Development of Novel Intramolecular FRET-Based ABC Transporter Biosensors to Identify New Substrates and Modulators

Abstract
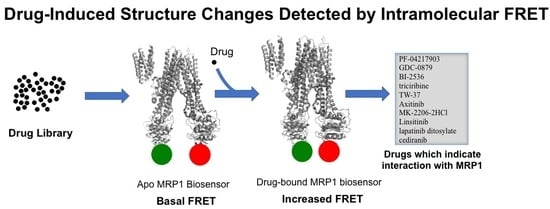
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
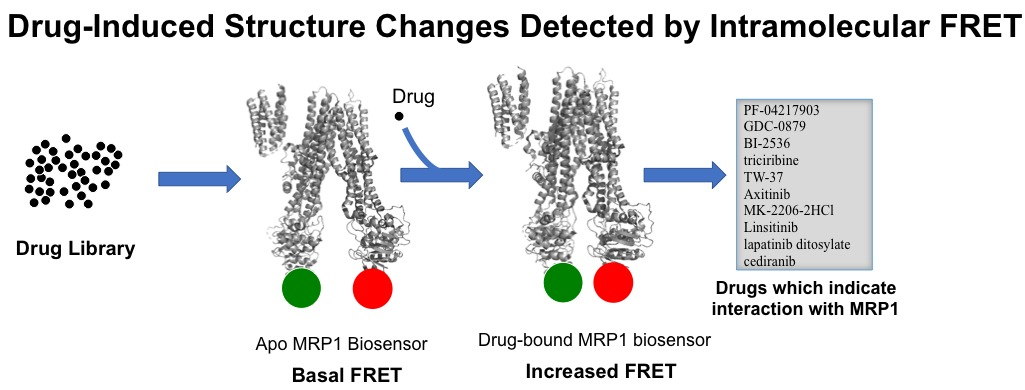
2.2. Engineering Two-Color Multidrug Resistance Protein 1 (MRP1) Constructs
2.3. Cell Lines and Cell Culture
2.4. Preparation of MRP1-Enriched Membrane Vesicles
