Genomic Prediction of Growth and Stem Quality Traits in Eucalyptus globulus Labill. at Its Southernmost Distribution Limit in Chile



Abstract
:1. Introduction
2. Materials and Methods
2.1. Genetic Material and Phenotypic Measurements
2.2. Genotyping and Estimation of Linkage Disequilibrium (LD)
2.3. Estimation of Pedigree-Based Breeding Values
2.4. Genomic Prediction Models
2.5. Cross-Validation and Prediction Ability
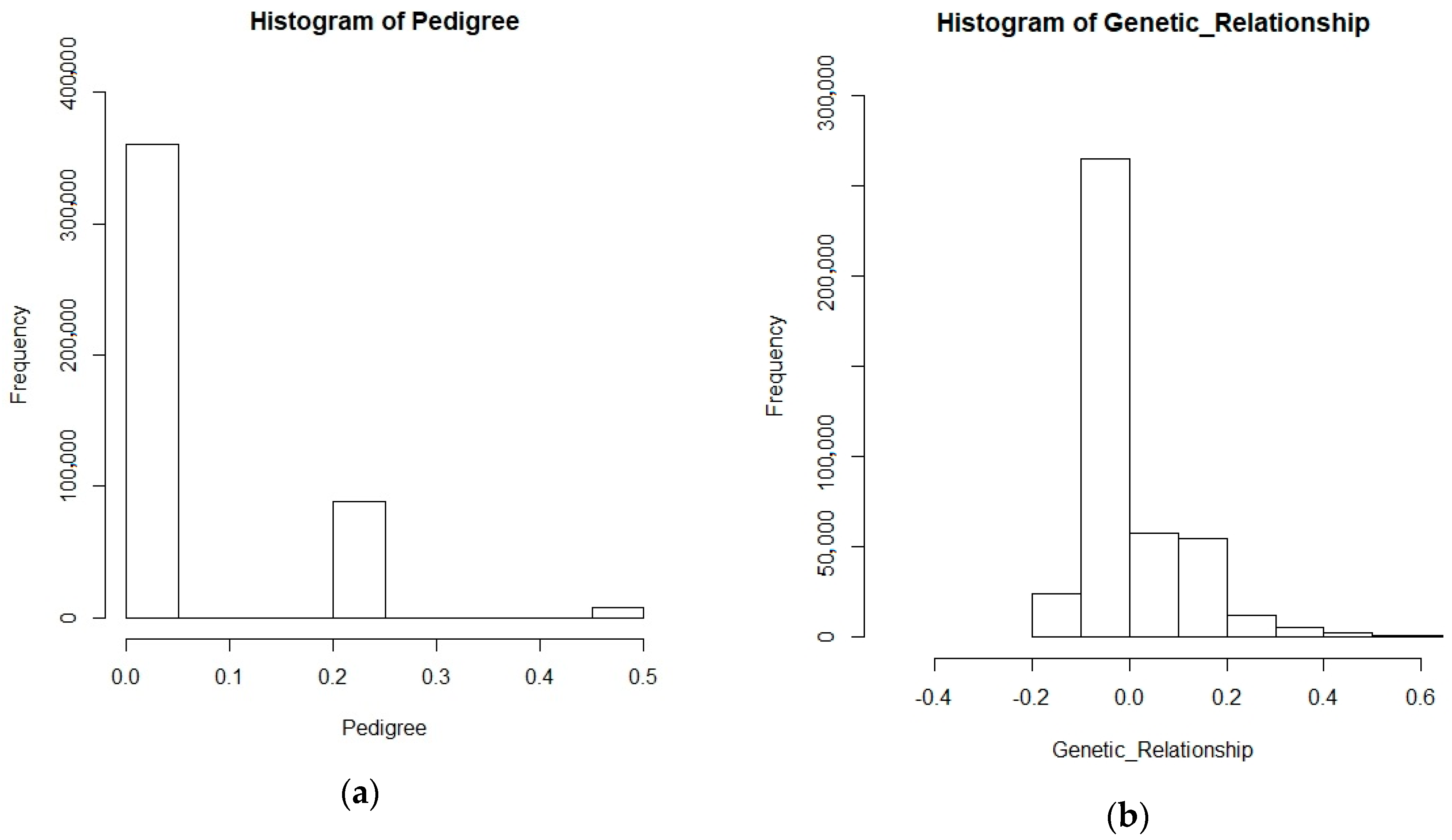
2.6. Validation of Pedigree Data
3. Results
3.1. Estimates of Variance Components and Heritability of Growth Traits, Branching Quality and Stem Straightness
3.2. Final Set of Qualified SNPs and Linkage Disequilibrium Decay
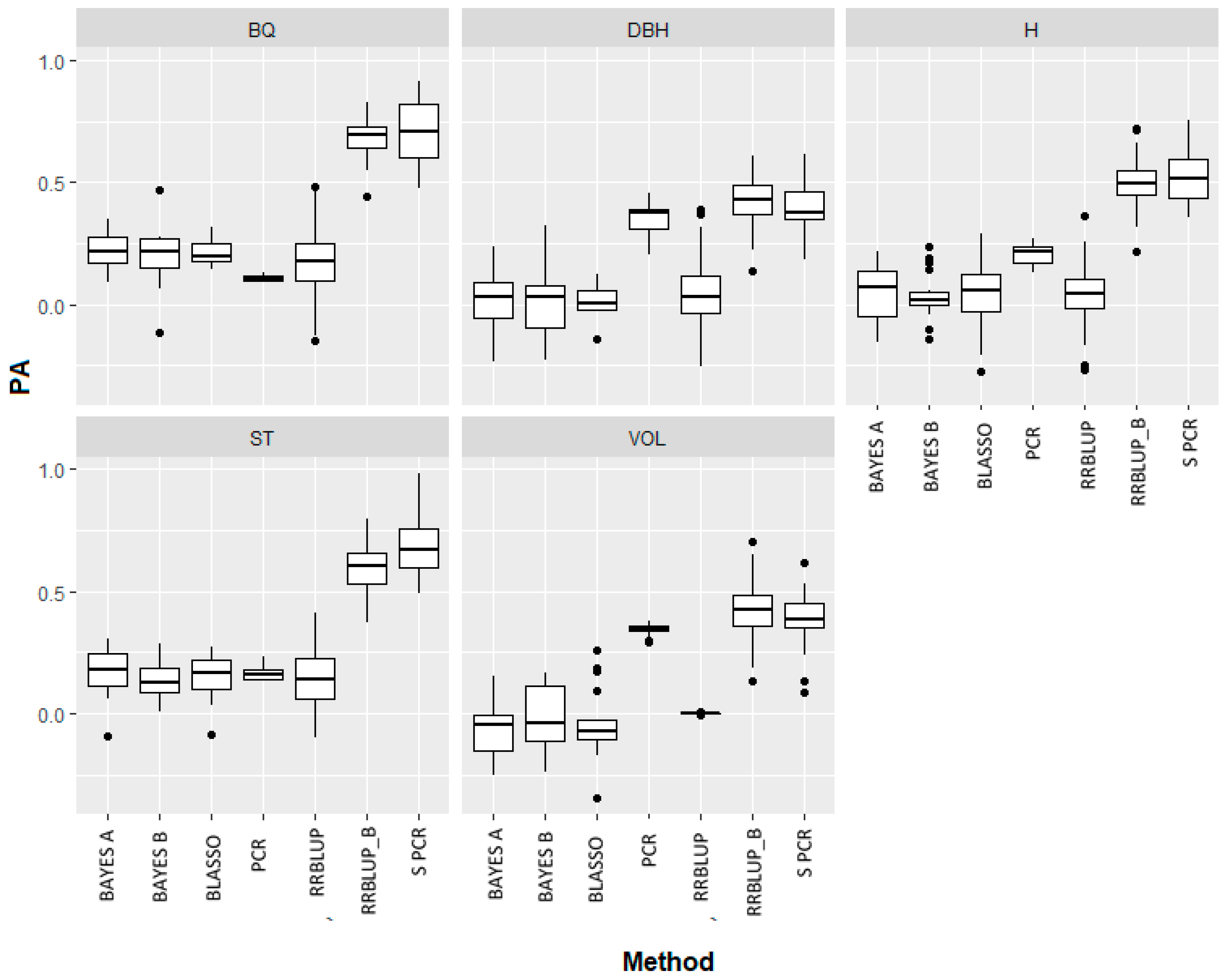
3.3. Predictive Ability of Frequentist, Bayesian and Dimension Reduction Methods
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Drake, J.E.; Aspinwall, M.J.; Pfautsch, S.; Rymer, P.D.; Reich, P.B.; Smith, R.A.; Crous, K.Y.; Tissue, D.T.; Ghannoum, O.; Tjoelker, M.G. The capacity to cope with climate warming declines from temperate to tropical latitudes in two widely distributed Eucalyptus species. Glob. Chang. Biol. 2015, 21, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Mora, F.; Arriagada, O.; Ballesta, P.; Ruiz, E. Genetic diversity and population structure of a drought-tolerant species of Eucalyptus, using microsatellite markers. J. Plant Biochem. Biotechnol. 2017, 26, 274–281. [Google Scholar] [CrossRef]
- Paiva, J.A.; Prat, E.; Vautrin, S.; Santos, M.D.; San Clemente, H.; Brommonschenkel, S.; Fonseca, P.G.S.; Grattapaglia, D.; Song, X.; Ammiraju, J.S.S.; et al. Advancing Eucalyptus genomics: Identification and sequencing of lignin biosynthesis genes from deep-coverage BAC libraries. BMC Genom. 2011, 12, 137. [Google Scholar] [CrossRef] [PubMed]
- Foster, S.A.; McKinnon, G.E.; Steane, D.A.; Potts, B.M. Parallel evolution of dwarf ecotypes in the forest tree Eucalyptus globulus. New Phytol. 2007, 175, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Dutkowski, G.W.; Potts, B.M. Geographic patterns of genetic variation in Eucalyptus globulus ssp. globulus and a revised racial classification. Aust. J. Bot. 1999, 47, 237–263. [Google Scholar] [CrossRef]
- Tibbits, W.N.; White, T.L.; Hodge, G.R.; Borralho, N.M. Genetic variation in frost resistance of Eucalyptus globulus ssp. globulus assessed by artificial freezing in winter. Aust. J. Bot. 2006, 54, 521–529. [Google Scholar] [CrossRef]
- Lanfranco, D.; Dungey, H.S. Insect damage in Eucalyptus: A review of plantations in Chile. Aust. Ecol. 2001, 26, 477–481. [Google Scholar] [CrossRef]
- Navarrete-Campos, D.; Bravo, L.A.; Rubilar, R.A.; Emhart, V.; Sanhueza, R. Drought effects on water use efficiency, freezing tolerance and survival of Eucalyptus globulus and Eucalyptus globulus × nitens cuttings. New For. 2013, 44, 119–134. [Google Scholar] [CrossRef]
- Fernández, M.; Valenzuela, S.A.; Arora, R.; Chen, K. Isolation and characterization of three cold acclimation-responsive dehydrin genes from Eucalyptus globulus. Tree Genet. Genom. 2012, 8, 149–162. [Google Scholar] [CrossRef]
