De Novo Transcriptome Sequencing in Passiflora edulis Sims to Identify Genes and Signaling Pathways Involved in Cold Tolerance

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Total RNA Extraction, cDNA Library Preparation and Transcriptome Sequencing
2.3. Transcriptome Assembly and Function Annotation
2.4. Differential Expression Analysis
2.5. Quantitative Real-Time PCR (qRT-PCR)
3. Results
3.1. Transcriptome Sequencing and De Novo Assembly
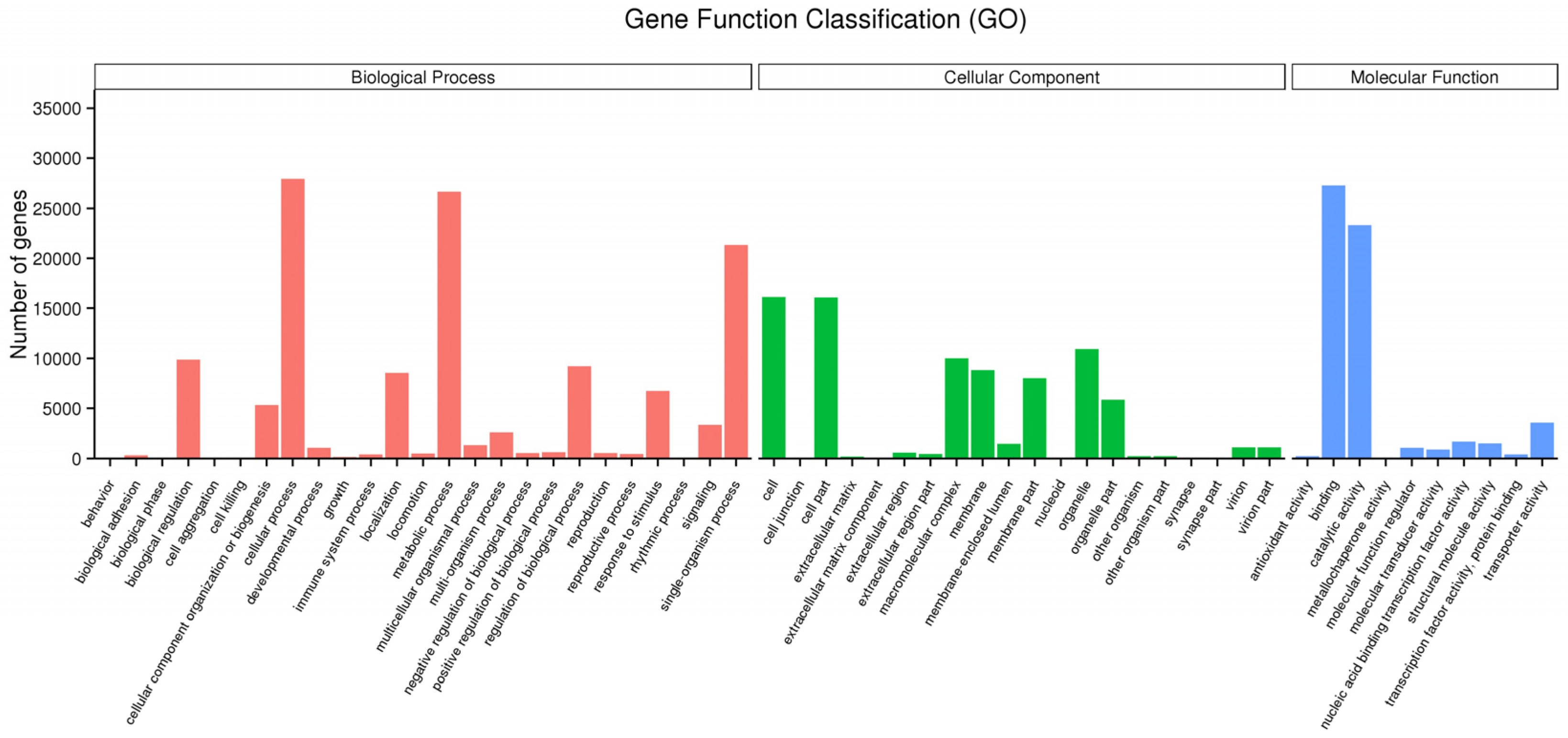
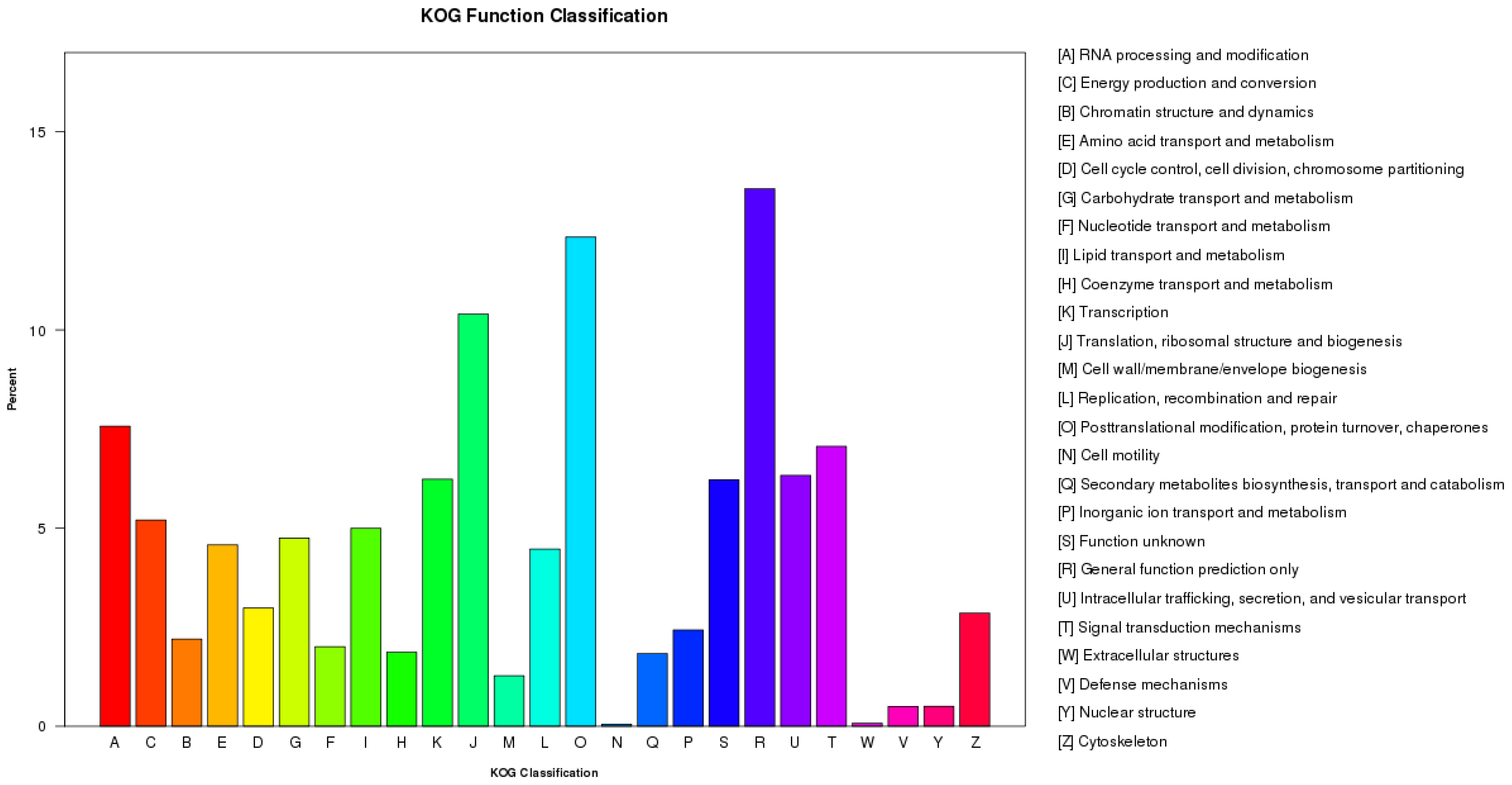
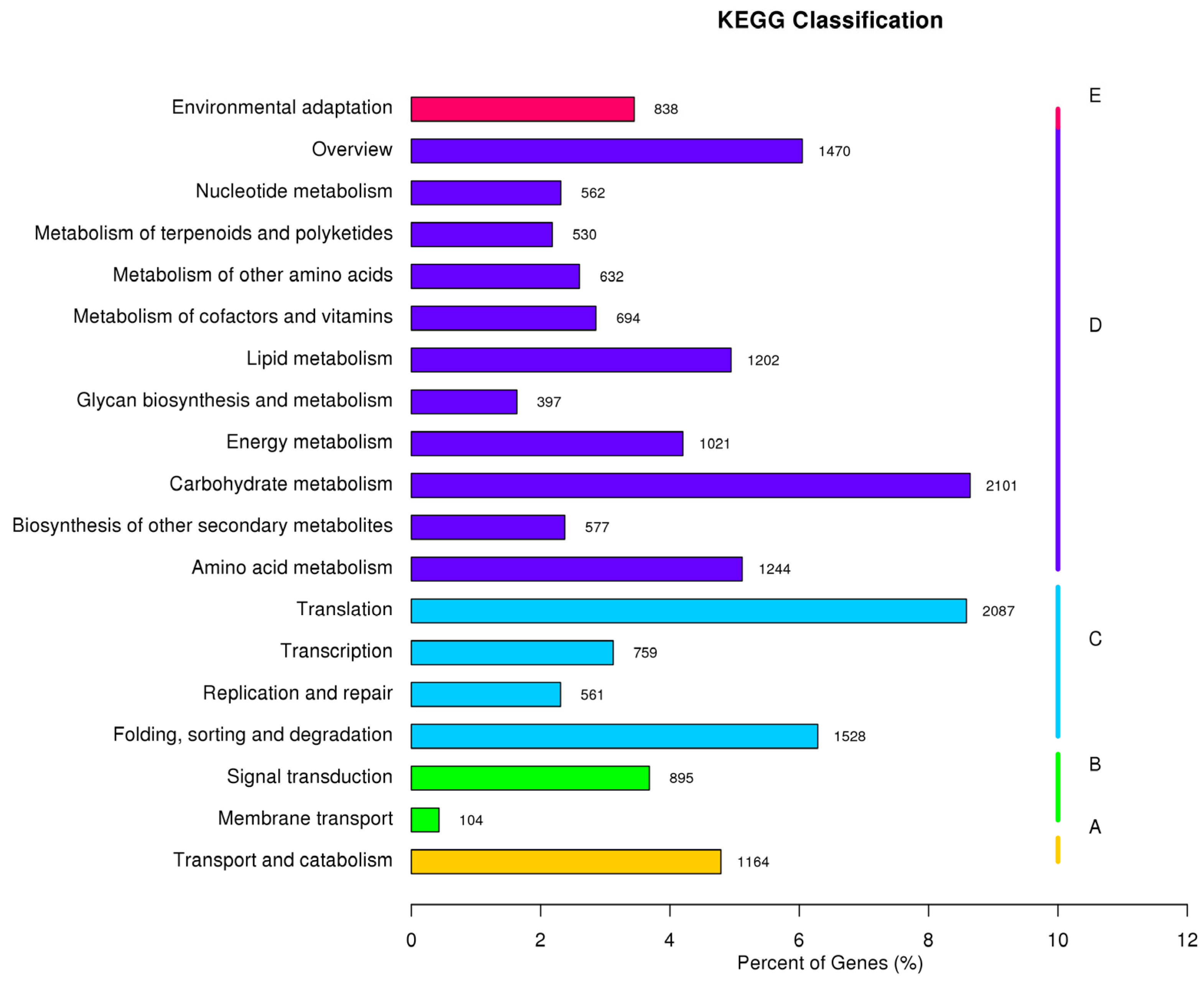
3.2. Function Annotation and Classification
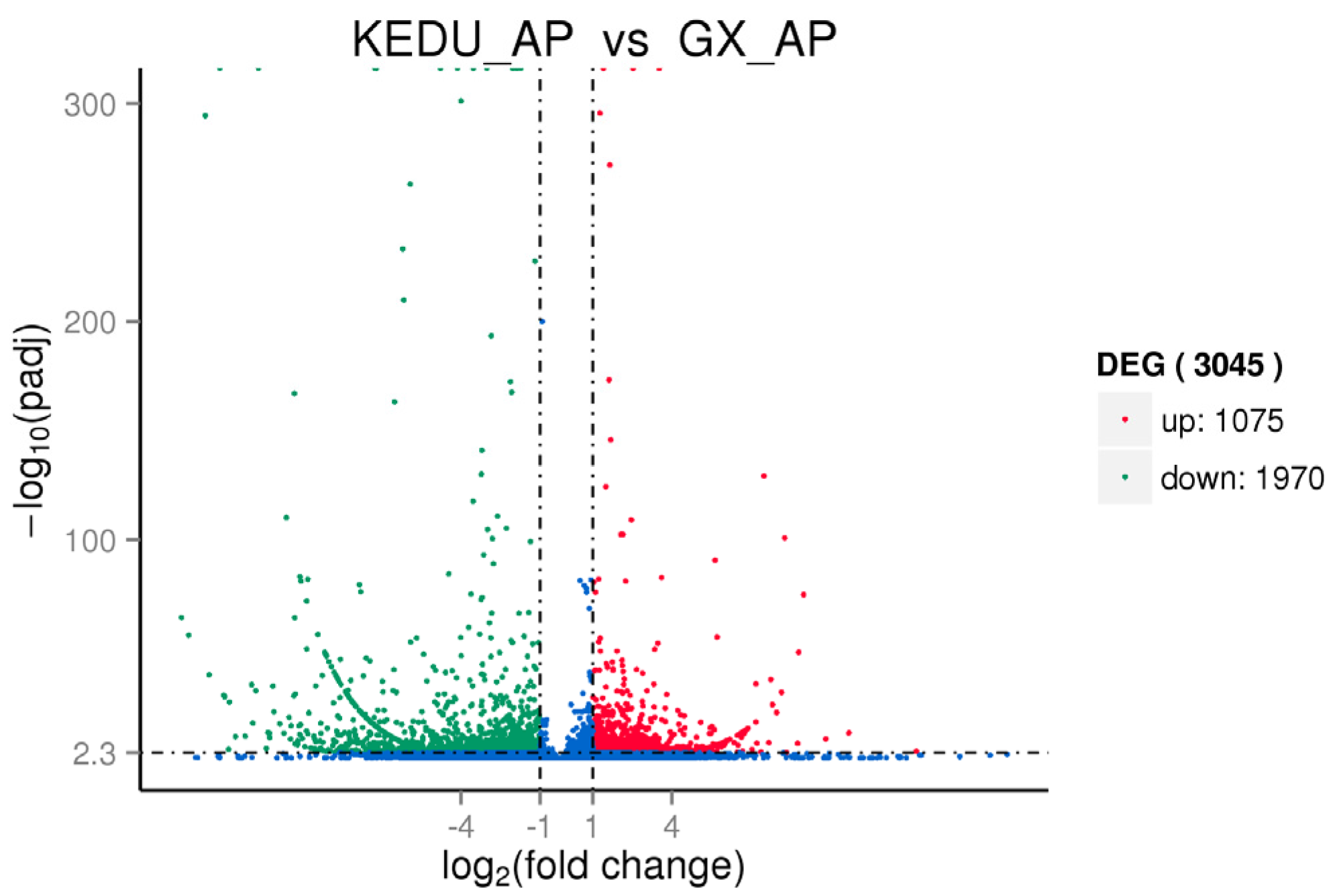
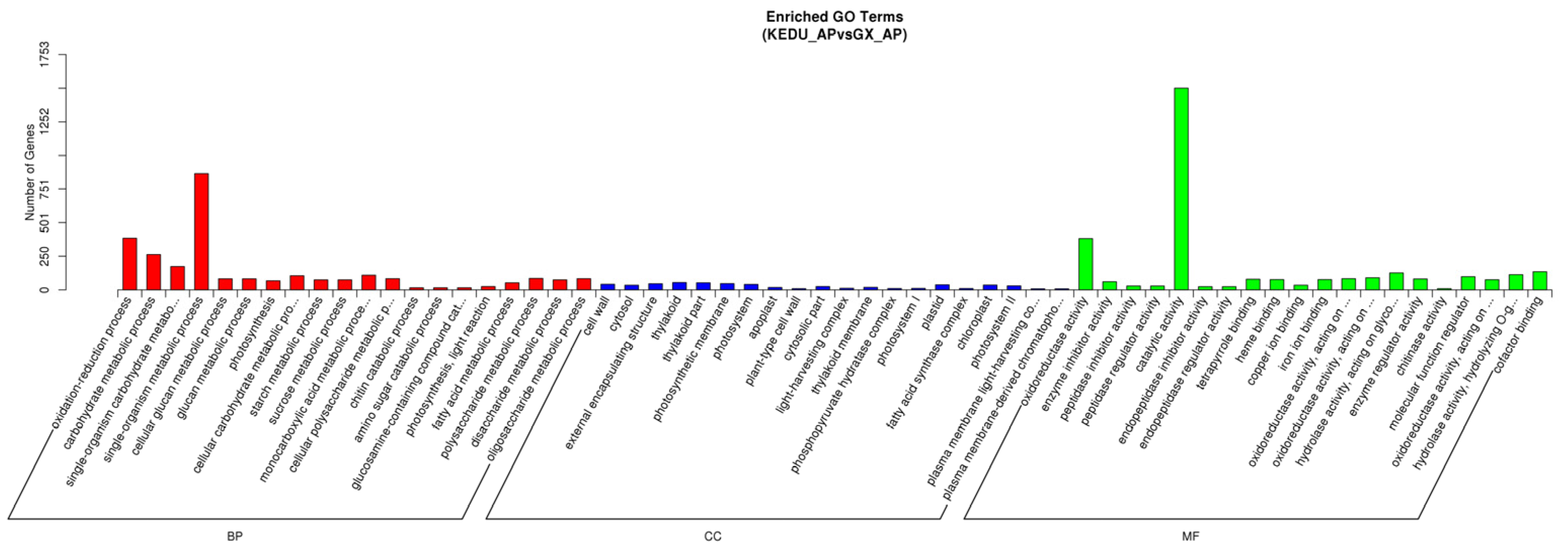
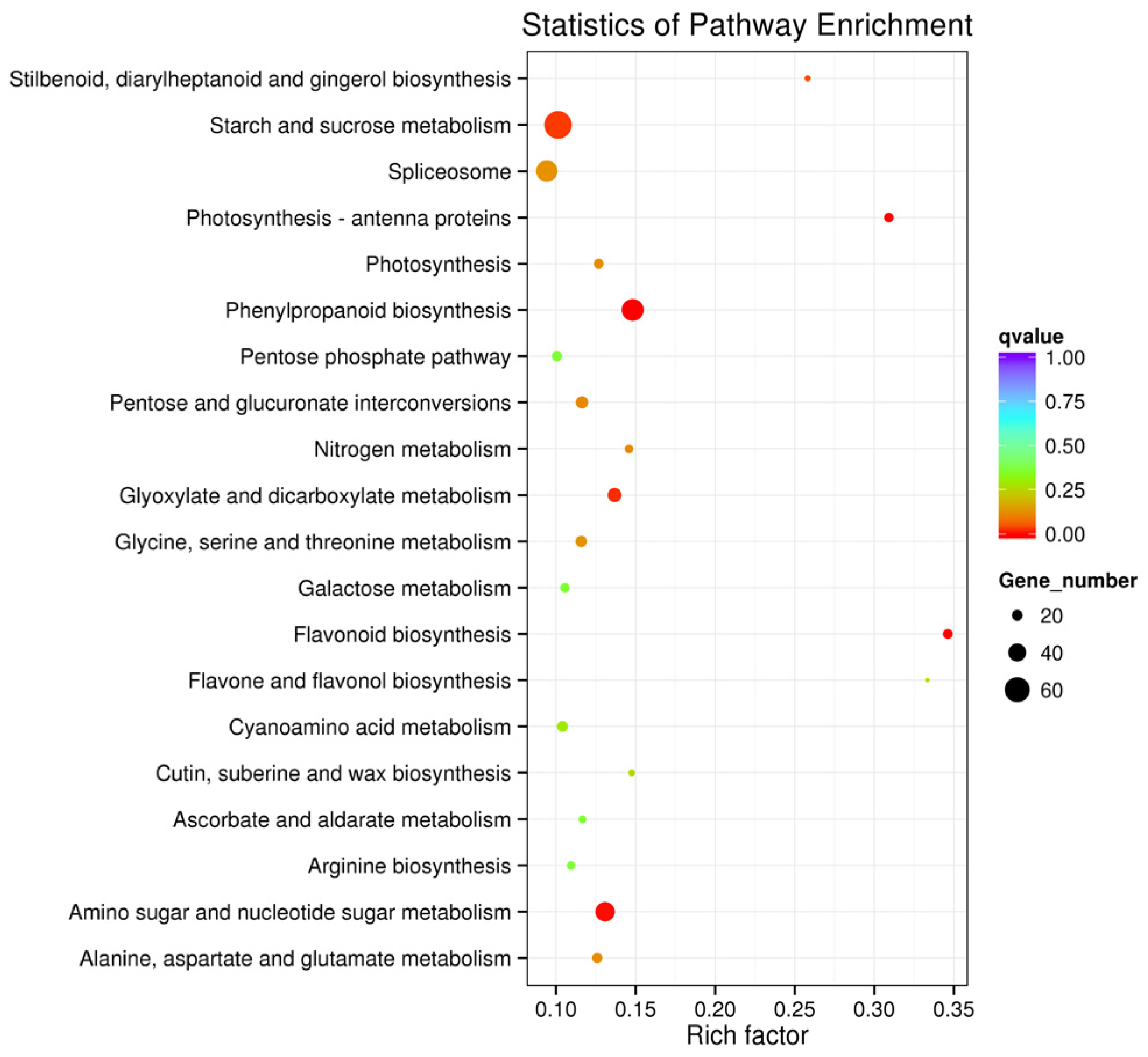
3.3. DEG Identification, GO and KEGG Enrichment Analysis
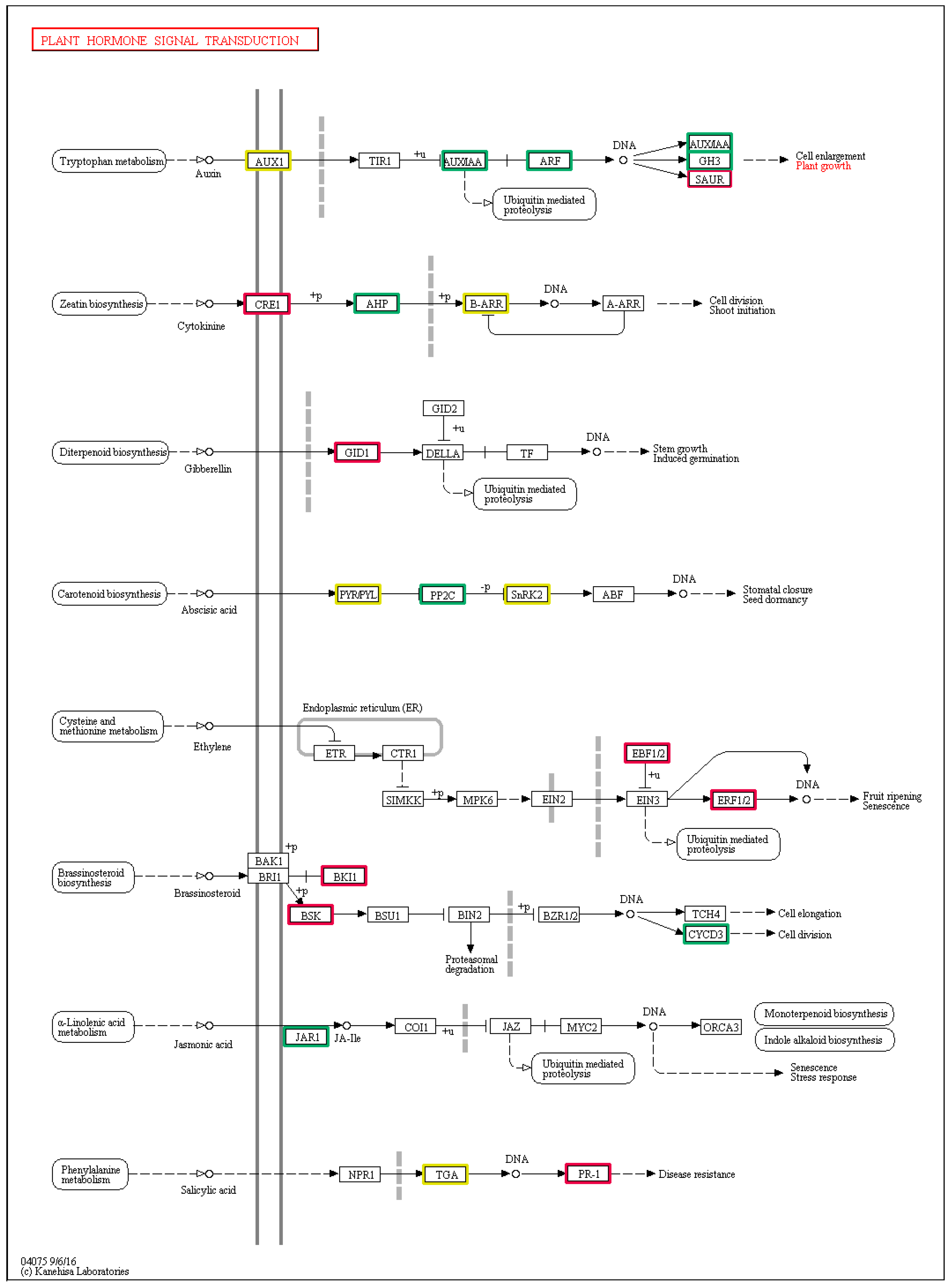
3.4. DEGs Involved in the Plant Hormone Signal Transduction Pathways
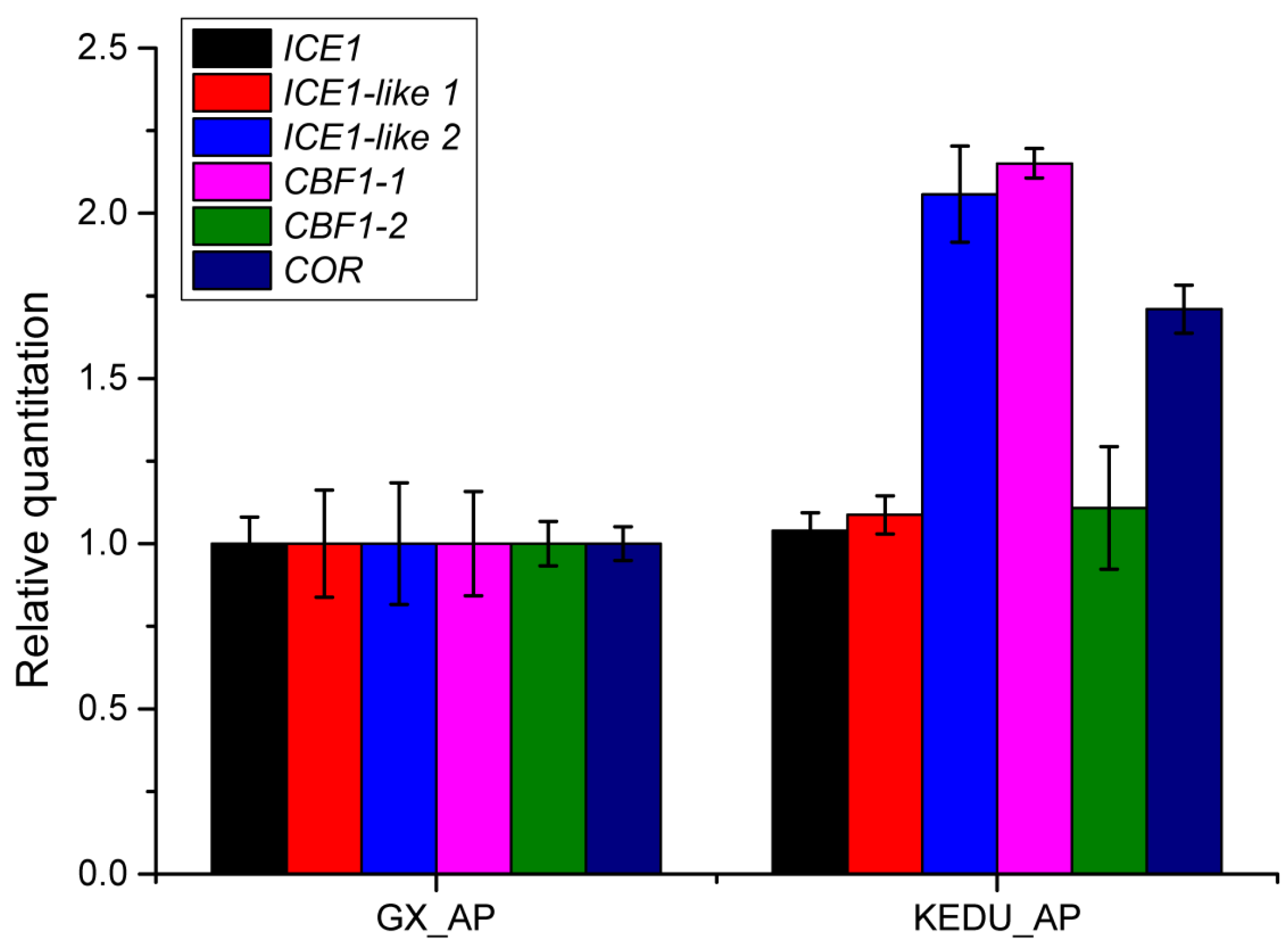
3.5. Verification by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Nr | NCBI non-redundant protein sequences |
| Nt | NCBI non-redundant nucleotide sequences |
| Pfam | Protein family |
| COG | Clusters of orthologous groups of proteins |
| KOG | EuKaryotic Orthologous Groups |
| Swiss-Prot | A manually annotated and reviewed protein sequence database |
| KEGG | Kyoto encyclopedia of genes and genomes |
| KO | KEGG ortholog database |
| GO | Gene ontology |
| FPKM | Fragments per kilobase of exon per million fragments mapped |
| DEGs | Differentially expressed genes |
References
- Rodriguez, R.R.; Dallas, M.P. Aislamientoy cultivo de protoplastos enmaracuyá isolation and cultive of protoplast in passion fruit. Acta Biol. Colomb. 2004, 9, 35–46. [Google Scholar]
