High Morphological Differentiation in Crown Architecture Contrasts with Low Population Genetic Structure of German Norway Spruce Stands

 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Sites and Sampling
2.2. Phenotypic Assessment
2.3. Marker Analysis
2.4. DNA Extraction
2.5. Phenotypic Variation
2.6. Genetic Variation—SSR Analyses
3. Results
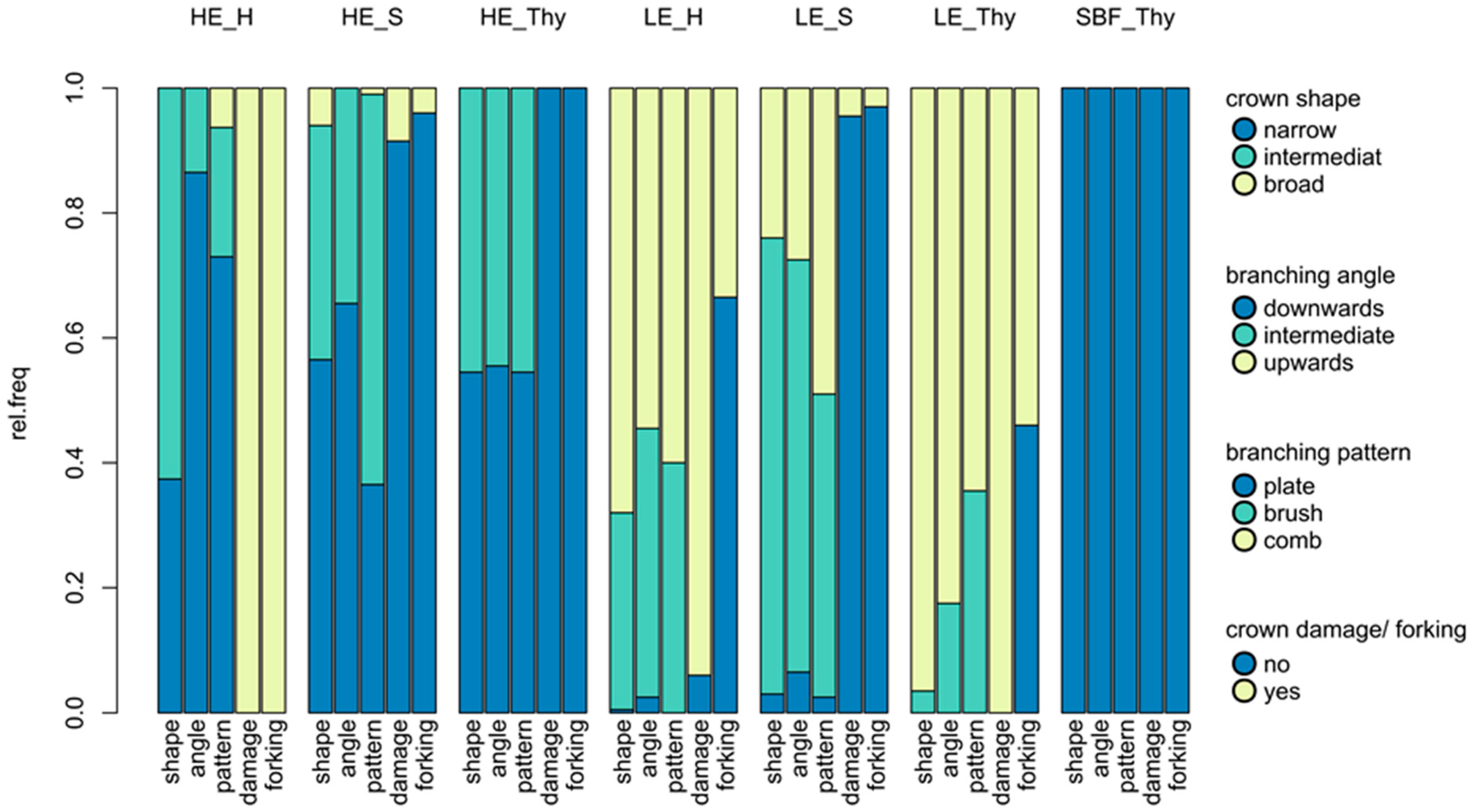
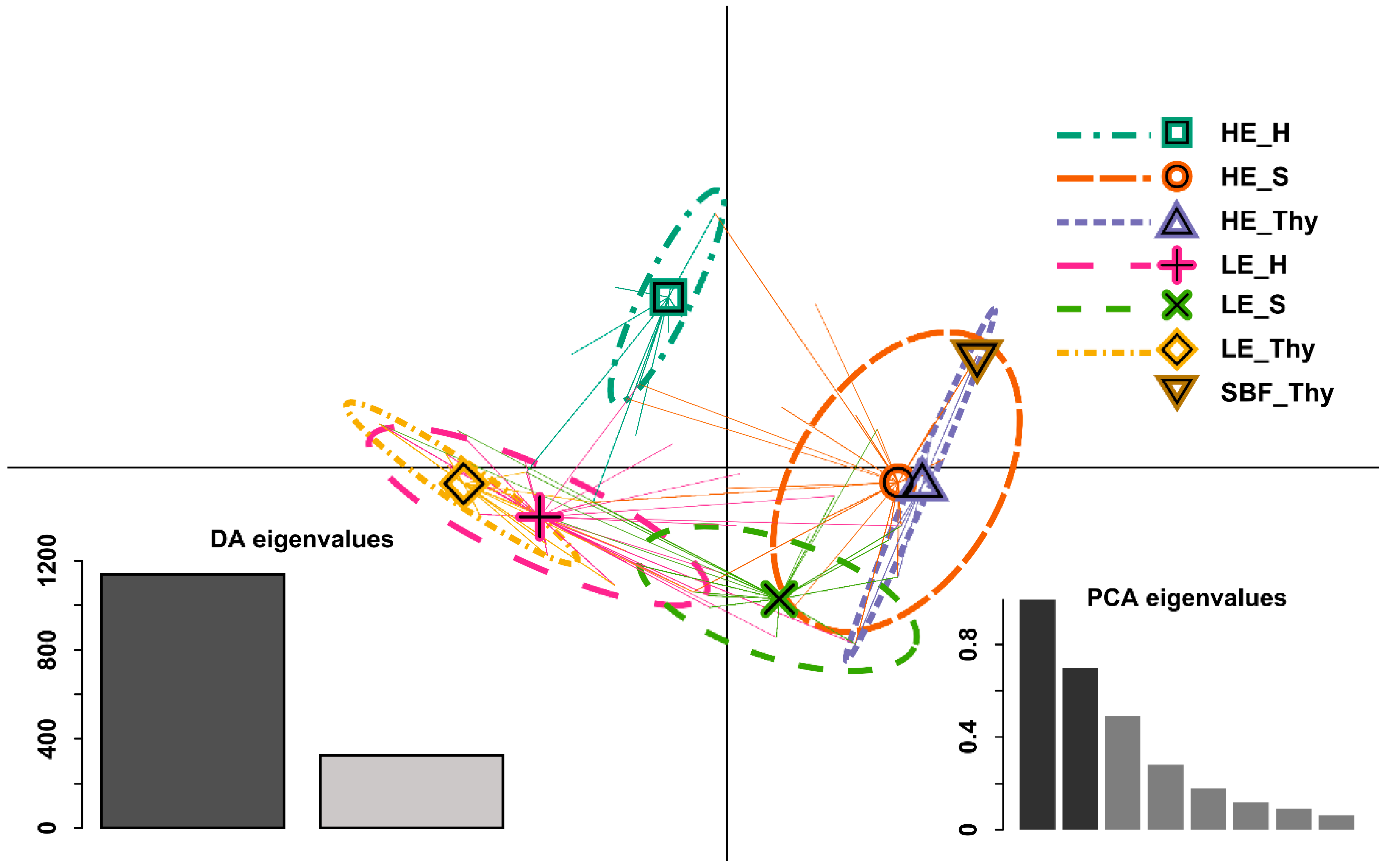
3.1. Phenotypic Differentiation between Low and High Elevation Types
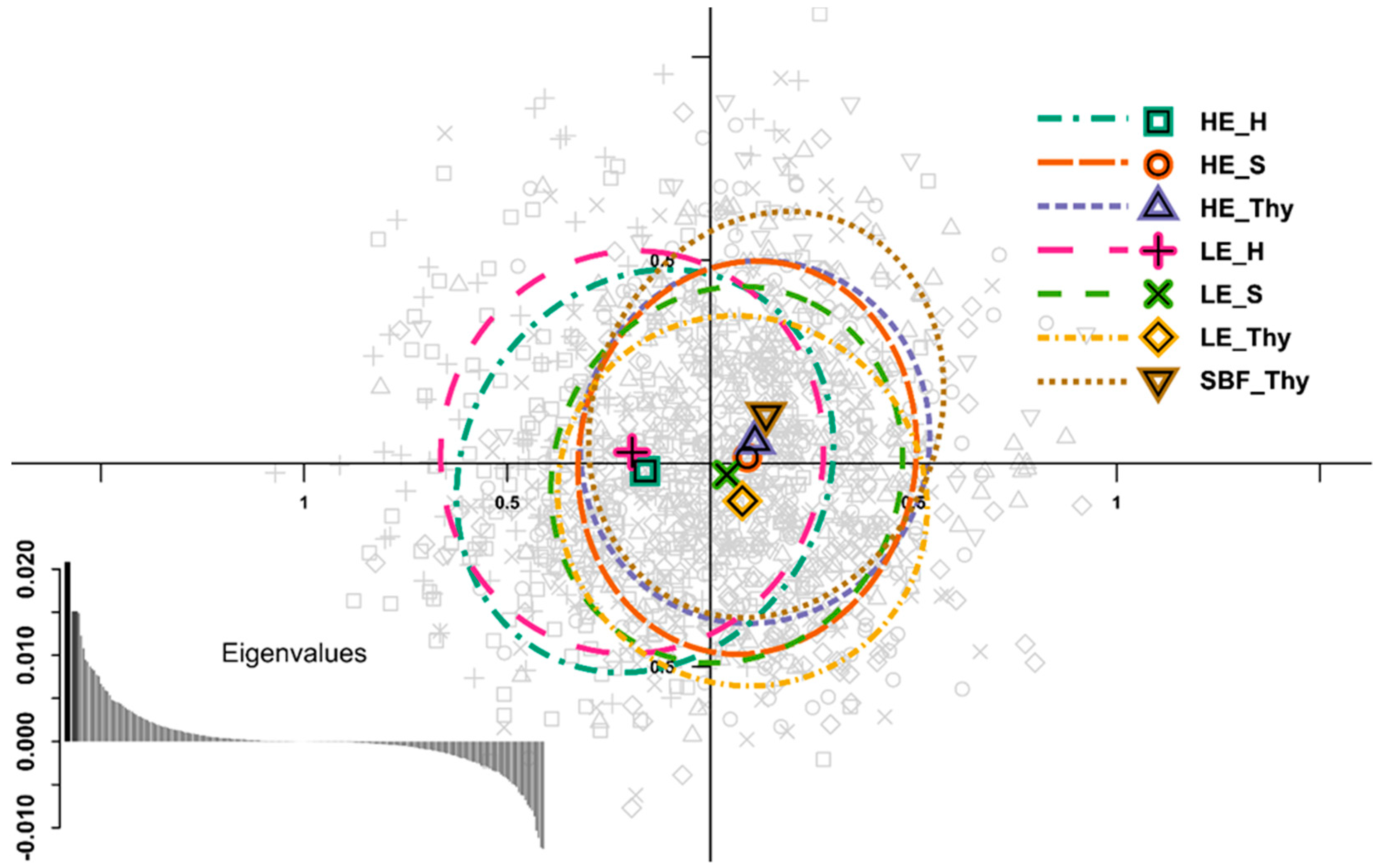
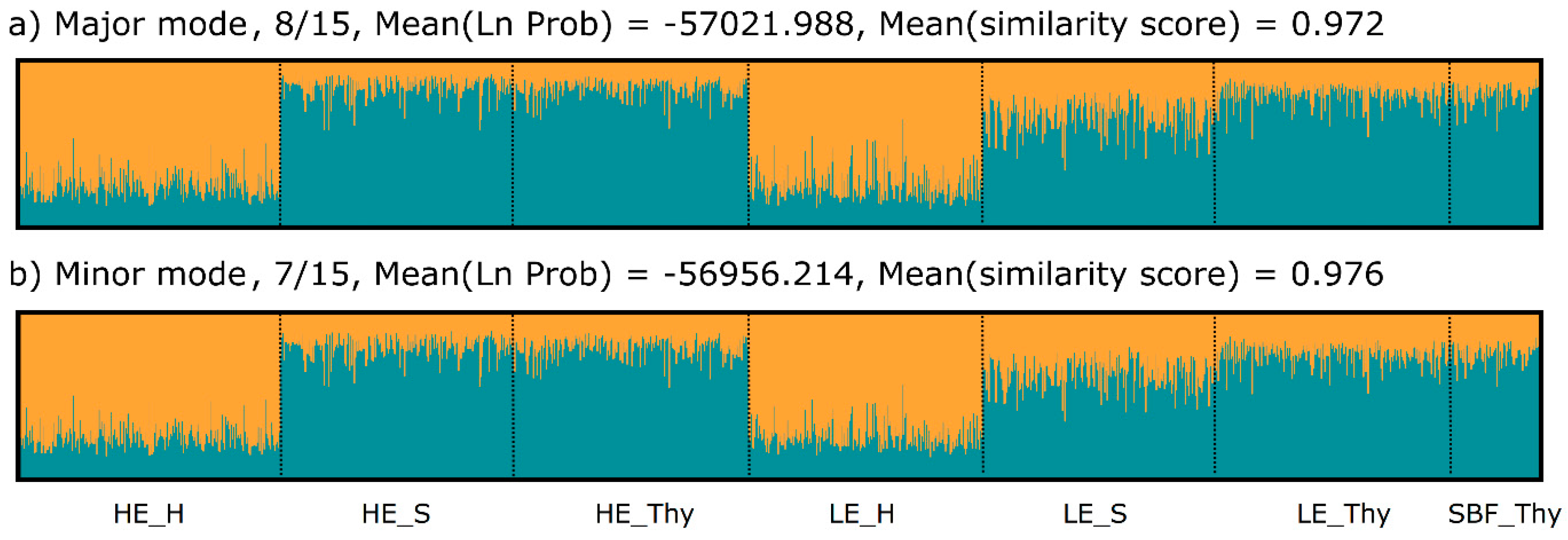
3.2. Genetic Variation and Differentiation
4. Discussion
4.1. Autochthonous and Allochthonous Stands
4.2. Phenotypic Differentiation
4.3. Genetic Variation and Differentiation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tollefsrud, M.M.; Sønstebø, J.H.; Brochmann, C.; Johnsen, Ø.; Skrøppa, T.; Vendramin, G.G. Combined analysis of nuclear and mitochondrial markers provide new insight into the genetic structure of North European Picea abies. Heredity 2009, 102, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Heuertz, M.; De Paoli, E.; Källman, T.; Larsson, H.; Jurman, I.; Morgante, M.; Lascoux, M.; Gyllenstrand, N. Multilocus patterns of nucleotide diversity, linkage disequilibrium and demographic history of Norway spruce (Picea abies (L.) Karst.). Genetics 2006, 174, 2095–2105. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Vogt, H. Studien zu morphologischen Variabilität der Fichte (Picea abies (L.) Karst.). Allg. Forst- u. J.-Ztg 1972, 142, 133–144. [Google Scholar]
- Priehäusser, G. Die Fichten-Variationen und -Kombinationen des Bayr. Waldes nach phänotypischen Merkmalen mit Bestimmungsschlüssel. Forstwiss. Cent. 1958, 77, 151–171. [Google Scholar] [CrossRef] [Green Version]
- Gruber, F. Phänotypen der Fichte (Picea abies (L.) Karst.) I. Verzweigungsphänotypen: Genotyp und Modifikation. Allg. Forst-u. J.-Ztg 1989, 160, 157–165. [Google Scholar]
- Merilä, J.; Crnokrak, P. Comparison of genetic differentaition at marker loci and quantitative traits. J. Evol. Biol. 2001, 14, 892–903. [Google Scholar] [CrossRef]
- Schmidt-Vogt, H. Taxonomie, verbreitung, morphologie, ökologie, waldgesellschaften. In Die Fichte—Ein Handbuch in Zwei Bänden; Parey: Hamburg, Berlin, Germany, 1977; p. 647. [Google Scholar]
- Jansen, S.; Konrad, H.; Geburek, T. The extent of historic translocation of Norway spruce forest reproductive material in Europe. Ann. For. Sci. 2017, 74, 56. [Google Scholar] [CrossRef]