- Castillo, R.; Otto, M.; Freer, J.; Valenzuela, S. Multivariate strategies for classification of Eucalyptus globulus genotypes using carbohydrates content and NIR spectra for evaluation of their cold resistance. J. Chem. Soc. 2008, 22, 268–280. [Google Scholar]
- Tambarussi, E.V.; Pereira, F.B.; Da Silva, P.H.M.; Lee, D.; Bush, D. Are tree breeders properly predicting genetic gain? A case study involving Corymbia species. Euphytica 2018, 214, 150. [Google Scholar] [CrossRef]
- Grattapaglia, D.; Resende, M.D. Genomic selection in forest tree breeding. Tree Genet. Genom. 2011, 7, 241–255. [Google Scholar] [CrossRef]
- Arriagada, O.; Mora, F.; Amaral Junior, A.T. Thirteen years under arid conditions: Exploring marker-trait associations in Eucalyptus cladocalyx for complex traits related to flowering, stem form and growth. Breed. Sci. 2018, 68, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Ballesta, P.; Mora, F.; Ruiz, E.; Contreras-Soto, R. Marker-trait associations for survival, growth, and flowering components in Eucalyptus cladocalyx under arid conditions. Biol. Plant. 2015, 59, 389–392. [Google Scholar] [CrossRef]
- Bartholomé, J.; Salmon, F.; Vigneron, P.; Bouvet, J.M.; Plomion, C.; Gion, J.M. Plasticity of primary and secondary growth dynamics in Eucalyptus hybrids: A quantitative genetics and QTL mapping perspective. BMC Plant Biol. 2013, 13, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Cappa, E.P.; El-Kassaby, Y.A.; Garcia, M.N.; Acuña, C.; Borralho, N.M.G.; Grattapaglia, D.; Marcucci-Poltri, S.N. Impacts of population structure and analytical models in genome-wide association studies of complex traits in forest trees: A case study in Eucalyptus globulus. PLoS ONE 2013, 8, e81267. [Google Scholar] [CrossRef] [PubMed]
- Carocha, V.; Soler, M.; Hefer, C.; Cassan-Wang, H.; Fevereiro, P.; Myburg, A.A.; Paiva, J.A.; Grima-Pettenati, J. Genome-wide analysis of the lignin toolbox of Eucalyptus grandis. New Phytol. 2015, 206, 1297–1313. [Google Scholar] [CrossRef] [PubMed]
- Isik, F. Genomic selection in forest tree breeding: The concept and an outlook to the future. New For. 2014, 45, 379–401. [Google Scholar] [CrossRef]
- Beaulieu, J.; Doerksen, T.; Clément, S.; MacKay, J.; Bousquet, J. Accuracy of genomic selection models in a large population of open-pollinated families in white spruce. Heredity 2014, 113, 343. [Google Scholar] [CrossRef] [PubMed]
- Heffner, E.L.; Lorenz, A.J.; Jannink, J.L.; Sorrells, M.E. Plant breeding with genomic selection: Gain per unit time and cost. Crop Sci. 2010, 50, 1681–1690. [Google Scholar] [CrossRef]
- Neale, D.B.; Kremer, A. Forest tree genomics: Growing resources and applications. Nat. Rev. Genet. 2011, 12, 111. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.; Doerksen, T.K.; MacKay, J.; Rainville, A.; Bousquet, J. Genomic selection accuracies within and between environments and small breeding groups in white spruce. BMC Genom. 2014, 15, 1048. [Google Scholar] [CrossRef] [PubMed]