- Benincá, J.P.; Montanher, A.B.; Zucolotto, S.M.; Schenkel, E.P.; Fröde, T.S. Evaluation of the anti-inflammatory efficacy of Passiflora edulis. Food Chem. 2007, 104, 1097–1105. [Google Scholar] [CrossRef]
- Bernacci, L.C.; Soares-Scott, M.D.; Junqueira, N.T.V.; Passos, I.R.D.S.; Meletti, L.M.M. Passiflora edulis sims the correct taxonomic way to cite the yellow passionfruit (and of others colors). Rev. Bras. Frutic. 2008, 30, 566–576. [Google Scholar] [CrossRef]
- Rudnicki, M.; de Oliveira, M.R.; Veiga Pereira, T.d.; Reginatto, F.H.; Dal-Pizzol, F.; Fonseca Moreira, J.C. Antioxidant and antiglycation properties of Passiflora alata and Passiflora edulis extracts. Food Chem. 2007, 100, 719–724. [Google Scholar] [CrossRef]
- Benigni, R.; Capra, C.; Cattorini, P. Plante medicinale; chimica, farmacologia e terapia. Milan Inverni Della Beffa 1962, 2, 1012–1022. [Google Scholar]
- Zibadi, S.; Watson, R.R. Passion fruit (Passiflora edulis) composition, efficacy and safety. Evid. Based Integr. Med. 2004, 1, 183–187. [Google Scholar] [CrossRef]
- Chinnusamy, V.; Zhu, J.K.; Sunkar, R. Gene regulation during cold stress acclimation in plants. Methods Mol. Biol. 2010, 639, 39–55. [Google Scholar] [PubMed]
- Chinnusamy, V.; Zhu, J.; Zhu, J.K. Cold stress regulation of gene expression in plants. Trends Plant Sci. 2007, 12, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Thomashow, M.F. Plant cold acclimation: Freezing tolerance genes and regulatory mechanisms. Annu. Rev. Plant Biol. 1999, 50, 571–599. [Google Scholar] [CrossRef] [PubMed]