- Beckstein, J.M. Forstbotanik oder vollständige Naturgeschichte der deutschen Holzgewächse und einiger Fremden: Zur Selbstbelehrung für Oberförster, Förster und Forstgehülfen; Henning: Gotha, Germany, 1810. [Google Scholar]
- Wahlenberg, G. Flora Lapponica. Available online: https://www.biodiversitylibrary.org/item/91422 (accessed on 23 May 2018).
- Nykänen, M.L.; Peltola, H.; Quine, C.; Kellomäki, S.; Broadgate, M. Factors affecting snow damage of trees with particular reference to European conditions. Silv. Fenn. 1997, 31, 193–213. [Google Scholar] [CrossRef]
- Gruber, F. Verzweigungssystem, Benadelung und Nadelfall der Fichte (Picea abies); Ecomed-Verlag: Landsberg am Lech, Germany, 1993. [Google Scholar]
- Geburek, T.; Robitschek, K.; Milasowszky, N. A tree of many faces: Why are there different crown types in Norway spruce (Picea abies (L.) Karst.)? Flora Morphol. Distrib. Funct. Ecol. Plants 2008, 203, 126–133. [Google Scholar] [CrossRef]
- König, A. Growth and snowbreak damage of Norway spruce provenances from Poland under harsh site conditions. In Forest Tree Breeding in an Ecologically Oriented Forest Management System; Meier-Dinkel, A., Steiner, W., Eds.; Schriften Forstlichen Fakultät Universität Göttingen und der Niedersächsischen Forstlichen Versuchsanstalt: Frankfurt am Main, Germany, 2002; pp. 139–150. [Google Scholar]
- Mátyás, C. Population, conservation and ecological genetics. In Encyclopedia of Forest Sciences; Burley, J., Evans, J., Youngquist, J.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; pp. 188–197. [Google Scholar]
- Kathke, S.; Bruelheide, H. Differences in frost hardiness of two Norway spruce morphotypes growing at Mt. Brocken, Germany. Flora Morphol. Distrib. Funct. Ecol. Plants 2011, 206, 120–126. [Google Scholar] [CrossRef]
- Sylvén, N. Studier Öfver Granens Formrikedom, Särskildt Dess Förgreningstyper Och Deras Skogliga Värde; Statens Skogsförsöksanstalt: Stockholm, Sweden, 1909. [Google Scholar]
- Kiellander, C.L. Picea, Abies, Pseudozuga. In Manual of Plant Breeding VI-Breeding of Legumes and Fruits, Viniculture and Silviculture; Kappert, H., Rudorf, W., Eds.; Parey: Berlin, Hamburg, Germany, 1962; pp. 854–873. [Google Scholar]
- Liesebach, M.; Rau, H.; König, A.O. Fichtenherkunftsversuch von 1962 und IUFRO-Fichtenherkunftsversuch von 1972—Ergebnisse von mehr als 30-Jähriger Beobachtung in Deutschland; Nordwestdeutsche Forstliche Versuchsanstalt (NW-FVA): Göttingen, Germany, 2010. [Google Scholar]