- Ratcliffe, B.; El-Dien, O.G.; Klápště, J.; Porth, I.; Chen, C.; Jaquish, B.; El-Kassaby, Y.A. A comparison of genomic selection models across time in interior spruce (Picea engelmannii× glauca) using unordered SNP imputation methods. Heredity 2015, 115, 547. [Google Scholar] [CrossRef] [PubMed]
- Ukrainetz, N.K.; Ritland, K.; Mansfield, S.D. Identification of quantitative trait loci for wood quality and growth across eight full-sib coastal Douglas-fir families. Tree Genet. Genom. 2008, 4, 159–170. [Google Scholar] [CrossRef]
- Mamani, E.M.; Bueno, N.W.; Faria, D.A.; Guimarães, L.M.; Lau, D.; Alfenas, A.C. Positioning of the major locus for Puccinia psidii rust resistance (Ppr1) on the Eucalyptus reference map and its validation across unrelated pedigrees. Tree Genet. Genom. 2010, 6, 953–962. [Google Scholar] [CrossRef]
- Heffner, E.L.; Jannink, J.L.; Sorrells, M.E. Genomic selection accuracy using multifamily prediction models in a wheat breeding program. Plant Genom. 2011, 4, 65–75. [Google Scholar] [CrossRef]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [PubMed]
- De Los Campos, G.; Sorensen, D.; Gianola, D. Genomic Heritability: What Is It? PLoS Genet. 2015, 11, e1005048. [Google Scholar] [CrossRef] [PubMed]
- Daetwyler, H.D.; Calus, M.P.L.; Pong-Wong, R.; De Los Campos, G.; Hickey, J.M. Genomic prediction in animals and plants: Simulation of data, validation, reporting, and benchmarking. Genetics 2013, 193, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Resende, M.D.; Resende, M.F.; Sansaloni, C.P.; Petroli, C.D.; Missiaggia, A.A.; Aguiar, A.M.; Abad, J.M.; Takahashi, E.K.; Rosado, A.M.; Faria, D.A.; et al. Genomic selection for growth and wood quality in Eucalyptus: Capturing the missing heritability and accelerating breeding for complex traits in forest trees. New Phytol. 2012, 194, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Suontama, M.; Klápště, J.; Telfer, E.; Graham, N.; Stovold, T.; Low, C.; McKinley, R.; Dungey, H. Efficiency of genomic prediction across two Eucalyptus nitens seed orchards with different selection histories. Heredity 2018. [Google Scholar] [CrossRef]
- Grattapaglia, D. Breeding forest trees by genomic selection: Current progress and the way forward. Genom. Plant Genet. Resour. 2014, 1, 651–682. [Google Scholar]
- Iwata, H.; Hayashi, T.; Tsumura, Y. Prospects for genomic selection in conifer breeding: A simulation study of Cryptomeria japonica. Tree Genet. Genom. 2011, 7, 747–758. [Google Scholar] [CrossRef]
- Zhong, S.; Dekkers, J.C.; Fernando, R.L.; Jannink, J.L. Factors affecting accuracy from genomic selection in populations derived from multiple inbred lines: A barley case study. Genetics 2009, 182, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Durán, R.; Isik, F.; Zapata-Valenzuela, J.; Balocchi, C.; Valenzuela, S. Genomic predictions of breeding values in a cloned Eucalyptus globulus population in Chile. Tree Genet. Genom. 2007, 13, 74. [Google Scholar] [CrossRef]