- Jaglo, K.R.; Kleff, S.; Amundsen, K.L.; Zhang, X.; Haake, V.; Zhang, J.Z.; Deits, T.; Thomashow, M.F. Components of the Arabidopsis C-repeat/dehydration-responsive element binding factor cold-response pathway are conserved in Brassica napus and other plant species. Plant Physiol. 2001, 127, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Pellegrineschi, A.; Reynolds, M.; Pacheco, M.; Brito, R.M.; Almeraya, R.; Yamaguchi-Shinozaki, K.; Hoisington, D. Stress-induced expression in wheat of the Arabidopsis thaliana DREB1a gene delays water stress symptoms under greenhouse conditions. Genome 2004, 47, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.H.; Lee, J.T.; Yang, P.T.; Chiu, L.H.; Charng, Y.Y.; Wang, Y.C.; Chan, M.T. Heterology expression of the arabidopsis C-repeat/dehydration response element binding factor 1 gene confers elevated tolerance to chilling and oxidative stresses in transgenic tomato. Plant Physiol. 2002, 129, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Song, S.I.; Kim, Y.S.; Jang, H.J.; Kim, S.Y.; Kim, M.; Kim, Y.K.; Nahm, B.H.; Kim, J.K. Arabidopsis CBF3/DREB1a and ABF3 in transgenic rice increased tolerance to abiotic stress without stunting growth. Plant Physiol. 2005, 138, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Teige, M.; Scheikl, E.; Eulgem, T.; Doczi, R.; Ichimura, K.; Shinozaki, K.; Dangl, J.L.; Hirt, H. The MKK2 pathway mediates cold and salt stress signaling in Arabidopsis. Mol. Cell 2004, 15, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, H.; Zhang, X.; Xie, Q.; Gong, Z.; Yang, S. Ost1 kinase modulates freezing tolerance by enhancing ice1 stability in arabidopsis. Dev. Cell 2015, 32, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Fowler, S. Arabidopsis transcriptome profiling indicates that multiple regulatory pathways are activated during cold acclimation in addition to the CBF cold response pathway. Plant Cell 2002, 14, 1675–1690. [Google Scholar] [CrossRef] [PubMed]