- Kalela, A. Zur Synthese er Experimentellen Untersuchungen über Klimarassen der Holzarten; Communications Instituti Forestalis Fenniae: Helsinki, Finland, 1937. [Google Scholar]
- Pulkkinen, P.; Pöykkö, T. Inherited narrow crown form, harvest index and stem biomass production in Norway spruce, Picea abies. Tree Physiol. 1990, 6, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Lehner, A.; Campbell, M.A.; Wheeler, N.C.; Piöykkiö, T.; Gliissl, J.; Kreike, J.; Neale, D.B. Identification of a RAPD marker linked to the pendula gene in Norway spruce (Picea abies (L.) Karst. f. pendula). Theor. Appl. Genet. 1995, 91, 1092–1094. [Google Scholar] [CrossRef] [PubMed]
- Latałowa, M.; van der Knaap, W.O. Late quaternary expansion of Norway spruce (Picea abies (L.) Karst.) in Europe according to pollen data. Quat. Sci. Rev. 2006, 25, 2780–2805. [Google Scholar] [CrossRef]
- Terhürne-Berson, R. Changing Distribution Patterns of Selected Conifers in the Quaternary of Europe Caused by Climatic Variations. Ph.D. Thesis, Friedrich-Wilhelms-Universität, Bonn, Germany, 16 June 2005. [Google Scholar]
- Acheré, V.; Favre, J.M.; Besnard, G.; Jeandroz, S. Genomic organization of molecular differentiation in Norway spruce (Picea abies). Mol. Ecol. 2005, 14, 3191–3201. [Google Scholar] [CrossRef] [PubMed]
- Vendramin, G.G.; Anzidei, M.; Madaghiele, A.; Sperisen, C.; Bucci, G. Chloroplast microsatellite analysis reveals the presence of population subdivision in Norway spruce (Picea abies K.). Genome 2000, 43, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Sperisen, C.; Büchler, U.; Gugerli, F.; Mátyás, G.; Geburek, T.; Vendramin, G.G. Tandem repeats in plant mitochondrial genomes: Application to the analysis of population differentiation in the conifer Norway spruce. Mol. Ecol. 2001, 10, 257–263. [Google Scholar] [CrossRef]
- Gugerli, F.; Sperisen, C.; Magni, F. Haplotype variation in a mitochondrial tandem repeat of Norway spruce (Picea abies) populations suggests a serious founder effect during postglacial re-colonization of the western Alps. Mol. Ecol. 2001, 1255–1263. [Google Scholar] [CrossRef]
- Scalfi, M.; Mosca, E.; Di Pierro, E.A.; Troggio, M.; Vendramin, G.G.; Sperisen, C.; La Porta, N.; Neale, D.B. Micro-and macro-geographic scale effect on the molecular imprint of selection and adaptation in Norway spruce. PLoS ONE 2014, 9, e115499. [Google Scholar] [CrossRef]
