Genetic Diversity and Structure of Natural Quercus variabilis Population in China as Revealed by Microsatellites Markers

Abstract
:1. Introduction
2. Materials and Methods
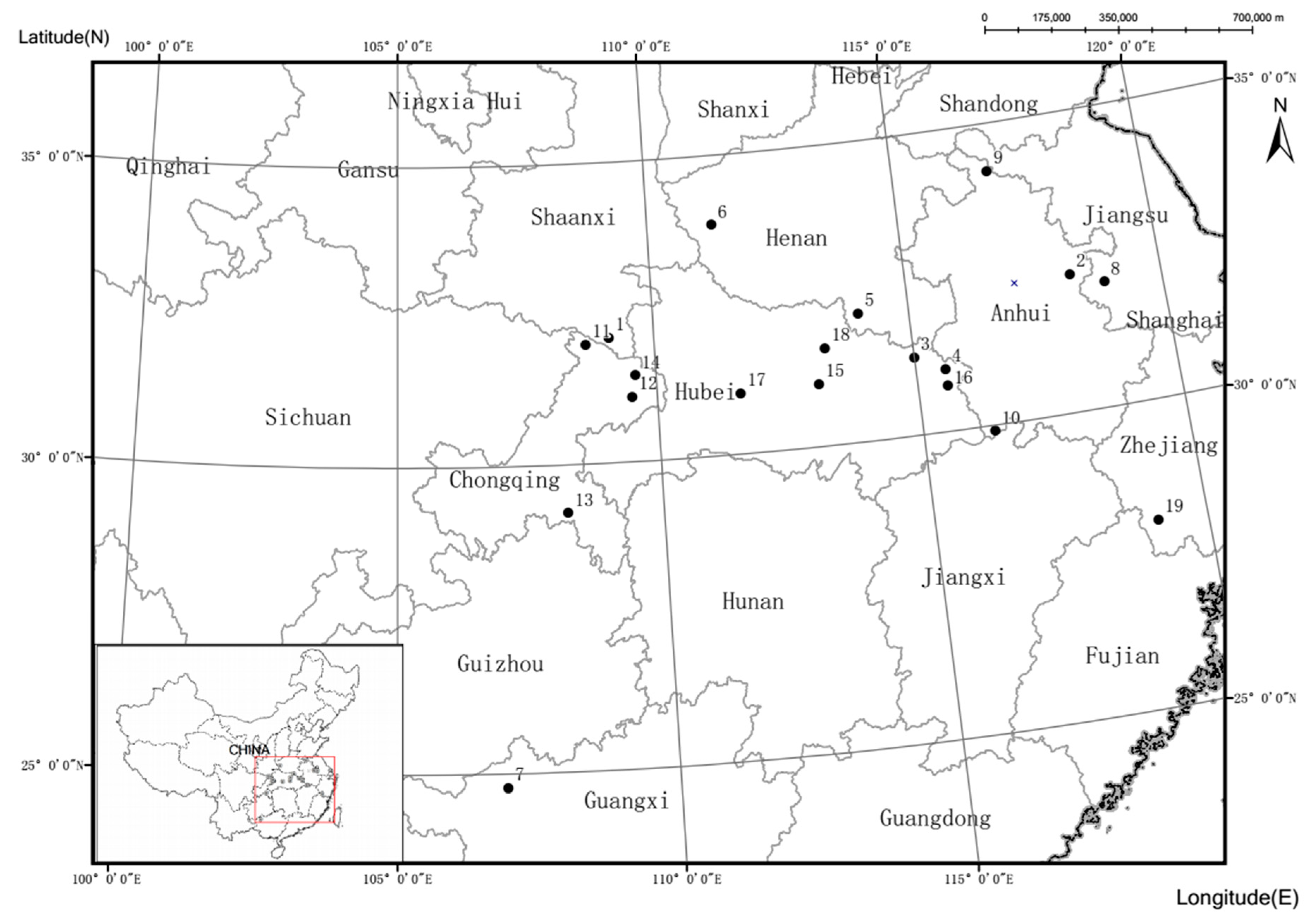
2.1. Population Sample Information
2.2. Experimental Methods
2.3. Data Analysis
3. Results
3.1. Genetic Diversity
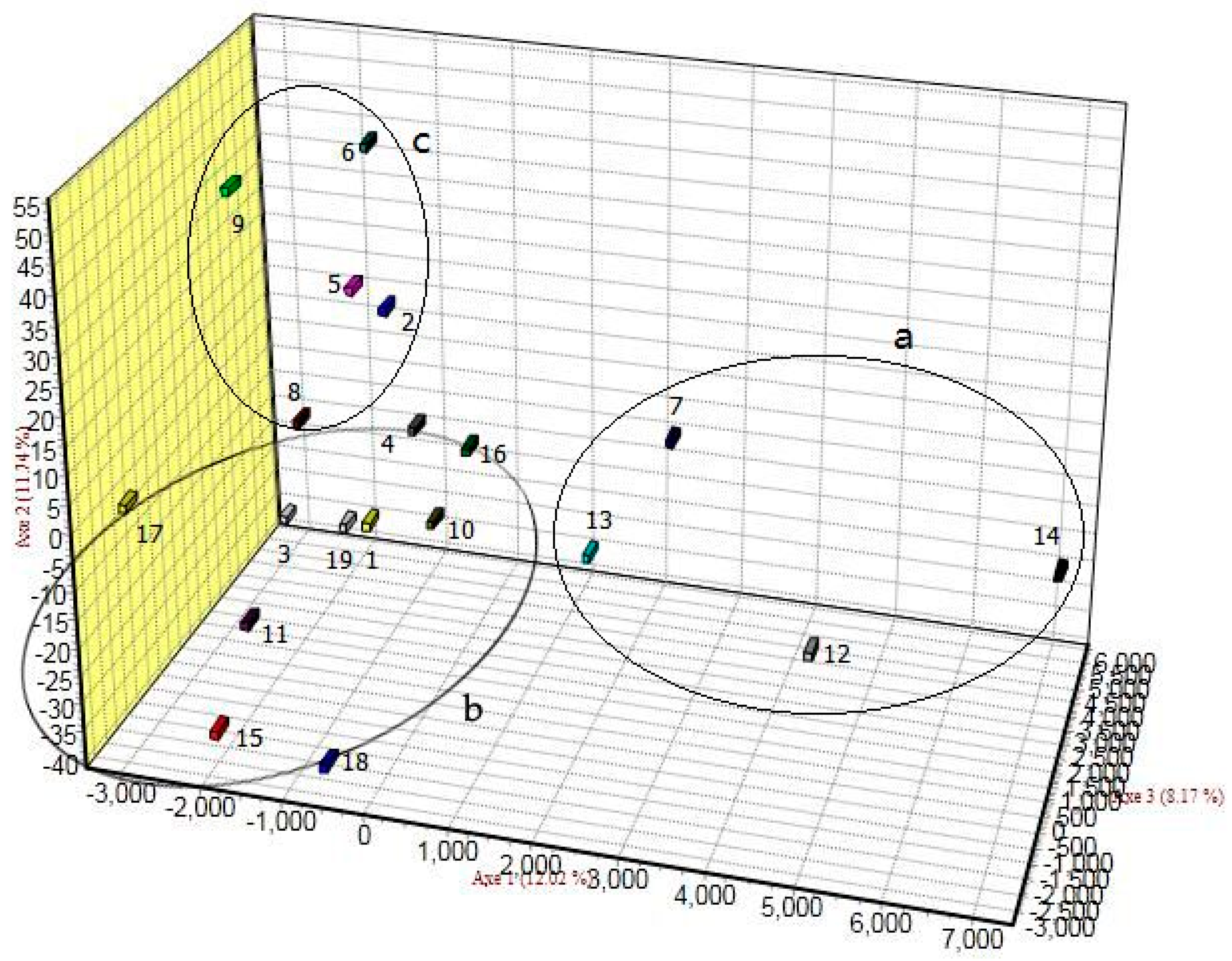