- Kreps, J.A.; Wu, Y.; Chang, H.S.; Zhu, T.; Wang, X.; Harper, J.F. Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol. 2002, 130, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Mantyla, E.; Lang, V.; Palva, E.T. Role of abscisic acid in drought-lnduced freezing tolerance, cold acclimation, and accumulation of lti78 and rabi8 proteins in Arabidopsis thaliana. Plant Physiol. 1995, 107, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, S.J.; Thomashow, M.F. Cold acclimation and cold-regulated gene expression in ABA mutants of Arabidopsis thaliana. Plant Mol. Biol. 1991, 17, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.P.; Luo, C.; Long, X.Q.; Hu, J.; Li, Y. Effects of low temperature stress on physiological indexes of cold resistance of Passiflora edulis. Plant Physiol. J. 2015, 51, 771–777. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, M.J.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the blast2go suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. Rsem: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.; McCarthy, D.; Smyth, G. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the kegg orthology (ko) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.N.; Ali, G.S.; Celesnik, H.; Day, I. Coping with stresses roles of calcium and calciumcalmodulin-regulated gene expression. Plant Cell 2011, 23, 2010–2032. [Google Scholar] [CrossRef] [PubMed]
- Janska, A.; Marsık, P.; Zelenkova, S.; Ovesna, J. Cold stress and acclimation—What is important for metabolic adjustment. Plant Biol. 2010, 12, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, K.N.; Trewavas, A.J. The effect of short-term low-temperature treatments on gene expression in arabidopsis correlates with changes in intracellular Ca2+ levels. Plant Cell Environ. 2003, 26, 485–496. [Google Scholar] [CrossRef]
- Reddy, V.S.; Reddy, A.S.N. Proteomics of calcium-signaling components in plants. Phytochemistry 2004, 65, 1745–1776. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, N.; Mahajan, S. Further characterization of calcineurin B-like protein and its interacting partner CBL-interacting protein kinase from Pisum sativum. Plant Signal. Behav. 2007, 2, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.K. Emergence of a novel calcium signaling pathway in plants CBL-CIPK signaling network. Physiol. Mol. Biol. Plants 2008, 14, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.; Cle’ment, C.; Barka, E.A. Physiological and molecular changes in plants grown at low temperatures. Planta 2012, 235, 1091–1105. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, V.; Örvar, B.L.; Beyerly, J.; Hirt, H.; Dhindsa, R.S. Opposite changes in membrane fluidity mimic cold andheat stress activation of distinct plant map kinase pathways. Plant J. 2002, 31, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Ali, G.S.; Yang, L.; Du, L.; Reddy, A.S.N.; Poovaiah, B.W. Calciumcalmodulin-regulated receptor-like kinase CRLK1 interacts with MEKK1 in plants. Plant Signal. Behav. 2010, 5, 991–994. [Google Scholar] [CrossRef] [PubMed]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. Wrky transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Jakoby, M.; Weisshaara, B.; Dröge-Laserb, W.; Vicente-Carbajosac, J.; Tiedemannd, J.; Kroje, T.; Parcye, F. Bzip transcription factors in Arabidopsis. Trends Plant Sci. 2002, 7, 106–111. [Google Scholar] [CrossRef]