- Tollefsrud, M.M.; Kissling, R.; Gugerli, F.; Johnsen, Ø.; Skrøppa, T.; Cheddadi, R.; Van Der Knaap, W.O.; Latałowa, M.; Terhürne-Berson, R.; Litt, T.; et al. Genetic consequences of glacial survival and postglacial colonization in Norway spruce: Combined analysis of mitochondrial DNA and fossil pollen. Mol. Ecol. 2008, 17, 4134–4150. [Google Scholar] [CrossRef]
- Di Pierro, E.A.; Mosca, E.; González-Martínez, S.C.; Binelli, G.; Neale, D.B.; La Porta, N. Adaptive variation in natural Alpine populations of Norway spruce (Picea abies (L.) Karst.) at regional scale: Landscape features and altitudinal gradient effects. For. Ecol. Manag. 2017, 405, 350–359. [Google Scholar] [CrossRef]
- Müller-Starck, G. Genetic variation in high elevated populations of Norway spruce (Picea abies (L.) Karst.) in Switzerland. Silv. Genet. 1995, 44, 356–362. [Google Scholar]
- Ndrosiuk, B.P.A.; Shimono, A.; Westin, J.; Lindgren, D.; Fries, A.; Wang, X. Genetic status of Norway spruce (Picea abies) breeding populations for northern Sweden. Silvae Genet. 2013, 63, 288–295. [Google Scholar] [CrossRef]
- Quack, M. Molekulargenetische Untersuchungen zur Variabilität der Fichte (Picea abies (L.) Karst.) in Deutschland. Ph.D. Thesis, Universität Trier, Trier, Germany, 28 April 2004. [Google Scholar]
- Greger, O. Erfassung von Relikten des Autochtonen Fichtenvorkommens im Hochharz. Ph.D. Thesis, Georg-August-Universität, Göttingen, Germany, 1992. [Google Scholar]
- Maghuly, F.; Pinsker, W.; Praznik, W.; Fluch, S. Genetic diversity in managed subpopulations of Norway spruce (Picea abies (L.) Karst.). For. Ecol. Manag. 2006, 222, 266–271. [Google Scholar] [CrossRef]
- Gömöry, D. Effect of stand origin on the genetic diversity of Norway spruce (Picea abies Karst.) populations. For. Ecol. Manag. 1992, 54, 215–223. [Google Scholar] [CrossRef]
- Deutscher Wetterdienst DWD Climate Data Center (CDC): Multi-Annual Means of Grids of Monthly Averaged Daily Mean Air Temperature (2m) over Germany, Version v1.0, Offenbach. 2017. Available online: ftp://ftp-cdc.dwd.de/pub/CDC/grids_germany/multi_annual/air_temperature_mean/8110/ (accessed on 2 November 2017).
- Deutscher Wetterdienst DWD Climate Data Center (CDC): Multi-Annual Grids of the Begin of the Vegetation Period in Germany, Version 0.x, Offenbach. 2017. Available online: ftp://ftp-cdc.dwd.de/pub/CDC/grids_germany/multi_annual/vegetation_begin/ (accessed on 2 November 2017).