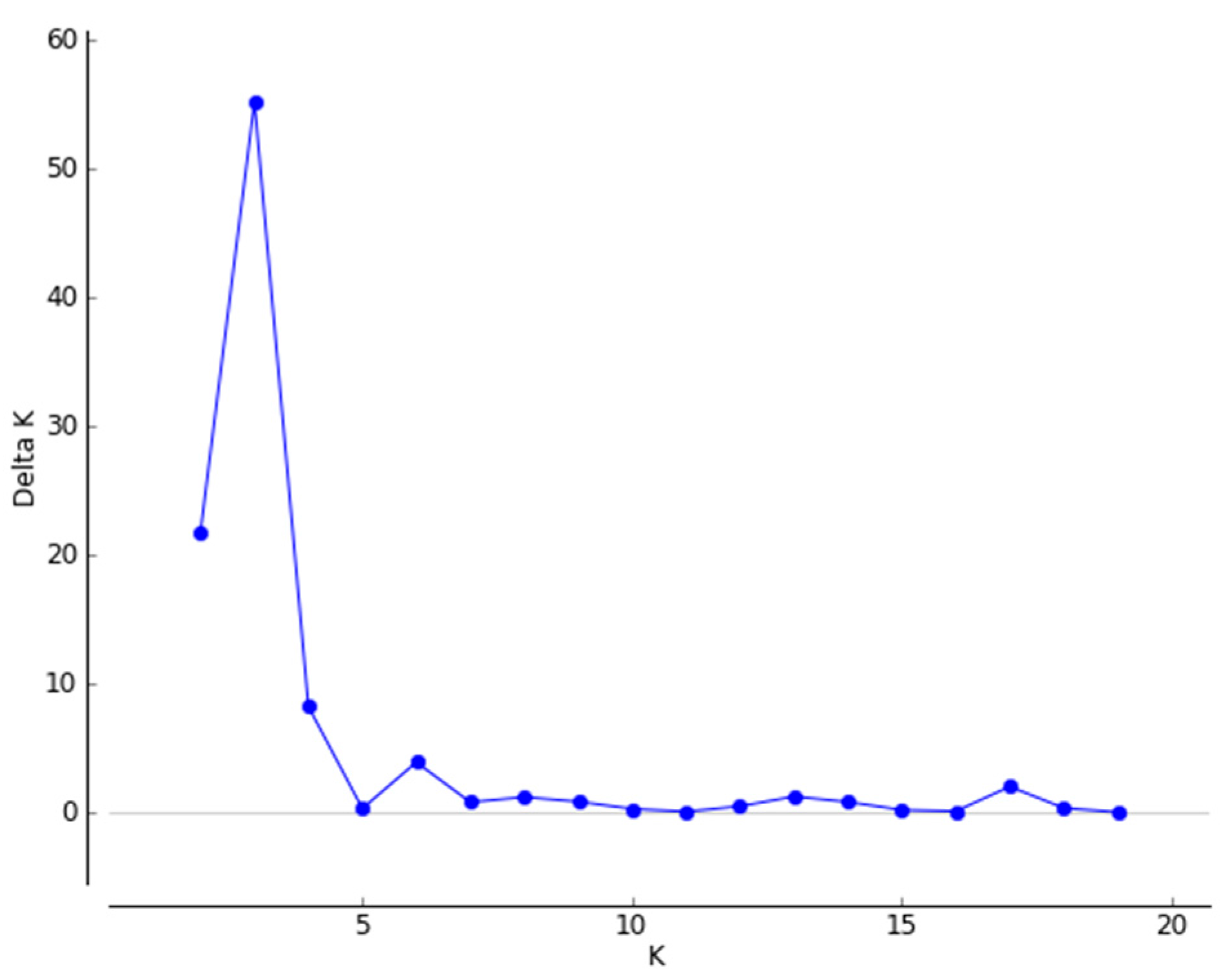
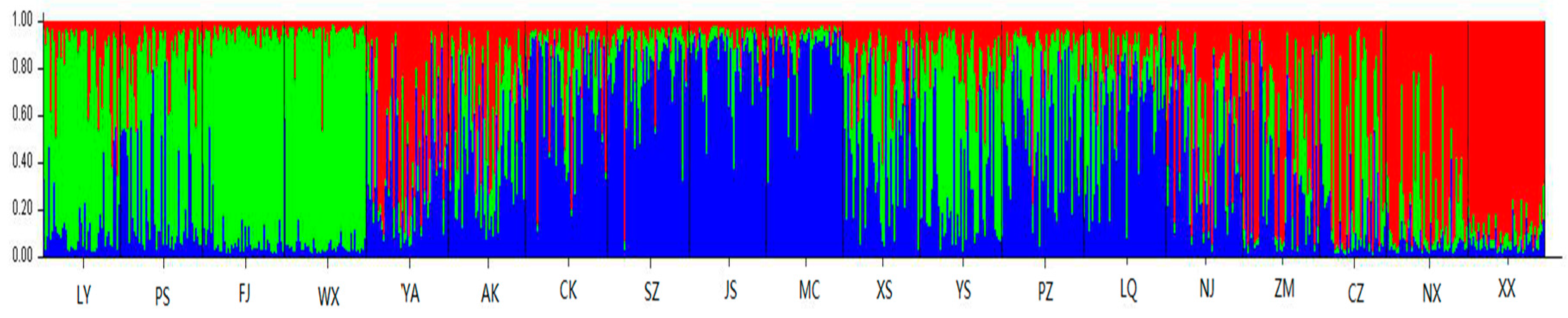
3.2. Genetic Differentiation and Genetic Structure
4. Discussion
4.1. Genetic Diversity
4.2. Genetic Differentiation
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Johnson, P.S.; Shifley, S.R. The Ecology and Silviculture of Oaks; CABI Publishing: New York, NY, USA, 2001; pp. 9–10. ISBN 9780851995700. [Google Scholar]
- Kremer, A.; Petit, R.J. Gene diversity in natural Population of oak species. Ann. For. Sci. 1993, 50, 186–202. [Google Scholar] [CrossRef]
- eFlora. Org., Flora of China, Quercus variabilis Blume. Available online: http://www.efloras.org/ (accessed on 10 October 2017).