- Resende-Junior, M.F.R.; Muñoz, P.; Resende, M.D.V.; Garrick, D.J.; Fernando, R.L.; Davis, J.M.; Jokela, E.J.; Martin, T.A.; Peter, G.F.; Kirst, M. Accuracy of genomic selection methods in a standard data set of loblolly pine (Pinus taeda L.). Genetics 2012, 190, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Macciotta, N.P.; Gaspa, G.; Steri, R.; Pieramati, C.; Carnier, P.; Dimauro, C. Pre-selection of most significant SNPS for the estimation of genomic breeding values. BMC Proc. 2009, 3, 14. [Google Scholar] [CrossRef]
- Arojju, S.K.; Conaghan, P.; Barth, S.; Milbourne, D.; Casler, M.D.; Hodkinson, T.R.; Michel, T.; Byrne, S.L. Genomic prediction of crown rust resistance in Lolium perenne. BMC Genet. 2018, 19, 35. [Google Scholar] [CrossRef] [PubMed]
- Long, N.; Gianola, D.; Rosa, G.J.M.; Weigel, K.A. Dimension reduction and variable selection for genomic selection: Application to predicting milk yield in Holsteins. J. Anim. Breed. Genet. 2011, 128, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Solberg, T.R.; Sonesson, A.K.; Woolliams, J.A.; Meuwissen, T.H. Reducing dimensionality for prediction of genome-wide breeding values. Genet. Sel. Evol. 2009, 41, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, C.; Wei, J.; Wang, S.; Jia, Z. Genomic selection using principal component regression. Heredity 2018, 121, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.F.; Silva, F.; Resende, M.D.; Lopes, M.S.; Duijvesteijn, S.E.; Lopes, P.S.; Kelly, M.J.; Viana, J.M.; Knol, E.F. Supervised independent component analysis as an alternative method for genomic selection in pigs. J. Anim. Breed. Genet. 2014, 131, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Doyle, J.L. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Silva-Junior, O.B.; Faria, D.A.; Grattapaglia, D.A. Flexible multi-species genome-wide 60K SNP chip developed from pooled resequencing of 240 Eucalyptus tree genomes across 12 species. New Phytol. 2015, 206, 1527–1540. [Google Scholar] [CrossRef] [PubMed]
- Mangin, B.; Siberchicot, A.; Nicolas, S.; Doligez, A.; This, P.; Cierco-Ayrolles, C. Novel measures of linkage disequilibrium that correct the bias due to population structure and relatedness. Heredity 2012, 108, 285. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.G.; Weir, B.S. Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 1988, 33, 54–78. [Google Scholar] [CrossRef]
- Myburg, A.A.; Grattapaglia, D.; Tuskan, G.A.; Hellsten, U.; Hayes, R.D.; Grimwood, J.; Jenkins, J.; Lindquist, E.; Tice, H.; Bauer, D.; et al. The genome of Eucalyptus grandis. Nature 2014, 510, 356. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, A.R.; Gogel, B.J.; Cullis, B.R.; Welham, S. ASReml User Guide Release 4.1 Structural Specification. 2015. Available online: https://www.vsni.co.uk/downloads/asreml/release4/UserGuideStructural.pdf (accessed on 13 April 2018).
- Wang, X.; Yang, Z.; Xu, C. A comparison of genomic selection methods for breeding value prediction. Sci. Bull. 2015, 60, 925–935. [Google Scholar] [CrossRef]