- Deutscher Wetterdienst DWD Climate Data Center (CDC): Multi-Annual Grids of the End of the Vegetation Period in Germany, Version 0.x, Offenbach. 2017. Available online: ftp://ftp-cdc.dwd.de/pub/CDC/grids_germany/multi_annual/vegetation_end/ (accessed on 2 November 2017).
- Deutscher Wetterdienst DWD Climate Data Center (CDC): Multi-Annual Grids of Numbers of Snowcover Days over Germany, Version v1.0, Offenbach. 2017. Available online: ftp://ftp-cdc.dwd.de/pub/CDC/grids_germany/multi_annual/snowcover_days/ (accessed on 2 November 2017).
- Deutscher Wetterdienst DWD Climate Data Center (CDC): Multi-Annual Grids of Precipitation Height over Germany 1981–2010, Version v1.0, Offenbach. 2017. Available online: ftp://ftp-cdc.dwd.de/pub/CDC/grids_germany/multi_annual/precipitation/8110/ (accessed on 2 November 2017).
- Deutscher Wetterdienst DWD Climate Data Center (CDC): 1 km × 1 km Gridded Mean of Annual Wind Speeds from 10 m to 100 m (in 10 m Steps) above Ground and Weibull Parameters, for Germany, Version V0.1, Offenbach. 2014. Available online: ftp://ftp-cdc.dwd.de/pub/CDC/grids_germany/wind_parameters/resol_1000x1000/ (accessed on 2 November 2017).
- Wilhelm, B. Die autochtonen Höhenfichten des Thüringer Waldes am Beispiel der Oberhofer Schlosßberg-Fichte. AFZ 1990, 33, 845–846. [Google Scholar]
- Jetschke, G. Oberhofer Schlossberg-Fichten und Methusalem-Kiefern von Paulinzella: Zwei Bemerkenswerte Altbestände von Nadelbäumen; ThüringenForst—Anstalt öffentlichen Rechts: Erfurt, Germany, 2019; (accepted); Mitteilungen 37/2019. [Google Scholar]
- Meyer, P.; Janda, P.; Mikoláš, M.; Trotsiuk, V.; Krumm, F.; Mrhalová, H.; Synek, M.; Lábusová, J.; Kraus, D.; Brandes, J.; et al. A matter of time: Self-regulated tree regeneration in a natural Norway spruce (Picea abies) forest at Mt. Brocken, Germany. Eur. J. For. Res. 2017, 136, 907–921. [Google Scholar] [CrossRef]
- Scotti, I.; Paglia, G.P.; Magni, F.; Morgante, M. Efficient development of dinucleotide microsatellite markers in Norway spruce (Picea abies Karst.) through dot-blot selection. Theor. Appl. Genet. 2002, 104, 1035–1041. [Google Scholar] [CrossRef]
- Scotti, I.; Magni, F.; Paglia, G.P.; Morgante, M. Trinucleotide microsatellites in Norway spruce (Picea abies): Their features and the development of molecular markers. Theor. Appl. Genet. 2002, 106, 40–50. [Google Scholar] [CrossRef]
- Rungis, D.; Bérubé, Y.; Zhang, J.; Ralph, S.; Ritland, C.E.; Ellis, B.E.; Douglas, C.; Bohlmann, J.; Ritland, K. Robust simple sequence repeat markers for spruce (Picea spp.) from expressed sequence tags. Theor. Appl. Genet. 2004, 109, 1283–1294. [Google Scholar] [CrossRef]
- Shi, Y.-Z.; Forneris, N.; Rajora, O.P. Highly informative single-copy nuclear microsatellite DNA markers developed using an AFLP-SSR approach in black spruce (Picea mariana) and red spruce (P. rubens). PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Pfeiffer, A.; Olivieri, A.M.; Morgante, M. Identification and characterization of microsatellites in Norway spruce (Picea abies K.). Genome 1997, 40, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Fluch, S.; Burg, A.; Kopecky, D.; Homolka, A.; Spiess, N.; Vendramin, G.G. Characterization of variable EST SSR markers for Norway spruce (Picea abies L.). BMC Res. Notes 2011, 4, 401. [Google Scholar] [CrossRef] [PubMed]
- Hodgetts, R.B.; Aleksiuk, M.A.; Brown, A.; Clarke, C.; Macdonald, E.; Nadeem, S.; Khasa, D.; Macdonald, E. Development of microsatellite markers for white spruce (Picea glauca) and related species. Theor. Appl. Genet. 2001, 102, 1252–1258. [Google Scholar] [CrossRef]
- Besnard, G.; Acheré, V.; Rampant, P.F.; Favre, J.M.; Jeandroz, S. A set of cross-species amplifying microsatellite markers developed from DNA sequence databanks in Picea (Pinaceae). Mol. Ecol. Resour. 2003, 3, 380–383. [Google Scholar] [CrossRef]
- De Sousa, S.N.; Finkeldey, R.; Gailing, O. Commentary: Experimental verification of microsatellite null alleles in norway spruce (Picea abies (L.) Karst.): Implications for population genetic studies. Plant Mol. Biol. Rep. 2005, 23, 113–119. [Google Scholar] [CrossRef]
- Yazdani, R.; Scotti, I.; Jansson, G.; Plomion, C.; Mathur, G. Inheritance and diversity of simple sequence repeat (SSR) microsatellite markers in various families of Picea abies. Hereditas 2003, 138, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Paglia, G.P.; Olivieri, A.M.; Morgante, M. Towards second-generation STS (sequence-tagged sites) linkage maps in conifers: A genetic map of Norway spruce (Picea abies K.). Mol. Gen. Genet. 1998, 258, 466–478. [Google Scholar] [CrossRef]
- Scotti, I.; Burelli, A.; Cattonaro, F.; Chagné, D.; Fuller, J.; Hedley, P.E.; Jansson, G.; Lalanne, C.; Madur, D.; Neale, D. Analysis of the distribution of marker classes in a genetic linkage map: A case study in Norway spruce (Picea abies Karst). Tree Genet. Genomes 2005, 1, 93–102. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acid. Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Spitze, K. Population structure in Daphnia obtusa: Quantitative genetic and allozymic variation. Genetics 1993, 135, 367–374. [Google Scholar] [CrossRef]
- Leinonen, T.; McCairns, R.J.S.; O’Hara, R.B.; Merilä, J. QST–FST comparisons: Evolutionary and ecological insights from genomic heterogeneity. Nat. Rev. Genet. 2013, 14, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Brommer, J.E. Whither PST? The approximation of QST by PST in evolutionary and conservation biology. J. Evol. Biol. 2011, 24, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, S.B.; Da Silva, A. Pstat: An R Package to Assess Population Differentiation in Phenotypic Traits. R J. 2018, 10, 447–454. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing 2018. Available online: https://www.R-project.org (accessed on 2 November 2018).