- Wang, J.; Wang, S.B.; Kang, H.Z.; Xin, Z.J.; Qian, Z.H.; Liu, C.J. Distribution Pattern of Oriental Oak (Quercus variabilis Blume) and the Characteristics of Climate of Distribution Area in Eastern Asia. J. Shanghai Jiaotong Univ. 2009, 27, 235–241. [Google Scholar]
- Wei, L. Preliminary investigation on the distribution of the cork oak. Sci. Silvae Sin. 1960, 6, 70–71. [Google Scholar]
- Xu, X.L.; Xu, L.A.; Huan, M.R.; Wang, Z.R. Genetic Diversity of Microsatellites (SSRs) of Natural Populations of Quercus variabilis. Hereditas 2004, 26, 683–688. [Google Scholar] [PubMed]
- Zhao, G.; Duan, X.F.; Guan, T.; Huang, L.H. Situation of Cork Utilization in the World and the Development Countermeasure of the China's Cork Industry. World For. Res. 2004, 17, 25–28. [Google Scholar]
- Flores, M.; Rosa, M.E. Properties and uses of consolidated cork dust. J. Mater. Sci. 1992, 27, 5629–5634. [Google Scholar] [CrossRef]
- Zhang, W.H.; Lu, Z.J. A study on the biological and ecological property and geographical distribution of Quercus variabilis population. Acta Bot. Boreali-occident. Sin. 2002, 22, 1093–1101. [Google Scholar]
- Lei, J.P.; Xiao, W.F.; Liu, J.F. Distribution of Quercus variabilis Blume and Its Ecological Research in China. World For. Res. 2013, 26, 57–62. [Google Scholar]
- Hamrick, J.L.; Godt, M.J.W. Allozyme diversity in plant species. In Plant Population Genetics, Breeding, and Genetic Resources; Brown, A.D.H., Clegg, M.T., Kahler, A.L., Weit, B.S., Eds.; Sinauer Associates: Sunderland, MA, USA, 1990; pp. 43–63. ISBN 9780878931170. [Google Scholar]
- Hamrick, J.L.; Godt, M.J.W.; Sherman-Broyles, S.L. Factors influencing levels of genetic diversity in woody plant species. New For. 1992, 6, 95–124. [Google Scholar] [CrossRef]
- Heuertz, M.; Hausman, J.F.; Hardy, O.J.; Vendramin, G.G.; Frascarialacoste, N. Nuclear microsatellites reveal contrasting patterns of genetic structure between western and southeastern European populations of the common ash (Fraxinus excelsior L.). Evolution 2004, 58, 976–988. [Google Scholar] [PubMed]
- Frankham, R.; Ballou, J.D.; Briscoe, D.A. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2004; Volume 190, pp. 385–386. ISBN 9780521702713. [Google Scholar]
- Sutherland, W.J.; Albon, S.D.; Allison, H.S.; Armstrong, B.; Bailey, M.J.; Brereton, T.; Boyd, I.L.; Carey, P.; Edwards, J.; Gill, M.; et al. The identification of priority policy options for UK nature conservation. J. Appl. Ecol. 2010, 47, 955–965. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, X.W.; Fang, Y.M. Predicting the impact of global warming on the geographical distribution pattern of Quercus variabilis in China. Chin. J. Appl. Ecol. 2014, 25, 3381–3389. [Google Scholar]
- Zhou, J.Y.; Lin, J.; He, J.F.; Zhang, W.H. Review and Perspective on Quercus variabilis Research. J. Northwest For. Univ. 2010, 25, 43–49. [Google Scholar]
- Yuan, Z.L.; Wang, T.; Zhu, X.L.; Sha, Y.Y.; Ye, Y.Z. Patterns of spatial distribution of Quercus variabilis in deciduous broadleaf forests in Baotianman Nature Reserve. Biodivers. Sci. 2011, 19, 224–231. [Google Scholar]
- Hu, X.J.; Zhang, W.H.; He, J.F.; Yin, Y.N. Architectural analysis of crown geometry of Quercus variablis BL. natural regenerative seedlings in different habitats. Acta Ecol. Sin. 2014, 35, 789–795. [Google Scholar]
- Li, Z.P.; Zhang, W.H.; Cui, Y.C. Effects of NaCl and Na2CO3 stresses on seed germination and seedling growth of Quercus variabilis. Acta Ecol. Sin. 2015, 35, 742–751. [Google Scholar]
- Ran, R.; Zhang, W.H.; He, J.F.; Zhou, J.Y. Effects of thinning intensities on population regeneration of natural Quercus variabilis forest on the south slope of Qinling Mountains. Chin. J. Appl. Ecol. 2014, 25, 695–701. [Google Scholar]
- Zhang, Y.Y.; Fang, Y.M.; Yu, M.K.; Li, X.X.; Xia, T. Molecular characterization and genetic structure of Quercus acutissima germplasm in China using microsatellites. Mol. Biol. Rep. 2013, 40, 4083–4090. [Google Scholar] [CrossRef] [PubMed]
- Jennifer, F.L.; Oliver, G. Genetic structure of Quercus rubra L. and Quercus ellipsoidalis E.J. Hill populations at gene-based EST-SSR and nuclear SSR markers. Tree Genet. Genomes 2013, 9, 707–722. [Google Scholar]
- Saneyoshi, U.; Yoshihiko, T. Development of ten microsatellite markers for Quercus mongolica var. crispula by database mining. Conserv. Genet. 2008, 9, 1083–1085. [Google Scholar]
- Neophytou, C.H.; Dounavi, A.; Aravanopoulos, F.A. Conservation of Nuclear SSR Loci Reveals High Affinity of Quercus infectoria ssp. veneris A. Kern (Fagaceae) to Section Robur. Plant Mol. Biol. Rep. 2008, 26, 133–141. [Google Scholar] [CrossRef]
- Chen, D.M.; Zhang, X.X.; Kang, H.Z.; Sun, X.; Yin, S.; Du, H.M.; Yamanaka, N.; Gapare, W.; Wu, H.X.; Liu, C.J. Phylogeography of Quercus variabilis Based on Chloroplast DNA Sequence in East Asia: Multiple Glacial Refugia and Mainland-Migrated Island Populations. PLoS ONE 2013, 7, e47268. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.H.; Pan, H.X.; Zhu, G.Q.; Yin, T.M.; Zou, H.Y.; Huang, M.R. Analysis of Genetic Structure of Natural Populations of Castanopsis fargesii by RAPDs. Acta Bot. Sin. 2002, 44, 1321–1326. [Google Scholar]
- BiOptic Inc. Available online: http://www.bioptic.com.tw (accessed on 5 July 2016).