- Pant, S.; Schenkel, F.S.; Verschoor, C.P.; You, Q. A principal component regression based genome wide analysis approach reveals the presence of a novel QTL on BTA7 for MAP resistance in holstein cattle. Genomics 2010, 95, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bair, E.; Hastie, T.; Paul, D.; Tibshirani, R. Prediction by supervised principal components. J. Am. Stat. Assoc. 2006, 101, 119–137. [Google Scholar] [CrossRef]
- Chun, H.; Keleş, S. Sparse partial least squares regression for simultaneous dimension reduction and variable selection. J. R. Stat. Soc. B 2010, 72, 3–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lê Cao, K.A.; Rossouw, D.; Robert-Granié, C.; Besse, P. A sparse PLS for variable selection when integrating omics data. Stat. Appl. Genet. Mol. B 2008, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Mevik, B.H.; Wehrens, R. The pls package: Principal component and partial least squares regression in R. J. Stat. Softw. 2007, 18, 1–24. [Google Scholar] [CrossRef]
- Endelman, J.B. Ridge regression and other kernels for genomic selection with R. package rrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef]
- Pérez, P.; de los Campos, G.; Crossa, J.; Gianola, D. Genomic-enabled prediction based on molecular markers and pedigree using the Bayesian linear regression package in R. Plant Genome 2010, 3, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dutkowski, G.W.; Apiolaza, L.A.; Pilbeam, D. The genetic architecture of a Eucalyptus globulus full-sib breeding population in Australia. For. Genet. 2007, 12, 167–179. [Google Scholar]
- Costa e Silva, J.C.; Hardner, C.; Potts, B.M. Genetic variation and parental performance under inbreeding for growth in Eucalyptus globulus. Ann. For. Sci. 2010, 67, 606. [Google Scholar] [CrossRef]
- Callister, A.N.; England, N.; Collins, S. Genetic analysis of Eucalyptus globulus diameter, straightness, branch size, and forking in Western Australia. Can. J. For. Res. 2011, 41, 1333–1343. [Google Scholar] [CrossRef]
- Mora, F.; Serra, N. Bayesian estimation of genetic parameters for growth, stem straightness, and survival in Eucalyptus globulus on an Andean Foothill site. Tree Genet. Genom. 2014, 10, 711–719. [Google Scholar] [CrossRef]
- Blackburn, D.; Farrell, R.; Hamilton, M.; Volker, P. Genetic improvement for pulpwood and peeled veneer in Eucalyptus nitens. Can. J. For. Res. 2012, 42, 1724–1732. [Google Scholar] [CrossRef]
- Blackburn, D.P.; Hamilton, M.G.; Harwood, C.E.; Baker, T.G. Assessing genetic variation to improve stem straightness in Eucalyptus globulus. Ann. For. Sci. 2013, 70, 461–470. [Google Scholar] [CrossRef]
- Burdon, R.D. Short note: Coefficients of variation in variables with bounded scales. Silvae Genet. 2008, 57, 179–180. [Google Scholar] [CrossRef]
- Thavamanikumar, S.; McManus, L.J.; Tibbits, J.F.; Bossinger, G. The significance of single nucleotide polymorphisms (SNPs) in Eucalyptus globulus breeding programs. Aust. For. 2013, 74, 23–29. [Google Scholar] [CrossRef]
- Brito, F.V.; Neto, J.B.; Sargolzaei, M.; Cobuci, J.A. Accuracy of genomic selection in simulated populations mimicking the extent of linkage disequilibrium in beef cattle. BMC Genet. 2011, 12, 80. [Google Scholar] [CrossRef] [PubMed]