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Devillard, S.; Dufour, A.; Pontier, D. Revealing cryptic spatial patterns in genetic variability by a new multivariate method. Heredity 2008, 101, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Engels, W.R. Exact tests for Hardy-Weinberg proportions. Genetics 2009, 183, 1431–1441. [Google Scholar] [CrossRef]
- Engels, W.R. HWxtest: Exact Tests for Hardy-Weinberg Proportions. R package version 1.1.7 2016. Available online: https://CRAN.R-project.org/package=HWxtest (accessed on 2 November 2018).
- Agapow, P.-M.; Burt, A. Indices of multilocus linkage disequilibrium. Mol. Ecol. Notes 2001, 1, 101–102. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014. [Google Scholar] [CrossRef] [Green Version]
- Dempster, A.P.; Laird, N.M.; Rubin, D.B. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. 1977, 39, 1–38. [Google Scholar]
- Rousset, F. GENEPOP’007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L. Maximum likelihood estimation of the frequency of null alleles at microsatellite loci. Conserv. Genet. 2006, 7, 991–995. [Google Scholar] [CrossRef]
- Dabrowski, M.J.; Bornelöv, S.; Kruczyk, M.; Baltzer, N.; Komorowski, J. “True” null allele detection in microsatellite loci: A comparison of methods, assessment of difficulties and survey of possible improvements. Mol. Ecol. Resour. 2015, 15, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, J. Effects of microsatellite null alleles on assignment testing. J. Hered. 2008, 99, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenALEx 6.5: Genetic analysis in Excel—Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Goudet, J.; Jombart, T. Hierfstat: Estimation and Tests of Hierarchical F-Statistics. R package version 0.04–22. 2015. Available online: https://CRAN.R-project.org/package=hierfstat (accessed on 2 November 2018).
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Ellis, J.R.; Burke, J.M. EST-SSRs as a resource for population genetic analyses. Heredity 2007, 99, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalia, R.K.; Rai, M.K.; Kalia, S.; Singh, R.; Dhawan, A.K. Microsatellite markers: An overview of the recent progress in plants. Euphytica 2011, 177, 309–334. [Google Scholar] [CrossRef]
- Antao, T.; Lopes, A.; Lopes, R.J.; Beja-Pereira, A.; Luikart, G. LOSITAN: A workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinform. 2008, 9, 323. [Google Scholar] [CrossRef]
- Foll, M.; Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Sundqvist, L.; Keenan, K.; Zackrisson, M.; Prodöhl, P.; Kleinhans, D. Directional genetic differentiation and relative migration. Ecol. Evol. 2016, 6, 3461–3475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef]
- Jost, L. GST and its relatives do not measure differentiation. Mol. Ecol. 2008, 17, 4015–4026. [Google Scholar] [CrossRef]
- Crawford, N.G. Smogd: Software for the measurement of genetic diversity. Mol. Ecol. Resour. 2010, 10, 556–557. [Google Scholar] [CrossRef] [PubMed]
- Alcala, N.; Goudet, J.; Vuilleumier, S. On the transition of genetic differentiation from isolation to panmixia: What we can learn from GST and D. Theor. Popul. Biol. 2014, 93, 75–84. [Google Scholar] [CrossRef]
- Keenan, K.; McGinnity, P.; Cross, T.F.; Crozier, W.W.; Prodöhl, P.A. diveRsity: An R package for the estimation of population genetics parameters and their associated errors. Method. Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef]
- Moran, P.A. The interpretation of statistical maps. J. R. Stat. Soc. 1948, 10, 243–251. [Google Scholar] [CrossRef]
- Moran, P.A. Notes on continuous stochastic phenomena. Biometrika 1950, 37, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Montano, V.; Jombart, T. An Eigenvalue test for spatial principal component analysis. BMC Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. BMC Bioinform. 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Ahmed, I. Adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. BMC Bioinform. 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [Green Version]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef] [PubMed]
- Porras-Hurtado, L.; Ruiz, Y.; Santos, C.; Phillips, C.; Carracedo, Á.; Lareu, M.V. An overview of STRUCTURE: Applications, parameter settings, and supporting software. Front. Genet. 2013, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Hanewinkel, M.; Cullmann, D.A.; Schelhaas, M.; Nabuurs, G.; Zimmermann, N.E. Climate change may cause severe loss in the economic value of European forest land. Nat. Clim. Chang. 2012, 3, 203–207. [Google Scholar] [CrossRef]
- Reif, A.; Brucke, U.; Kratzer, R.; Schmiedinger, A.; Bauhus, J. Waldbau und Baumartenwahl in Zeiten des Klimawandels aus Sicht des Naturschutzes; Bundesamt für Naturschutz (BfN): Bonn, Germany, 2010. [Google Scholar]
- Kaplan, J.O.; Krumhardt, K.M.; Zimmermann, N. The prehistoric and preindustrial deforestation of Europe. Quat. Sci. Rev. 2009, 28, 3016–3034. [Google Scholar] [CrossRef]
- Farrell, E.P.; Führer, E.; Ryan, D.; Andersson, F.; Hüttl, R.; Piussi, P. European forest ecosystems: Buildind the future on the legacy of the past. For. Ecol. Manag. 2000, 132, 5–20. [Google Scholar] [CrossRef]
- Finkeldey, R.; Ziehe, M. Genetic implications of silvicultural regimes. For. Ecol. Manag. 2004, 197, 231–244. [Google Scholar] [CrossRef]