- Alexis, R.S.; Jennifer, F.L.; Tim, S.M.; Jeanne, R.S.; Oliver, G. Development and Characterization of Genomic and Gene-Based Microsatellite Markers in North American Red Oak Species. Plant Mol. Biol. Rep. 2012, 31, 231–239. [Google Scholar]
- Qin, Y.Y.; Han, H.R.; Kang, F.F.; Zhao, Q. Genetic diversity in natural populations of Quercus liaotungensis in Shanxi Province based on nuclear SSR markers. J. Beijing For. Univ. 2012, 34, 61–65. [Google Scholar]
- Durand, J.; Bodenes, C.; Chancerel, E.; Frigerio, J.M.; Vendramin, G.; Sebastiani, F.; Buonamici, A.; Gailing, O.; Koelewijn, H.P.; Villani, F.; et al. A fast and cost-effective approach to develop and map EST-SSR markers: Oak as a case study. BMC Genom. 2010, 11, 570. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987; pp. 73–95. ISBN 9780231063210. [Google Scholar]
- Mousadik, E.L.; Petit, R.J. High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L.) Skeels endemic to Morocco. Theor. Appl. Genet. 1996, 92, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef] [PubMed]
- Goudet, J. FSTAT Version 2.9.3, A Program to Estimate and Test Gene Diversities and Fixation Indices. 2002. Available online: http://www.unil.ch/izea/softwares/fstat.html (accessed on 5 July 2016).
- Liu, J. POWERMARKER: A Powerful Software For Marker Data Analysis; North Carolina State University, Bioinformatics Research Center: Raleigh, NC, USA, 2002. [Google Scholar]
- Peakall, R.; Smouse, P.E. GENEALEX: Genetic analysis in excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Curtu, A.L.; Gailing, O.; Leinemann, L.; Finkeldey, R. Genetic variation and differentiation within a natural community of five oak species (Quercus spp.). Plant Biol. 2007, 9, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Slatkin, M.; Barton, N.H. Methods for estimating geneflow. Evolution 1989, 43, 1349–1368. [Google Scholar] [CrossRef] [PubMed]
- Iwaizumi, M.G.; Tsuda, Y.; Ohtani, M.; Ohtani, M.; Tsumura, Y.; Takahashi, M. Recent distribution changes affect geographic clines in genetic diversity and structure of Pinus densiflora natural populations in Japan. For. Ecol. Manag. 2013, 304, 407–416. [Google Scholar] [CrossRef]
- Tian, C.; Mu, N.; Zhu, Z.Y.; Wang, C.J. Method for the Rapid Obtaining of Climate Data Based on DIVA-GIS. J. Agric. 2015, 5, 109–113. [Google Scholar]
- Piry, S.; Luikart, G.; Cornuet, J.M. BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Luikart, G.; Cornuet, J.M. Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv. Biol. 1998, 12, 228–237. [Google Scholar] [CrossRef]
- Belkhir, K.; Borsa, P.; Chikhi, L.; Raufaste, N.; Bonhomme, F. GENETIX (ver.4.02): Logiciel sous Windows TM pour la Genetique des Populations, Laboratoire Genome, Populations, Interactions; CNRSUMR 5000; Universite Montpellier II: Montpellier, France, 2004. [Google Scholar]
- Pritchard, J.K.; Wen, X.; Falush, D. STRUCTURE Version 2.3.1. 2009. Available online: http://pritch.bsd.uchicago.edu/structure (accessed on 5 July 2016).