- Habier, D.; Fernando, R.; Garrick, D. Genomic BLUP decoded: A look into the black box of genomic prediction. Genetics 2013, 194, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhou, H.; Wu, Y.; Li, X.; Zhao, J.; Zuo, T.; Zhang, X.; Zhang, Y.; Liu, S.; Shen, Y.; et al. The impact of genetic relationship and linkage disequilibrium on genomic selection. PLoS ONE 2015, 10, e0132379. [Google Scholar] [CrossRef] [PubMed]
- Long, N.; Gianola, D.; Rosa, G.J.M.; Weigel, K.A. Machine learning classification procedure for selecting SNPs in genomic selection: Application to early mortality in broilers. J. Anim. Breed. Genet. 2007, 124, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Usai, M.G.; Goddard, M.E.; Hayes, B.J. LASSO with cross-validation for genomic selection. Genet. Res. 2009, 91, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Weigel, K.A.; de los Campos, G.; González-Recio, O.; Naya, H.; Wu, X.L.; Long, N.; Rosa, G.J.; Gianola, D. Predictive ability of direct genomic values for lifetime net merit of Holstein sires using selected subsets of single nucleotide polymorphism markers. J. Dairy Sci. 2009, 92, 5248–5257. [Google Scholar] [CrossRef] [PubMed]
- Spindel, J.; Begum, H.; Akdemir, D.; Virk, P.; Collard, B.; Redoña, E.; Atlin, G.; Jannink, J.L.; McCouch, S.R. Genomic selection and association mapping in rice (Oryza sativa): Effect of trait genetic architecture, training population composition, marker number and statistical model on accuracy of rice genomic selection in elite, tropical rice breeding lines. PLoS Genet. 2015, 11, e1004982. [Google Scholar]
- Wimmer, V.; Lehermeier, C.; Albrecht, T.; Auinger, H.J.; Wang, Y.; Schon, C.C. Genome-wide prediction of traits with different genetic architecture through efficient variable selection. Genetics 2013, 113, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Stackpole, D.J.; Vaillancourt, R.E.; de Aguigar, M.; Potts, B.M. Age trends in genetic parameters for growth and wood density in Eucalyptus globulus. Tree Genet. Genom. 2010, 6, 179–193. [Google Scholar] [CrossRef]
- Stackpole, D.J.; Vaillancourt, R.E.; Alves, A.; Rodrigues, J.; Potts, B.M. Genetic variation in the chemical components of Eucalyptus globulus wood. G3: Genes Genom. Genet. 2011, 1, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Daetwyler, H.D.; Pong-Wong, R.; Villanueva, B.; Woolliams, J.A. The impact of 539 genetic architecture on genome-wide evaluation methods. Genetics 2010, 185, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Jannink, J.L.; Lorenz, A.J.; Iwata, H. Genomic selection in plant breeding: From 566 theory to practice. Brief. Funct. Genom. 2010, 9, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Colombani, C.; Croiseau, P.; Fritz, S.; Guillaume, F.; Legarra, A.; Ducrocq, V.; Robert-Granié, C. Comparison of partial least squares (PLS) and sparse PLS regressions in genomic selection in French dairy cattle. J. Dairy Sci. 2012, 95, 2120–2131. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.S.; Potts, B.M.; Downes, G.M.; Pilbeam, D. Stability of quantitative trait loci for growth and wood properties across multiple pedigrees and environments in Eucalyptus globulus. New Phytol. 2013, 198, 1121–1134. [Google Scholar] [CrossRef] [PubMed]