2.5. Two-Color MRP1-Expressing Stable Cell Lines
2.6. Immunoblot Analysis
2.7. Detection of MRP1 Localization
2.8. Doxorubicin Accumulation Assay
2.9. Ensemble Fluorescence Spectroscopy
2.10. Anti-Cancer Drug Screening Using Fluorescence Spectroscopy-Based FRET Approach
3. Results and Discussion
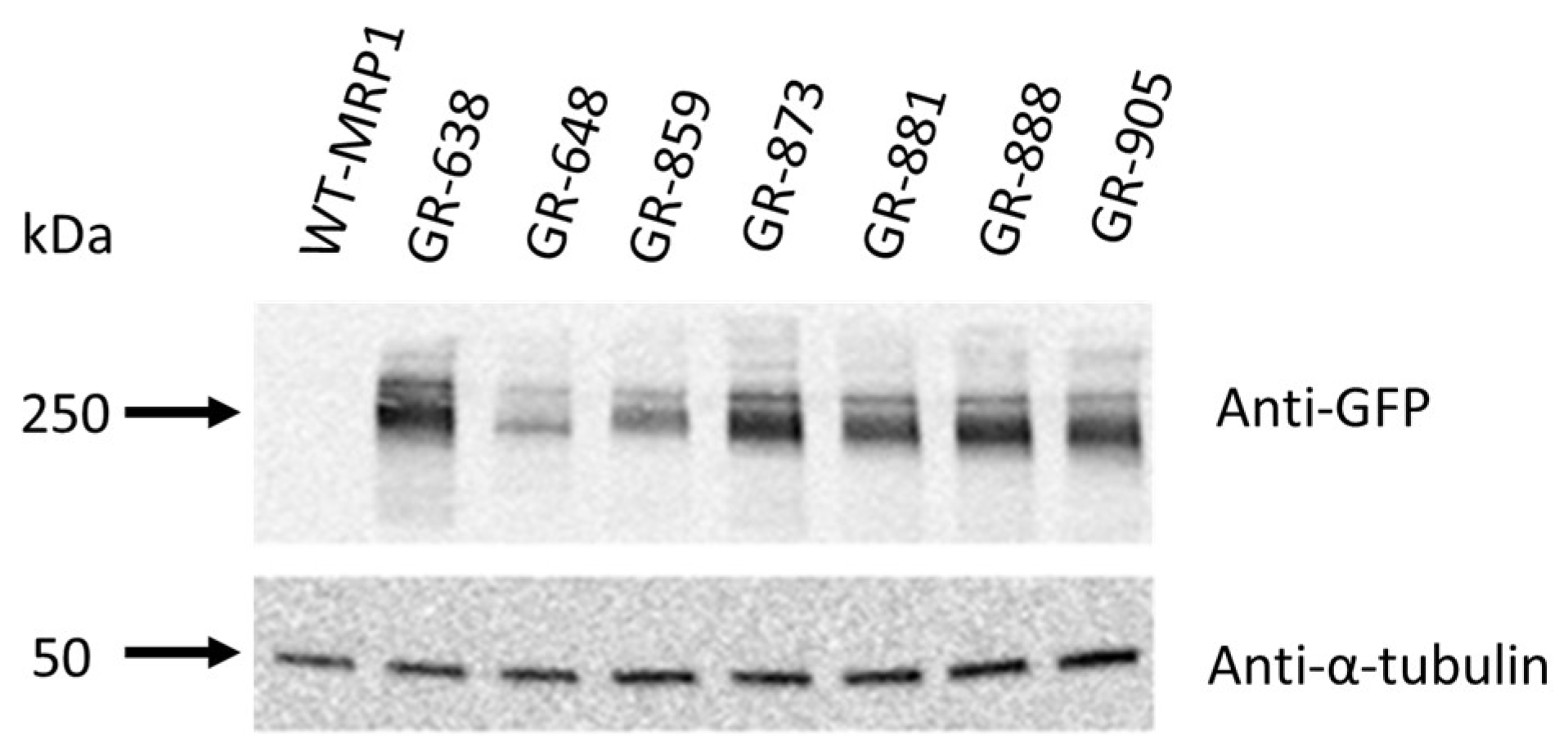
3.1. Genetic Engineering and Expression of Two-Color MRP1 Recombinant Proteins