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Laval, G.; Schneider, S. ARLEQIN (ver.3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2007, 1, 47–50. [Google Scholar] [PubMed]
- Excoffier, L.; Lischer, H. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Slatkin, M. A measure of population subdivision based on microsatellite allele frequencies. Genetics 1995, 139, 1463. [Google Scholar]
- Meirmans, P.G. Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution 2010, 60, 2399–2402. [Google Scholar] [CrossRef]
- Yan, B.Q.; Wang, T.; Hu, L.L. Population genetic diversity and structure of Schisandra sphenanthera, a medicinal plant in China. Chin. J. Ecol. 2009, 28, 811–819. [Google Scholar]
- Gaudeul, M.; Taberlet, P.; Till, B.I. Genetic diversity in an endangered alpine plant, Eryngium alpinum L. (Apiaceae), inferred from a mplified fragment length polymorphism markers. Mol. Ecol. 2000, 9, 1625–1637. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Jin, Z.X.; Lou, W.Y.; LI, J.M. Genetic diversity of Lithocarpus harlandii populations in three forest communities with different succession stage. Chin. J. Ecol. 2007, 26, 509–514. [Google Scholar] [CrossRef]
- Hellmanna, J.J.; Pineda, K.M. Constraints and reinforcement on adaptation under climate change: Selection of genetically correlated traits. Biol. Conserv. 2007, 13, 599–609. [Google Scholar] [CrossRef]
- Ge, Y.Q.; Qiu, Y.X.; Ding, B.Y.; Fu, C.X. An ISSR analysis on population genetic diversity of the relict plant Ginkgo biloba. Chin. Biodivers. 2003, 4, 276–287. [Google Scholar]
- Cornuet, J.M.; Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 1996, 144, 2001–2014. [Google Scholar] [PubMed]
- Mayr, E. Animal Species and Evolution; Harvard University Press: Cambridge, UK, 1963; pp. 425–457. ISBN 9780674037502. [Google Scholar]
- Hampe, A.; Petit, R.J. Conserving biodiversity under climate change: The rear edge matters. Ecol. Lett. 2005, 8, 461–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamrick, J.L. IsoZymes ananysis of genetic structure of plant population. In Isozymes in Plant Biology; Soltis, D., Soltis, P., Eds.; Dioscorids Press: Washington, DC, USA, 1989; pp. 87–105. ISBN 9780412365003. [Google Scholar]
- Ohsawa, T.; Tsuda, Y.; Saito, Y.; Ide, Y. The genetic structure of Quercus crispula in northeastern Japan as revealed by nuclear simple sequence repeat loci. J. Plant Res. 2011, 124, 645–654. [Google Scholar] [CrossRef] [PubMed]
- He, C.R.; Chen, F.Q. The endemic genera of Chinese seed plants distributed in Three Gorges Reservoir Area of Changjiang River. Guihaia 1999, 19, 43–46. [Google Scholar]
- Jin, J.H.; Liao, W.B.; Wang, B.S.; Peng, S.L. Global change in Cenozoic and evolution of flora in China. Guihaia 2003, 23, 217–225. [Google Scholar]
- Knowles, P.; Perry, D.J.; Foster, H.A. Spatial genetic structure in two tamarack (Larix laricina) populations with differing establish menthi stories. Evolution 1992, 46, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Young, A.C.; Merriam, H.G. Effect of forest fragmentation on the spatial genetic structure of Acer saccharum Marsh. (sugar maple) populations. Heredity 1994, 72, 201–208. [Google Scholar] [CrossRef]
- Aldrich, P.R.; Hamrick, J.L.; Chavarriaga, P. Microsatellite analysis of de mographic genetic structure in fragmented populations of the tropical tree Symphonia globulifera. Mol. Ecol. 1998, 7, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.K. Origian, Phylogeny and Disperal of Quercus from China. Acta Bot. Yunnanica 1992, 14, 227–236. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Population | Code | Latitude (°N) | Longitude (°E) | N | Annual Temperature (°C) | Weather Patterns | Annual Precipitation (mm) |
|---|---|---|---|---|---|---|---|---|