- Thumma, B.R.; Baltunis, B.S.; Bell, J.C.; Emebiri, L.C. Quantitative trait locus (QTL) analysis of growth and vegetative propagation traits in Eucalyptus nitens full-sib families. Tree Genet. Genom. 2010, 6, 877–889. [Google Scholar] [CrossRef]
- Bundock, P.C.; Potts, B.M.; Vaillancourt, R.E. Detection and stability of quantitative trait loci (QTL) in Eucalyptus globulus. Tree Genet. Genom. 2008, 24, 85–95. [Google Scholar] [CrossRef]
- Gion, J.M.; Carouché, A.; Deweer, S.; Bedon, F.; Pichavant, F.; Charpentier, J.P.; Bailleres, H.; Rozenberg, P.; Carocha, V.; Ognouabi, N.; et al. Comprehensive genetic dissection of wood properties in a widely-grown tropical tree: Eucalyptus. BMC Genom. 2011, 12, 301. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Lin, Z.; Li, H.; Li, X.; Li, J.; Wang, Y.; Zhang, X.; Zhu, Z.; Zhai, W.; Wang, X.; et al. TAC1, a major quantitative trait locus controlling tiller angle in rice. Plant J. 2007, 52, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Dardick, C.; Callahan, A.; Horn, R.; Ruiz, K.B.; Zhebentyayeva, T.; Hollender, C.; Whitaker, M.; Abbott, A.; Scorza, R. PpeTAC1 promotes the horizontal growth of branches in peach trees and is a member of a functionally conserved gene family found in diverse plants species. Plant J. 2013, 75, 618–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, B.S.; Neves, L.G.; de Almeida Filho, J.E.; Resende, M.F.; Muñoz, P.R.; Dos Santos, P.E.T.; Filho, E.P.; Kirst, M.; Grattapaglia, D. Genomic prediction in contrast to a genome-wide association study in explaining heritable variation of complex growth traits in breeding populations of Eucalyptus. BMC Genom. 2017, 18, 524. [Google Scholar] [CrossRef] [PubMed]
- Isik, F.; Bartholomé, J.; Farjat, A.; Chancerel, E.; Raffin, A.; Sanchez, L.; Plomion, C.; Bouffier, L. Genomic selection in maritime pine. Plant Sci. 2016, 242, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Iwata, H.; Ebana, K.; Uga, Y.; Hayashi, T. Genomic prediction of biological shape: Elliptic fourier analysis and kernel partial least squares (PLS) regression applied to grain shape prediction in rice (Oryza sativa L.). PLoS ONE 2015, 10, e0120610. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Contreras, I.; Equihua, M.; Castillo-Campos, G.; Rojas-Soto, O. Climate change and effects on vegetation in Veracruz, Mexico: An approach using ecological niche modelling. Acta Bot. Mex. 2015, 112, 73–93. [Google Scholar] [CrossRef]
- Woillez, M.N.; Kageyama, M.; Combourieu-Nebout, N.; Krinner, G. Simulating the vegetation response in western Europe to abrupt climate changes under glacial background conditions. Biogeosciences 2013, 10, 1561–1582. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site Conditions | Metrics |
|---|---|
| Coordinates | 40°57′ S, 73°30′ W |