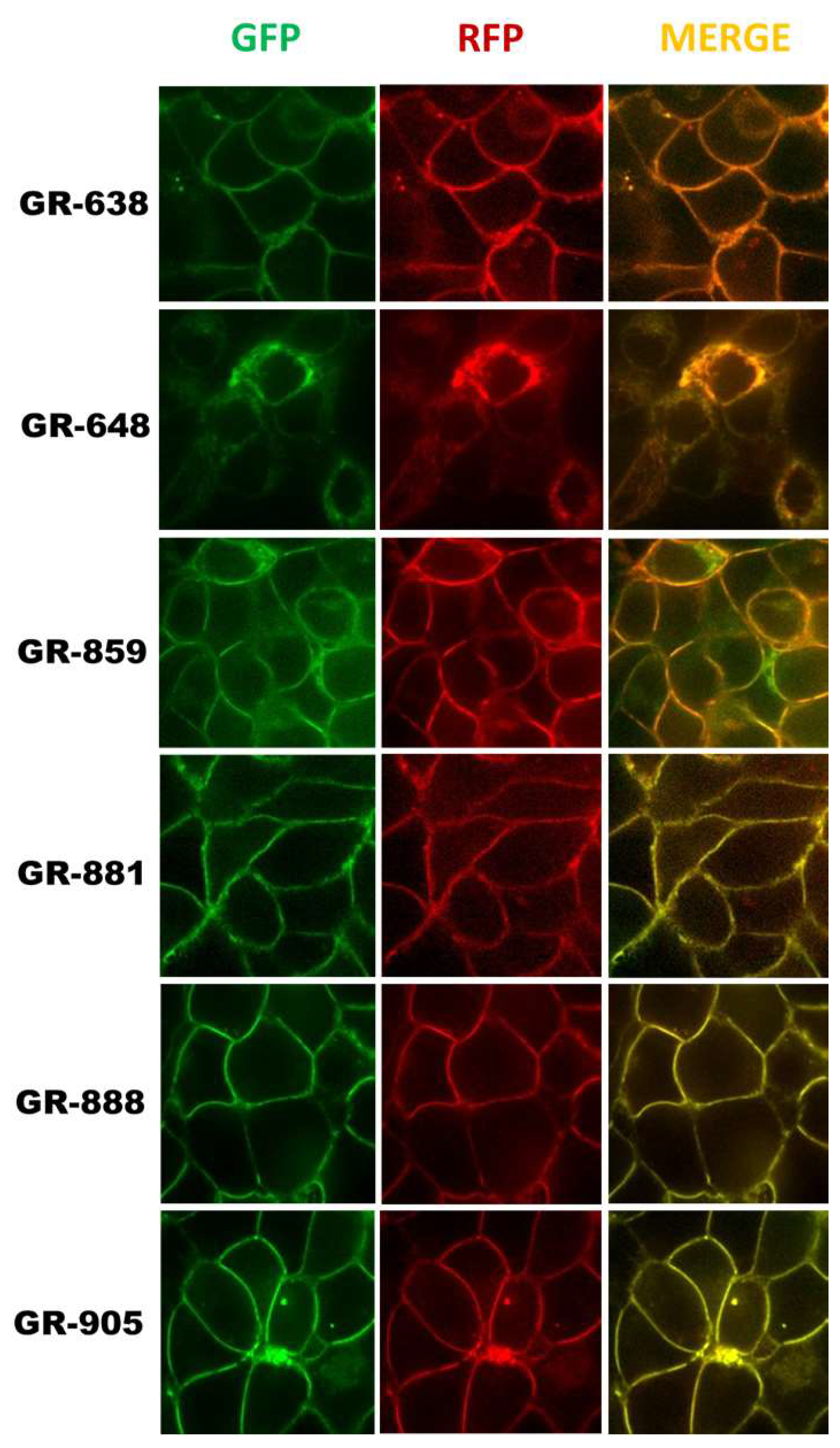
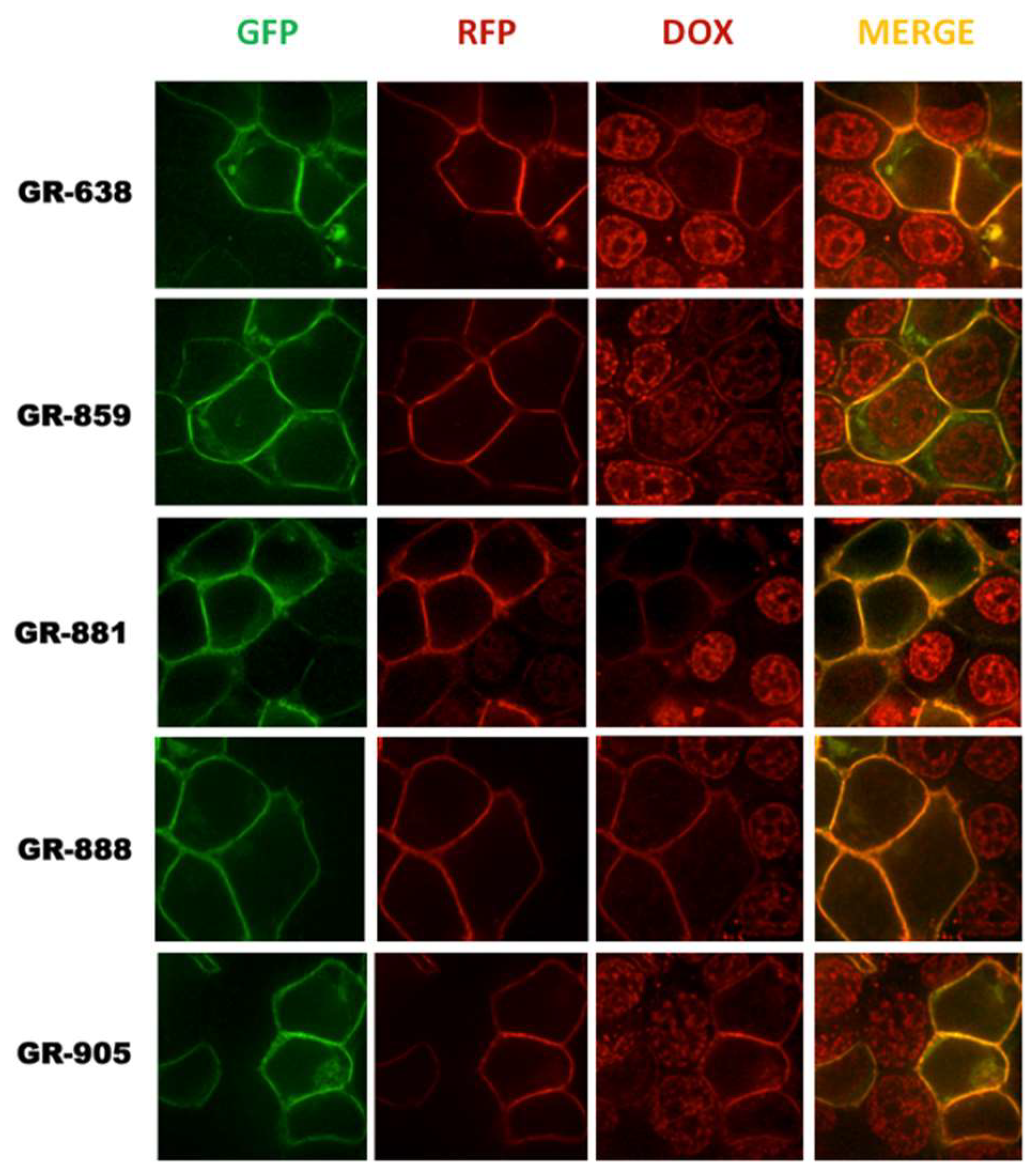
3.2. Localization and Transport Activity of Two-Color MRP1 Proteins in Live Cells
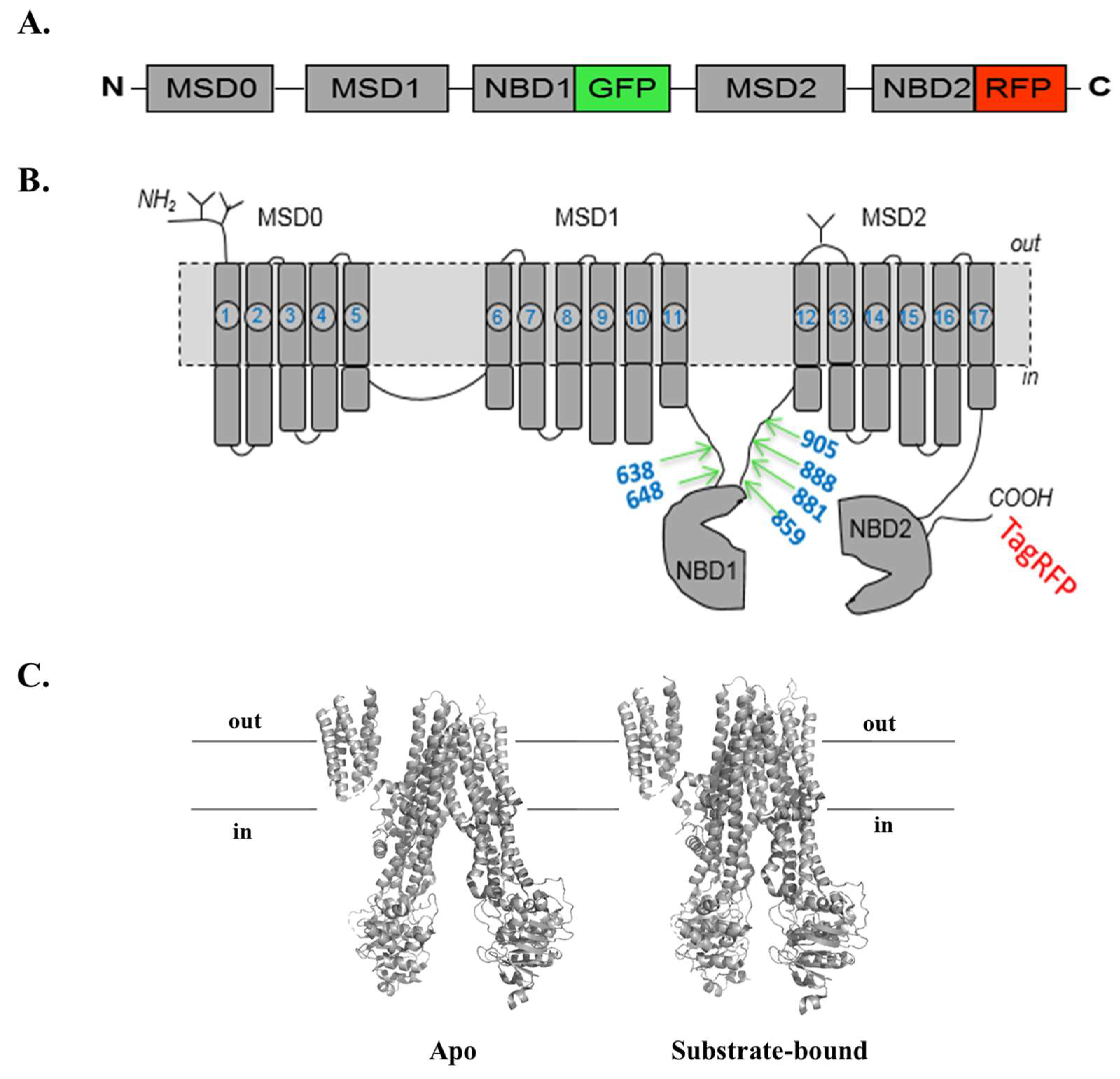