- Van Buskirka, H.A.; Thomashow, M.F. Arabidopsis transcription factors regulating cold acclimation. Physiol. Plant. 2006, 126, 72–80. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, G.; Jia, J.; Liu, X.; Kong, X. Molecular characterization of isolated wheat MYB genes and analysis of their expression during abiotic stress. J. Exp. Bot. 2012, 63, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, E.J.; Gilmour, S.J.; Thomashow, M.F. Arabidopsis thaliana cbf1 encodes an ap2 domain-containing transcriptional activator that binds to the C-repeatydre, a cis-acting DNA regulatory element that stimulates transcription in response to low temperature and water deficit. Proc. Natl. Acad. Sci. USA 1997, 94, 1035–1040. [Google Scholar] [CrossRef] [PubMed]
- Medina, J.N.; Bargues, M.N.; Terol, J.; Pe’rez-Alonso, M.; Salinas, J. The Arabidopsis CBF gene family is composed of three genes encoding AP2 domain-containing proteins whose expression is regulated by low temperature but not by abscisic acid or dehydration. Plant Physiol. 1999, 119, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, S.J.; Zarka, D.G.; Stockinger, E.J.; Salazar, M.P.; Houghton, J.M.; Thomashow, M.F. Low temperature regulation of the arabidopsis cbf family of ap2 transcriptional activators as an early step in coldinduced cor gene expression. Plant J. 1998, 16, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-F.; Wang, Y.-C.; Hsieh, T.-H.; Lu, C.-A.; Tseng, T.-H.; Yu, S.