| 1 | Ankang, Shaanxi | AK | 32°40′ | 109°08′ | 45 | 15.3 | S | 803 |
| 2 | Chuzhou, Anhui | CZ | 32°17′ | 118°17′ | 39 | 15.3 | S | 1009 |
| 3 | Macheng, Hubei | MC | 31°17′ | 115°00′ | 45 | 16.4 | S | 1217 |
| 4 | Xingshan, Hubei | XS | 31°02′ | 110°07′ | 45 | 16.5 | S | 1498 |
| 5 | Zhumadian, Henan | ZM | 32°08′ | 114°01′ | 45 | 15.4 | S | 1098 |
| 6 | Neixiang, Henan | NX | 33°50′ | 111°18′ | 48 | 8.3 | T | 871 |
| 7 | Leye, Guangxi | LY | 24°47′ | 106°57′ | 45 | 18.4 | S | 1314 |
| 8 | Nanjing, Jiangsu | NJ | 32°03′ | 118°52′ | 45 | 15.6 | S | 1017 |
| 9 | Xiaoxian, Anhui | XX | 34°12′ | 116°56′ | 45 | 9.9 | T | 756 |
| 10 | Pengze, Jiangxi | PZ | 29°54′ | 116°34′ | 48 | 17.4 | S | 1460 |
| 11 | Chengkou, Chongqing | CK | 31°59′ | 108°40′ | 48 | 13.1 | S | 1165 |
| 12 | Fengjie, Chongqing | FJ | 31°04′ | 109°31′ | 48 | 13.6 | S | 1071 |
| 13 | Pengshui, Chongqing | PS | 29°12′ | 108°12′ | 48 | 15.6 | S | 1296 |
| 14 | Wuxi, Chongqing | WX | 31°25′ | 109°36′ | 48 | 12.9 | S | 1131 |
| 15 | Jingshan, Hubei | JS | 31°02′ | 113°07′ | 45 | 16.3 | S | 1035 |
| 16 | Yingshan, Hubei | YS | 30°45′ | 115°34′ | 48 | 17.1 | S | 1459 |
| 17 | Yuanshan, Hube | YA | 31°00′ | 111°36′ | 48 | 16.6 | S | 1018 |
| 18 | Suizhou, Hubei | SZ | 31°36′ | 113°18′ | 48 | 16.1 | S | 1007 |
| 19 | Longquan, Zhejiang | LQ | 28°02′ | 119°05′ | 48 | 17.6 | S | 1793 |
| ALL | 879 |
| Locus | Repeat Motif | Na | Ho | He | PIC | Source |
|---|---|---|---|---|---|---|
| 2p24 | (CA)14 | 8 | 0.429 | 0.717 | 0.794 | Alexis, R.S. et al. [29] |
| E71-72 | (GA)46 | 8 | 0.212 | 0.771 | 0.803 | Qin, Y.Y. et al. [30] |
| PIE040 | (TTC)8 | 10 | 0.157 | 0.724 | 0.747 | Alexis, R.S. et al. [29] |
| GOT040 | (GA)11 | 5 | 0.389 | 0.468 | 0.782 | Durand, J. et al. [31] |
| G0T009 | (TC)7 | 6 | 0.208 | 0.590 | 0.673 | Durand, J. et al. [31] |
| FIR053 | (GTG)7 | 8 | 0.294 | 0.751 | 0.786 | Durand, J. et al. [31] |
| FIR039 | (CT)7 | 9 | 0.459 | 0.754 | 0.782 | Durand, J. et al. [31] |
| FIR004 | (CT)18 | 8 | 0.472 | 0.760 | 0.778 | Alexis, R.S. et al. [29] |
| G11 | (TC)22 | 4 | 0.324 | 0.551 | 0.615 | Xu, X.L. et al. [6] |
| PL111-112 | (TC)9 | 6 | 0.534 | 0.694 | 0.720 | Qin, Y.Y. et al. [30] |
| PL229-230 | (AG)15 | 9 | 0.387 | 0.669 | 0.689 | Qin, Y.Y. et al. [30] |
| VIT107 | (TA)13 | 5 | 0.306 | 0.452 | 0.524 | Durand, J. et al. [31] |
| DN949726 | (GAT)6 | 15 | 0.384 | 0.861 | 0.878 | Saneyoshi, U. et al. [24] |
| E11-12 | (GA)32 | 14 | 0.578 | 0.851 | 0.887 | Qin, Y.Y. et al. [30] |
| E79-80 | (TC)18 | 24 | 0.626 | 0.841 | 0.889 | Qin, Y.Y. et al. [30] |
| EE812 | (AG)7 | 20 | 0.393 | 0.804 | 0.856 | Zhang, Y.Y. et al. [22] |
| G7 | (TC)17 | 21 | 0.447 | 0.784 | 0.883 | Xu, X.L. et al. [6] |
| G16 | (AG)21 | 20 | 0.606 | 0.829 | 0.860 | Xu, X.L. et al. [6] |
| PL127-128 | (AG)12 | 18 | 0.408 | 0.846 | 0.874 | Qin, Y.Y. et al. [30] |
| Q16 | (GA)18 | 26 | 0.704 | 0.875 | 0.911 | Xu, X.L. et al. [6] |
| EE802 | (CT)8 | 7 | 0.427 | 0.748 | 0.790 | Zhang, Y.Y. et al. [22] |
| EE856 | (GGT)6 | 4 | 0.308 | 0.418 | 0.423 | Zhang, Y.Y. et al. [22] |
| FIR048 | (CT)9 | 8 | 0.438 | 0.758 | 0.784 | Durand, J. et al. [31] |
| FIR110 | (TC)20 | 6 | 0.186 | 0.552 | 0.602 | Alexis, R.S. et al. [29] |
| PIE125 | (GGAAGC)3 | 8 | 0.353 | 0.619 | 0.664 | Durand, J. et al. [31] |
| mean | 11 | 0.401 | 0.707 | 0.760 | ||
| min | 4 | 0.157 | 0.418 | 0.423 | ||
| max | 26 | 0.704 | 0.875 | 0.911 |
| Code | Na | AR | He | FIS | TPM | SMM | FST | GST | RST |
|---|---|---|---|---|---|---|---|---|---|
| AK | 8.20 | 8.03 | 0.710 | 0.038 | ns | ns | |||
| CZ | 7.12 | 7.12 | 0.683 | 0.054 | ns | 0.037 * | |||
| MC | 8.32 | 8.16 | 0.720 | 0.033 | ns | ns | |||
| XS | 8.68 | 8.56 | 0.723 | 0.045 | ns | ns | |||
| ZM | 7.84 | 7.65 | 0.711 | 0.043 | ns | ns | |||
| NX | 7.92 | 7.75 | 0.709 | 0.042 | ns | ns | |||