| Climate Type | Oceanic or marine |
| Annual Temperature | 13 °C |
| Average temperature in coldest months | 6 °C |
| Average temperature in warmest months | 16 °C |
| Annual accumulated rainfall | 1282 mm |
| Altitude | 326 masl |
| REML Estimates | ST | BQ | H | DBH | VOL |
|---|---|---|---|---|---|
| Additive variance | 0.194 | 0.073 | 0.107 | 0.693 | 0.00001 |
| Residual variance | 1.000 | 1.000 | 0.777 | 3.01 | 0.00006 |
| Heritability | 0.162 (0.03) | 0.068 (0.02) | 0.121 (0.04) | 0.187 (0.06) | 0.156 (0.05) |
| CVa | 17.6 * | 11.0 * | 5.03 | 8.5 | 18.7 |
| CH | r2 | Max. r2 | Min. r2 | Distance (Mbp) | Dist. Max (Mbp) | Dist. Min (Mbp) |
|---|---|---|---|---|---|---|
| 1 | 0.03 | 0.37 | 0.0057 | 0.96 | 3.65 | 0.00003 |
| 2 | 0.03 | 0.37 | 0.0057 | 0.78 | 3.19 | 0.00003 |
| 3 | 0.09 | 0.36 | 0.0057 | 1.11 | 5.85 | 0.00003 |
| 4 | 0.09 | 0.37 | 0.0057 | 0.99 | 5.31 | 0.000031 |
| 5 | 0.09 | 0.36 | 0.0057 | 1.04 | 5.5 | 0.00003 |
| 6 | 0.09 | 0.36 | 0.0137 | 0.81 | 3.19 | 0.000039 |
| 7 | 0.09 | 0.37 | 0.0137 | 0.97 | 4.59 | 0.000033 |
| 8 | 0.09 | 0.36 | 0.0137 | 0.9 | 3.85 | 0.000036 |
| 9 | 0.09 | 0.37 | 0.0137 | 0.93 | 3.92 | 0.00003 |
| 10 | 0.09 | 0.37 | 0.0137 | 0.81 | 3.4 | 0.000049 |
| 11 | 0.11 | 0.37 | 0.0217 | 0.83 | 3.43 | 0.000038 |
| Trait/NPV | RRBLUP | RRBLUP-B | BAYES-A | BAYES-B | BLASSO | PCR | S PCR |
|---|---|---|---|---|---|---|---|
| BQ | 0.17 | 0.68 | 0.21 | 0.28 | 0.23 | 0.1 | 0.69 |
| NPV | 14,422 | 950 | 14,422 | 14,422 | 14,422 | 573 | 71 |
| ST | 0.14 | 0.59 | 0.17 | 0.14 | 0.1 | 0.16 | 0.62 |
| NPV | 14,422 | 800 | 14,422 | 14,422 | 14,422 | 579 | 95 |
| VOL | 0.13 | 0.42 | 0.07 | 0.04 | 0.04 | 0.35 | 0.35 |
| NPV | 14,422 | 900 | 14,422 | 14,422 | 14,422 | 575 | 148 |
| DBH | 0.04 | 0.43 | 0.02 | 0.01 | 0.01 | 0.35 | 0.43 |
| NPV | 14,422 | 450 | 14,422 | 14,422 | 14,422 | 579 | 62 |
| H | 0.04 | 0.5 | 0.05 | 0.03 | 0.04 | 0.21 | 0.54 |
| NPV | 14,422 | 850 | 14,422 | 14,422 | 14,422 | 570 | 338 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ballesta, P.; Serra, N.; Guerra, F.P.; Hasbún, R.; Mora, F. Genomic Prediction of Growth and Stem Quality Traits in Eucalyptus globulus Labill. at Its Southernmost Distribution Limit in Chile. Forests 2018, 9, 779. https://doi.org/10.3390/f9120779
Ballesta P, Serra N, Guerra FP, Hasbún R, Mora F. Genomic Prediction of Growth and Stem Quality Traits in Eucalyptus globulus Labill. at Its Southernmost Distribution Limit in Chile. Forests. 2018; 9(12):779. https://doi.org/10.3390/f9120779
Chicago/Turabian StyleBallesta, Paulina, Nicolle Serra, Fernando P. Guerra, Rodrigo Hasbún, and Freddy Mora. 2018. "Genomic Prediction of Growth and Stem Quality Traits in Eucalyptus globulus Labill. at Its Southernmost Distribution Limit in Chile" Forests 9, no. 12: 779. https://doi.org/10.3390/f9120779
APA StyleBallesta, P., Serra, N., Guerra, F. P., Hasbún, R., & Mora, F. (2018). Genomic Prediction of Growth and Stem Quality Traits in Eucalyptus globulus Labill. at Its Southernmost Distribution Limit in Chile. Forests, 9(12), 779. https://doi.org/10.3390/f9120779