-M. A novel mybs3-dependent pathway confers cold tolerance in rice. Plant Physiol. 2010, 153, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Vannini, C.; Locatelli, F.; Bracale, M.; Magnani, E.; Marsoni, M.; Osnato, M.; Mattana, M.; Baldoni, E.; Coraggio, I. Overexpression of the rice osmyb4 gene increases chilling and freezing tolerance of arabidopsis thaliana plants. Plant J. 2004, 37, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Dai, X.; Zhang, W.-H. A r2r3-type myb gene, osmyb2, is involved in salt, cold, and dehydration tolerance in rice. J. Exp. Bot. 2012, 63, 2541–2556. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef] [PubMed]
- Szabados, L.; Savour, A. Proline: A multifunctional amino acid. Trends Plant Sci. 2010, 15, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Griffith, M.; Yaish, M. Antifreeze proteins in overwintering plants: A tale of two activities. Trends Plant Sci. 2004, 9, 399–405. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
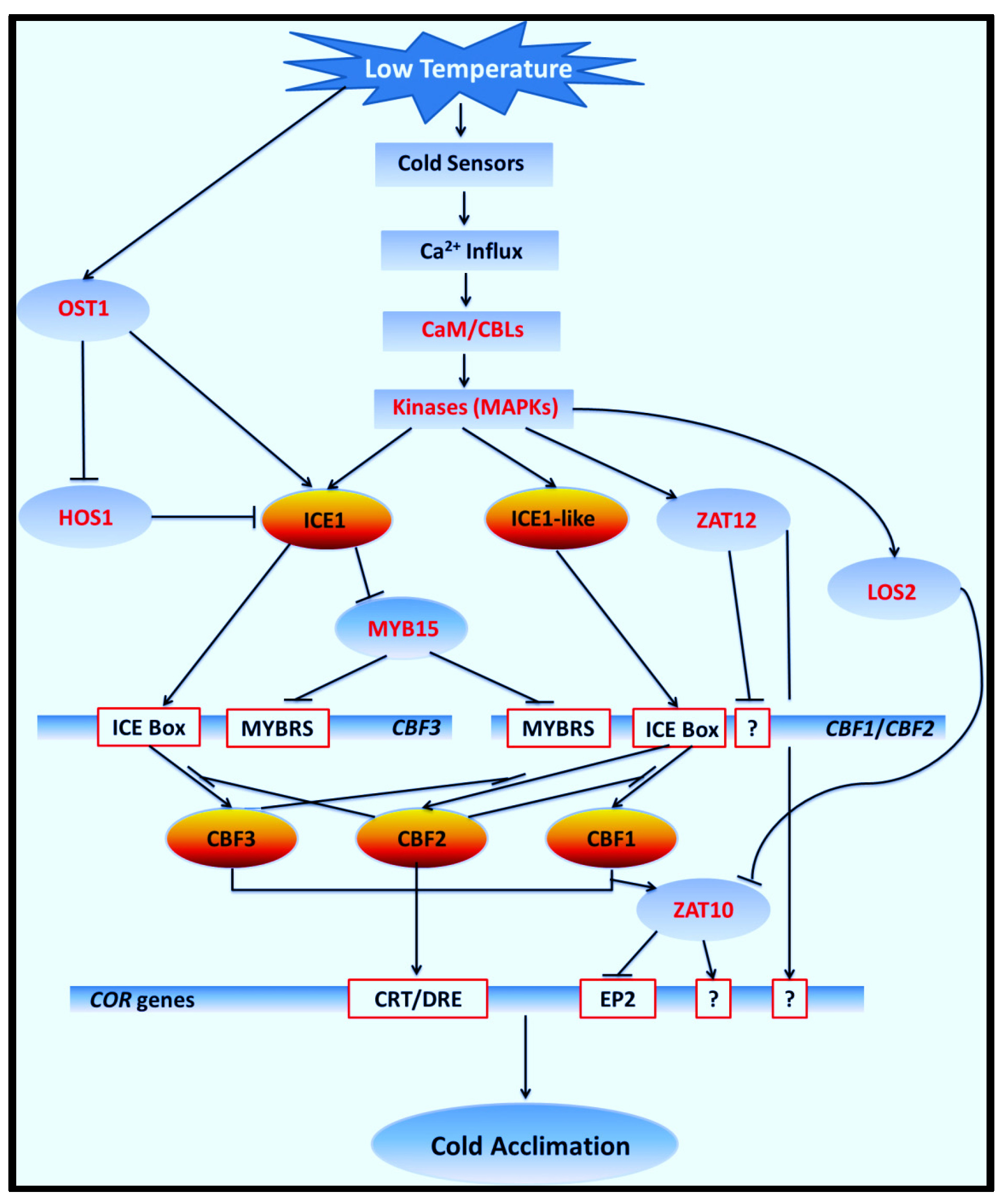{kind=link}
| Nucleotide Length | Transcripts | Unigenes |
|---|---|---|
| 200–500 bp | 60,598 | 20,947 |
| 0.5–1k bp | 22,605 | 21,684 |
| 1–2k bp | 22,073 | 22,051 |
| >2k bp | 22,198 | 22,198 |
| Total | 127,474 | 86,880 |
| Min length (bp) | 201 | 201 |
| Mean length (bp) | 1077 | 1449 |
| Max length (bp) | 13,397 | 13,397 |
| N50 (bp) | 2057 | 2222 |
| Component | Number of Unigenes | Percentage (%) |
|---|---|---|
| Annotated in NCBI non-redundant protein sequences (NR) | 60,028 | 69.09 |
| Annotated in NCBI non-redundant nucleotide sequences (NT) | 46,093 | 53.05 |
| Annotated in KEGG ortholog database (KO) | 24,312 | 27.98 |
| Annotated in Swiss-Prot | 47,717 | 54.92 |