| LY | 8.44 | 8.23 | 0.707 | 0.046 | ns | ns | |||
| NJ | 7.96 | 7.81 | 0.725 | 0.049 | ns | ns | |||
| XX | 7.04 | 6.90 | 0.690 | 0.041 | ns | ns | |||
| PZ | 8.76 | 8.54 | 0.725 | 0.042 | ns | ns | |||
| CK | 8.48 | 7.54 | 0.745 | 0.044 | ns | ns | |||
| FJ | 7.12 | 6.95 | 0.623 | 0.045 | 0.045 * | 0.002 * | |||
| PS | 8.68 | 7.73 | 0.699 | 0.050 | ns | 0.019 * | |||
| WX | 7.44 | 7.29 | 0.701 | 0.055 | ns | ns | |||
| JS | 7.92 | 8.45 | 0.705 | 0.039 | ns | ns | |||
| YS | 8.24 | 8.00 | 0.694 | 0.049 | ns | ns | |||
| YA | 8.04 | 7.86 | 0.726 | 0.041 | ns | ns | |||
| SZ | 8.16 | 7.93 | 0.728 | 0.043 | ns | ns | |||
| LQ | 7.76 | 7.54 | 0.716 | 0.039 | ns | ns | |||
| mean | 8.01 | 7.79 | 0.707 | 0.044 | 0.063 | 0.060 | 0.073 | ||
| min | 7.04 | 6.90 | 0.623 | 0.033 | |||||
| max | 8.76 | 8.56 | 0.745 | 0.055 |
| Code | AK | CZ | MC | XS | ZM | NX | LY | NJ | XX | PZ | CK | FJ | PS | WX | JS | YS | YA | SZ | LQ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AK | 0.000 | ||||||||||||||||||
| CZ | 0.051 | 0.000 | |||||||||||||||||
| MC | 0.067 | 0.079 | 0.000 | ||||||||||||||||
| XS | 0.040 | 0.042 | 0.049 | 0.000 | |||||||||||||||
| ZM | 0.059 | 0.044 | 0.062 | 0.048 | 0.000 | ||||||||||||||
| NX | 0.062 | 0.033 | 0.078 | 0.064 | 0.044 | 0.000 | |||||||||||||
| LY | 0.055 | 0.040 | 0.073 | 0.040 | 0.058 | 0.061 | 0.000 | ||||||||||||
| NJ | 0.063 | 0.048 | 0.051 | 0.035 | 0.050 | 0.053 | 0.050 | 0.000 | |||||||||||
| XX | 0.076 | 0.068 | 0.111 | 0.072 | 0.077 | 0.061 | 0.082 | 0.075 | 0.000 | ||||||||||
| PZ | 0.059 | 0.055 | 0.047 | 0.047 | 0.051 | 0.076 | 0.050 | 0.048 | 0.099 | 0.000 | |||||||||
| CK | 0.027 | 0.050 | 0.049 | 0.040 | 0.050 | 0.057 | 0.047 | 0.041 | 0.077 | 0.039 | 0.000 | ||||||||
| FJ | 0.077 | 0.081 | 0.105 | 0.078 | 0.108 | 0.112 | 0.059 | 0.091 | 0.135 | 0.082 | 0.079 | 0.000 | |||||||
| PS | 0.039 | 0.056 | 0.069 | 0.040 | 0.064 | 0.074 | 0.047 | 0.057 | 0.081 | 0.057 | 0.050 | 0.068 | 0.000 | ||||||
| WX | 0.078 | 0.082 | 0.101 | 0.066 | 0.091 | 0.096 | 0.053 | 0.078 | 0.101 | 0.076 | 0.079 | 0.082 | 0.053 | 0.000 | |||||
| JS | 0.058 | 0.076 | 0.067 | 0.071 | 0.086 | 0.093 | 0.084 | 0.065 | 0.113 | 0.049 | 0.045 | 0.093 | 0.079 | 0.111 | 0.000 | ||||
| YS | 0.040 | 0.040 | 0.049 | 0.039 | 0.041 | 0.058 | 0.043 | 0.035 | 0.088 | 0.029 | 0.033 | 0.065 | 0.036 | 0.070 | 0.060 | 0.000 | |||
| YA | 0.048 | 0.047 | 0.068 | 0.054 | 0.063 | 0.061 | 0.054 | 0.050 | 0.082 | 0.057 | 0.036 | 0.082 | 0.066 | 0.099 | 0.064 | 0.054 | 0.000 | ||
| SZ | 0.052 | 0.079 | 0.056 | 0.052 | 0.077 | 0.089 | 0.062 | 0.051 | 0.105 | 0.046 | 0.028 | 0.085 | 0.062 | 0.083 | 0.054 | 0.053 | 0.049 | 0.000 | |
| LQ | 0.052 | 0.050 | 0.050 | 0.031 | 0.052 | 0.069 | 0.049 | 0.043 | 0.082 | 0.039 | 0.037 | 0.085 | 0.054 | 0.084 | 0.060 | 0.038 | 0.045 | 0.053 | 0.000 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, X.; Wen, Q.; Cao, M.; Guo, X.; Xu, L.-a. Genetic Diversity and Structure of Natural Quercus variabilis Population in China as Revealed by Microsatellites Markers. Forests 2017, 8, 495. https://doi.org/10.3390/f8120495
Shi X, Wen Q, Cao M, Guo X, Xu L-a. Genetic Diversity and Structure of Natural Quercus variabilis Population in China as Revealed by Microsatellites Markers. Forests. 2017; 8(12):495. https://doi.org/10.3390/f8120495
Chicago/Turabian StyleShi, Xiaomeng, Qiang Wen, Mu Cao, Xin Guo, and Li-an Xu. 2017. "Genetic Diversity and Structure of Natural Quercus variabilis Population in China as Revealed by Microsatellites Markers" Forests 8, no. 12: 495. https://doi.org/10.3390/f8120495
APA StyleShi, X., Wen, Q., Cao, M., Guo, X., & Xu, L. -a. (2017). Genetic Diversity and Structure of Natural Quercus variabilis Population in China as Revealed by Microsatellites Markers. Forests, 8(12), 495. https://doi.org/10.3390/f8120495