 2007, 53, 317–327. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, S.E.; Coorevits, A.; Willems, A. Tardiphaga robiniae gen. nov., sp. nov., a new genus in the family Bradyrhizobiaceae isolated from Robinia pseudoacacia in Flanders (Belgium). Syst. Appl. Microbiol. 2012, 35, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Barka, E.A.; Vatsa, P.; Sanchez, L.; Gaveau-Vaillant, N.; Jacquard, C.; Klenk, H.P.; Clément, C.; Ouhdouch, Y.; van Wezel, G.P. Taxonomy, physiology, and natural products of Actinobacteria. Microbiol. Mol. Biol. Rev. 2016, 80, 1–43. [Google Scholar] [CrossRef] [PubMed]
- Hruska, K.; Kaevska, M. Mycobacteria in water, soil, plants and air: A review. Vet. Med. 2012, 57, 623–679. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | LAF | HAF | XZF |
|---|---|---|---|
| Number of reads | 173,277,220 | 171,236,955 | 192,242,088 |
| Number of contigs after assembly | 205,779 | 205,095 | 246,663 |
| Contig average length (bp) | 319.85 | 350.92 | 342.55 |
| Number of annotated genes in KEGG database | 193,696 | 184,352 | 184,881 |
| Size of largest contig (bp) | 6529 | 18,521 | 91,563 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cobo-Díaz, J.F.; Fernández-González, A.J.; Villadas, P.J.; Toro, N.; Tringe, S.G.; Fernández-López, M. Taxonomic and Functional Diversity of a Quercus pyrenaica Willd. Rhizospheric Microbiome in the Mediterranean Mountains. Forests 2017, 8, 390. https://doi.org/10.3390/f8100390
Cobo-Díaz JF, Fernández-González AJ, Villadas PJ, Toro N, Tringe SG, Fernández-López M. Taxonomic and Functional Diversity of a Quercus pyrenaica Willd. Rhizospheric Microbiome in the Mediterranean Mountains. Forests. 2017; 8(10):390. https://doi.org/10.3390/f8100390
Chicago/Turabian StyleCobo-Díaz, José F., Antonio J. Fernández-González, Pablo J. Villadas, Nicolás Toro, Susannah G. Tringe, and Manuel Fernández-López. 2017. "Taxonomic and Functional Diversity of a Quercus pyrenaica Willd. Rhizospheric Microbiome in the Mediterranean Mountains" Forests 8, no. 10: 390. https://doi.org/10.3390/f8100390
APA StyleCobo-Díaz, J. F., Fernández-González, A. J., Villadas, P. J., Toro, N., Tringe, S. G., & Fernández-López, M. (2017). Taxonomic and Functional Diversity of a Quercus pyrenaica Willd. Rhizospheric Microbiome in the Mediterranean Mountains. Forests, 8(10), 390. https://doi.org/10.3390/f8100390