- Hosius, B.; Leinemann, L.; Konnert, M.; Bergmann, F. Genetic aspects of forestry in the central Europe. Eur. J. For. Res. 2006, 125, 407–417. [Google Scholar] [CrossRef]
- Wegener, U.; Kison, H. Die Vegetation des Brockens im Nationalpark Hochharz (Exkursion G). Tuexenia 2002, 22, 243–267. [Google Scholar]
- Meirmans, P.G.; Hedrick, P.W. Assessing population structure: FST and related measures. Mol. Ecol. Resour. 2011, 11, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Whitlock, M.C. G′ST and D do not replace FST. Mol. Ecol. 2011, 20, 1083–1091. [Google Scholar] [CrossRef]
- Deleuze, C.; Hervé, J.C.; Colin, F.; Ribeyrolles, L. Modelling crown shape of Picea abies: Spacing effects. Can. J. For. Res. 1996, 26, 1957–1966. [Google Scholar] [CrossRef]
- Grassi, G.; Giannini, R. Influence of light and competition on crown and shoot morphological parameters of Norway spruce and silver fir saplings. Ann. For. Sci. 2005, 62, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Hertel, D.; Schöling, D. Below-ground response of Norway spruce to climate conditions at Mt. Brocken (Germany)—A re-assessment of Central Europe’s northernmost treeline. Flora Morphol. Distrib. Funct. Ecol. Plants 2011, 206, 127–135. [Google Scholar] [CrossRef]
- Schmidt-Vogt, H. Monographie der Picea abies (L.) Karst. unter Berücksichtigung genetischer und züchterischer Aspekte. Forstwiss. Cent. 1978, 97, 281–302. [Google Scholar] [CrossRef]
- Bergmann, F. The allelic distribution at an acid phosphatase locus in Norway spruce (Picea abies) along similar climatic gradients. Theor. Appl. Genet. 1978, 52, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Lagercrantz, U.; Ryman, N. Genetic structure of Norway spruce (Picea abies): Concordance of Morphological and Allozymic Variation. Evolution 1990, 44, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Källman, T.; Ma, X.; Gyllenstrand, N.; Zaina, G.; Morgante, M.; Bousquet, J.; Eckert, A.; Wegrzyn, J.; Neale, D.; et al. Disentangling the roles of history and local selection in shaping clinal variation of allele frequencies and gene expression in Norway spruce (Picea abies). Genetics 2012, 191, 865–881. [Google Scholar] [CrossRef] [PubMed]
- Trujillo-Moya, C.; George, J.-P.; Fluch, S.; Geburek, T.; Grabner, M.; Karanitsch-Ackerl, S.; Konrad, H.; Mayer, K.; Sehr, E.M.; Wischnitzki, E.; et al. Drought sensitivity of Norway Spruce at the species’ warmest fringe: Quantitative and molecular analysis reveals high genetic variation among and within provenances. G3 Genes Genomes Genet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ganthaler, A.; Stöggl, W.; Mayr, S.; Kranner, I.; Schüler, S.; Wischnitzki, E.; Sehr, E.M.; Fluch, S.; Trujillo-Moya, C. Association genetics of phenolic needle compounds in Norway spruce with variable susceptibility to needle bladder rust. Plant Mol. Biol. 2017, 94, 229–251. [Google Scholar] [CrossRef] [PubMed]
- Radu, R.G.; Curtu, L.A.; Spârchez, G.; Şofletea, N. Genetic diversity of Norway spruce (Picea abies (L.) Karst.) in Romanian Carpathians. Ann. For. Res. 2014, 57, 19–29. [Google Scholar] [CrossRef]
- Unger, G.M.; Konrad, H.; Geburek, T. Does spatial genetic structure increase with altitude? An answer from Picea abies in Tyrol, Austria. Plant Syst. Evol. 2011, 292, 133–141. [Google Scholar] [CrossRef]
- Scotti, I.; Paglia, G.; Magni, F.; Morgante, M. Population genetics of Norway spruce (Picea abies Karst.) at regional scale: Sensitivity of different microsatellite motif classes in detecting differentiation. Ann. For. Sci. 2006, 63, 485–491. [Google Scholar] [CrossRef]
- Muona, O.; Paule, L.; Szmidt, A.E.; Kärkkäinen, K. Mating system analysis in a central and northern European population of Picea abies. Scand. J. For. Res. 1990, 5, 97–102. [Google Scholar] [CrossRef]
- Chevarria, M.V. Genetic Characterisation of Populations from the European Natural Range of Norway Spruce (Picea abies (L.) Karst.) by Means of EST Markers. Ph.D. Thesis, Technische Universität München, Munich, Germany, 9 March 2005. [Google Scholar]
- Geburek, T. Genetic variation of Norway spruce (Picea abies (L.) Karst.) populations in Austria. III. Macrospatial allozyme patterns of high elevation populations. Int. J. For. Genet. 1999, 6, 201–211. [Google Scholar]
- Máchová, P.; Trčková, O.; Cvrčková, H. Use of nuclear microsatellite loci for evaluating genetic diversity of selected populations of Picea abies (L.) Karsten in the Czech Republic. Forests 2018, 9, 92. [Google Scholar] [CrossRef]
- Jeandroz, S.; Collignon, A.M.; Favre, J.M. RAPD and mtDNA variation among autochthonous and planted populations of Picea abies from the Vosges mountains (France) in reference to other French populations. For. Ecol. Manag. 2004, 197, 225–229. [Google Scholar] [CrossRef]
- Janes, J.K.; Miller, J.M.; Dupuis, J.R.; Malenfant, R.M.; Gorrell, J.C.; Cullingham, C.I.; Andrew, R.L. The K = 2 conundrum. Mol. Ecol. 2017, 3594–3602. [Google Scholar] [CrossRef] [PubMed]
- Ennos, R.A. Estimating the relative rates of pollen and seed migratin among plant populations. Heredity 1994, 72, 250–259. [Google Scholar] [CrossRef]
- QGIS Development Team. QGIS Geographic Information System. Open Source Geospatial Found. Project. Available online: http://www.qgis.org/ (accessed on 18 December 2017).