| Annotated in Pfam | 45,544 | 52.42 |
| Annotated in Gene ontology (GO) | 46,754 | 53.81 |
| Annotated in EuKaryotic Orthologous Groups (KOG) | 17,742 | 20.42 |
| Annotated in all databases | 10,457 | 12.03 |
| Annotated in at least one database | 64,067 | 73.74 |
| Total unigenes | 86,880 |
| Component | Unigenes | DEGs | Ratio (%) |
|---|---|---|---|
| AP2/EREBP | 216 | 8 | 3.7 |
| WRKY | 161 | 12 | 7.5 |
| bZIP | 181 | 8 | 4.4 |
| MYB | 387 | 14 | 3.6 |
| NAC | 221 | 11 | 5.0 |
| B-ARR | 20 | 3 | 15 |
| CaM/CBLs | 332 | 14 | 4.2 |
| MAPK | 93 | 11 | 11.8 |
| LEA | 38 | 4 | 10.5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Li, A.; Chen, C.; Cai, G.; Zhang, L.; Guo, C.; Xu, M. De Novo Transcriptome Sequencing in Passiflora edulis Sims to Identify Genes and Signaling Pathways Involved in Cold Tolerance. Forests 2017, 8, 435. https://doi.org/10.3390/f8110435
Liu S, Li A, Chen C, Cai G, Zhang L, Guo C, Xu M. De Novo Transcriptome Sequencing in Passiflora edulis Sims to Identify Genes and Signaling Pathways Involved in Cold Tolerance. Forests. 2017; 8(11):435. https://doi.org/10.3390/f8110435
Chicago/Turabian StyleLiu, Sian, Anding Li, Caihui Chen, Guojun Cai, Limin Zhang, Chunyan Guo, and Meng Xu. 2017. "De Novo Transcriptome Sequencing in Passiflora edulis Sims to Identify Genes and Signaling Pathways Involved in Cold Tolerance" Forests 8, no. 11: 435. https://doi.org/10.3390/f8110435
APA StyleLiu, S., Li, A., Chen, C., Cai, G., Zhang, L., Guo, C., & Xu, M. (2017). De Novo Transcriptome Sequencing in Passiflora edulis Sims to Identify Genes and Signaling Pathways Involved in Cold Tolerance. Forests, 8(11), 435. https://doi.org/10.3390/f8110435