- OpenStreetMap Contributors. OpenStreetMap. Available online: http://tile.openstreetmap.org (accessed on 18 October 2018).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stand | N | Age of Upper Story Trees, years | Mean Multi Annual Air Temperature 1981–2010, °C | Mean Vegetation Period in 1992–2015, days | Mean Snow Cover Days in 1981–2010, days | Annual Mean Precipitation in 1981–2010, mm | Mean Wind in 20 m a.g.l. m/s in 1981–2000 | Elevation Range Of Sampled Individuals, m a.s.l. |
|---|---|---|---|---|---|---|---|---|
| LE_H | 200 | 180 | 4.7 | 181.0 | 141.5 | 1668.5 | 56 | 889–915 |
| HE_H | 250 | 300 * | 3.9 | 177.0 | 158.0 | 1793.0 | 79 | 1036–1065 |
| LE_S | 200 | 162 | 5.2 | 184.0 | 127.0 | 1210.0 | 55.5 | 972–1009 |
| HE_S | 200 | 142 | 4.9 | 182.0 | 136.0 | 1255.0 | 58 | 988–1014 |
| LE_Thy | 200 | 90 | 5.7 | 190.0 | 122.0 | 1331.0 | 57 | 899–912 |
| HE_Thy | 200 | 151 | 6.3 | 195.5 | 100.5 | 1211.0 | 39 | 761–776 |
| Schloss-bergfichte | 75 | 280 * | 5.8 | 191.0 | 116.0 | 1331.0 | 50 | 818–840 |
| Mean | 5.2 | 185.8 | 128.7 | 1399.9 | 56.4 | 921 |
| Trait | Phenotype | ||
|---|---|---|---|
| Mountainous | Intermediate | Lowland | |
| overall crown architecture; structural appearance | narrow shaped crown | equivocal/intermediate shape | broad shape |
| angle of the first order branches | clearly downwards facing branches, stem and branches | no clear branch orientation up- or downwards | straight and upwards facing branches, all angles between stem and branches right or obtuse |
| branching pattern of the second order branches | plate or plate brush like | brush like | comb or comb brush like |
| SSR | Allele Size | Dye Label and the PCR Primer Sequences (5′–3′) | Repeat Motif | Reference | |
|---|---|---|---|---|---|
| Min, bp | Max, bp | ||||
| EATC1B2a | 197 | 219 | F: FAM-TGGCATGAGATTTATGTGGTT R: GTGTGCCACTCAACCTCAC | (ATC)7(AT)3 | [48] |
| EATC1D2a | 180 | 236 | F: FAM-TTGTCATCGTCGTCATTGTC R: TTTAGCCTCTGTTTTCTAGCG | (ATC)3AT(ATC)6 | [48] |
| EATC1E03a | 130 | 175 | F: FAM-CCCCTTATTCCTAACGTCAAA R: TACCAGTGGTGACAACGATG | (CAT)4CGT(CAT)8CGT-(CAT)4CGT(CT)4CGT(CAT)4 | [48] |
| EATC2G05a | 193 | 254 | F: HEX-TGGAGCATGGGTAAATCG R: TACCTCACACCCGTGAGAAT | (AAT)5(CAT)16CAA(CAT)4 | [48] |
| PaGB3b | 109 | 150 | F: FAM-AGTGATTAAACTCCTGACCAC R: CACTGAATACACCCATTATCC | (AT)11 | [54] |
| PaGB8b | 95 | 203 | F: FAM-AGCATGTACAAAATGAAGATTCTC R: CCCTTTAGTGTTTTCTCTTTCTAC | (AC)12 | [54] |
| SpAG2a | 88 | 122 | F: FAM-GCTCTTCACGTGTACTTGATC R: TTCGAAGATCCTCCAAGATAC | (TC)16 | [41] |
| SpAGC1a | 71 | 121 | F: HEX-TTCACCTTAGCCGAGAACC R: CACTGGAGATCTTCGTTCTGA | (TC)5TT(TC)10 | [41] |
| SpAGG3a | 109 | 149 | F: HEX-AGCATGTTGTCCCATATAGACC R: CTCCAACATTCCCATGTAGC | (GA)24 | [41] |
| WS00016.O09b | 386 | 402 | F: HEX-CTTTGGGGGCTAGCAAGTTT R: ATTCGGGCTTCATAGCACAA | (AT)9 | [49] |
| WS00111.K13b | 212 | 272 | F: HEX-GACTGAAGATGCCGATATGC R: GGCCATATCATCTCAAAATAAAGAA | (AT)9 | [49] |
| EST-SSR | GenBank Accession Number | Annotation | Location of SSR in the EST |
|---|---|---|---|
| PaGB3 | AJ133748 | P. abies mRNA for major intrinsic protein (aquaporin) | 3′UTR |
| PaGB8 | AF100429 | P. abies clone PA12H2 repetitive DNA sequence | |
| WS00016.O09 * | CN480894 | NP 197764—expressed protein (A. thaliana) | 3′UTR |
| WS00111.K13 * | CN480897 | BAB86071—putative beta-glucosidase (O. sativa (japonica cultivar-group)) | 3′UTR |
| Stand | Ho | He | A | Ar | Private Alleles | FIS | p-Value |
|---|---|---|---|---|---|---|---|
| HE_H | 0.655 | 0.733 | 17.09 | 16.51 | 0 | 0.142 | 0.000 |
| HE_S | 0.640 | 0.751 | 16.90 | 16.83 | 2 | 0.124 | 0.000 |
| HE_Thy | 0.702 | 0.757 | 17.00 | 16.67 | 4 | 0.129 | 0.006 |
| LE_H | 0.630 | 0.730 | 16.46 | 16.13 | 4 | 0.041 | 0.099 |
| LE_S | 0.673 | 0.746 | 16.64 | 16.64 | 0 | 0.108 | 0.000 |
| LE_Thy | 0.668 | 0.734 | 16.73 | 16.46 | 5 | 0.137 | 0.000 |
| SBF_Thy | 0.692 | 0.749 | 14.55 | 16.79 | 0 | 0.079 | 0.007 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caré, O.; Müller, M.; Vornam, B.; Höltken, A.M.; Kahlert, K.; Krutovsky, K.V.; Gailing, O.; Leinemann, L. High Morphological Differentiation in Crown Architecture Contrasts with Low Population Genetic Structure of German Norway Spruce Stands. Forests 2018, 9, 752. https://doi.org/10.3390/f9120752
Caré O, Müller M, Vornam B, Höltken AM, Kahlert K, Krutovsky KV, Gailing O, Leinemann L. High Morphological Differentiation in Crown Architecture Contrasts with Low Population Genetic Structure of German Norway Spruce Stands. Forests. 2018; 9(12):752. https://doi.org/10.3390/f9120752
Chicago/Turabian StyleCaré, Oliver, Markus Müller, Barbara Vornam, Aki M. Höltken, Karina Kahlert, Konstantin V. Krutovsky, Oliver Gailing, and Ludger Leinemann. 2018. "High Morphological Differentiation in Crown Architecture Contrasts with Low Population Genetic Structure of German Norway Spruce Stands" Forests 9, no. 12: 752. https://doi.org/10.3390/f9120752
APA StyleCaré, O., Müller, M., Vornam, B., Höltken, A. M., Kahlert, K., Krutovsky, K. V., Gailing, O., & Leinemann, L. (2018). High Morphological Differentiation in Crown Architecture Contrasts with Low Population Genetic Structure of German Norway Spruce Stands. Forests, 9(12), 752. https://doi.org/10.3390/f9120752