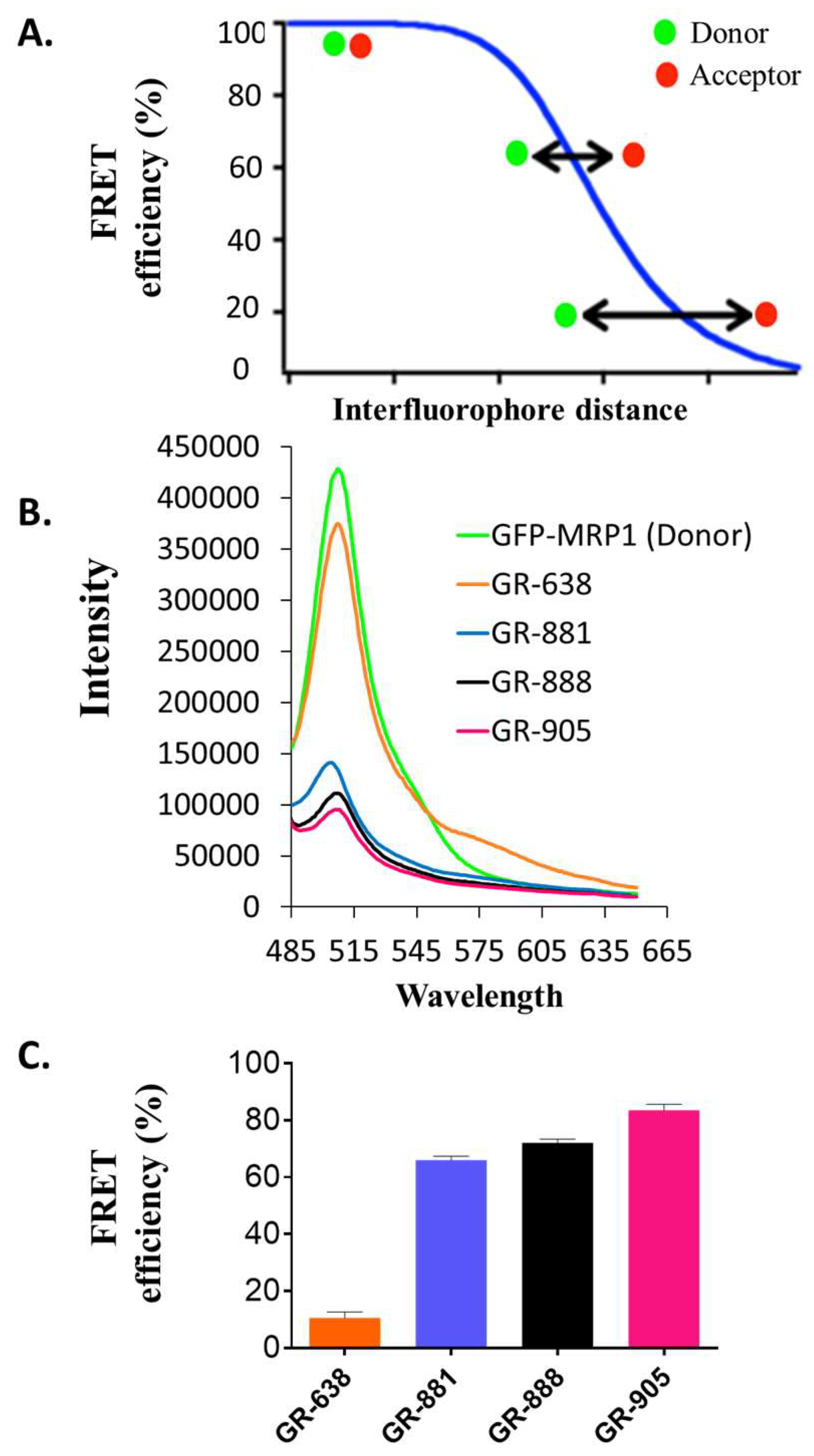
3.3. Substrate-Free FRET Efficiencies of the Two-Color MRP1 Proteins
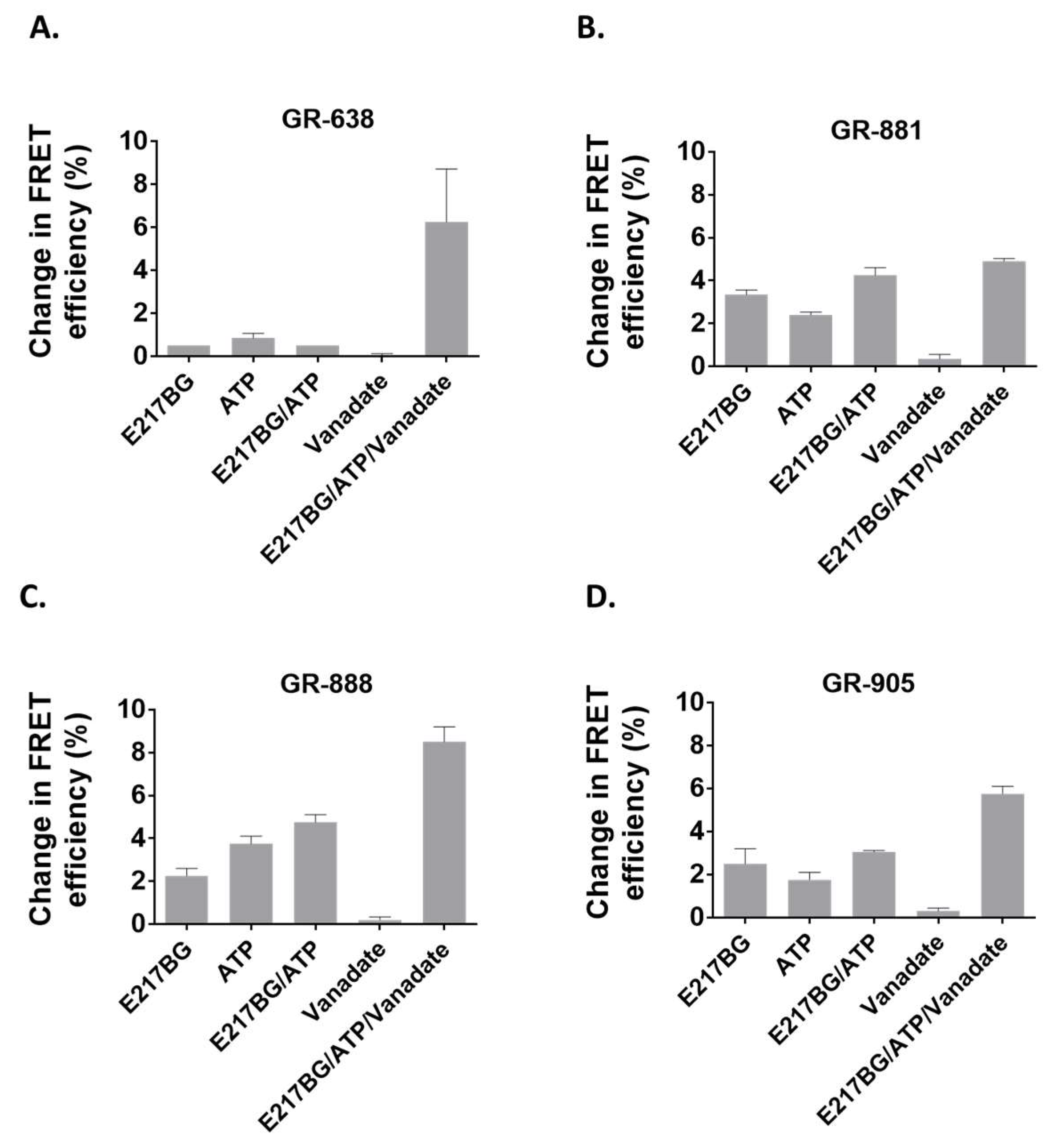
3.4. Evaluation of Two-Color MRP1 Proteins as FRET-Based Biosensors
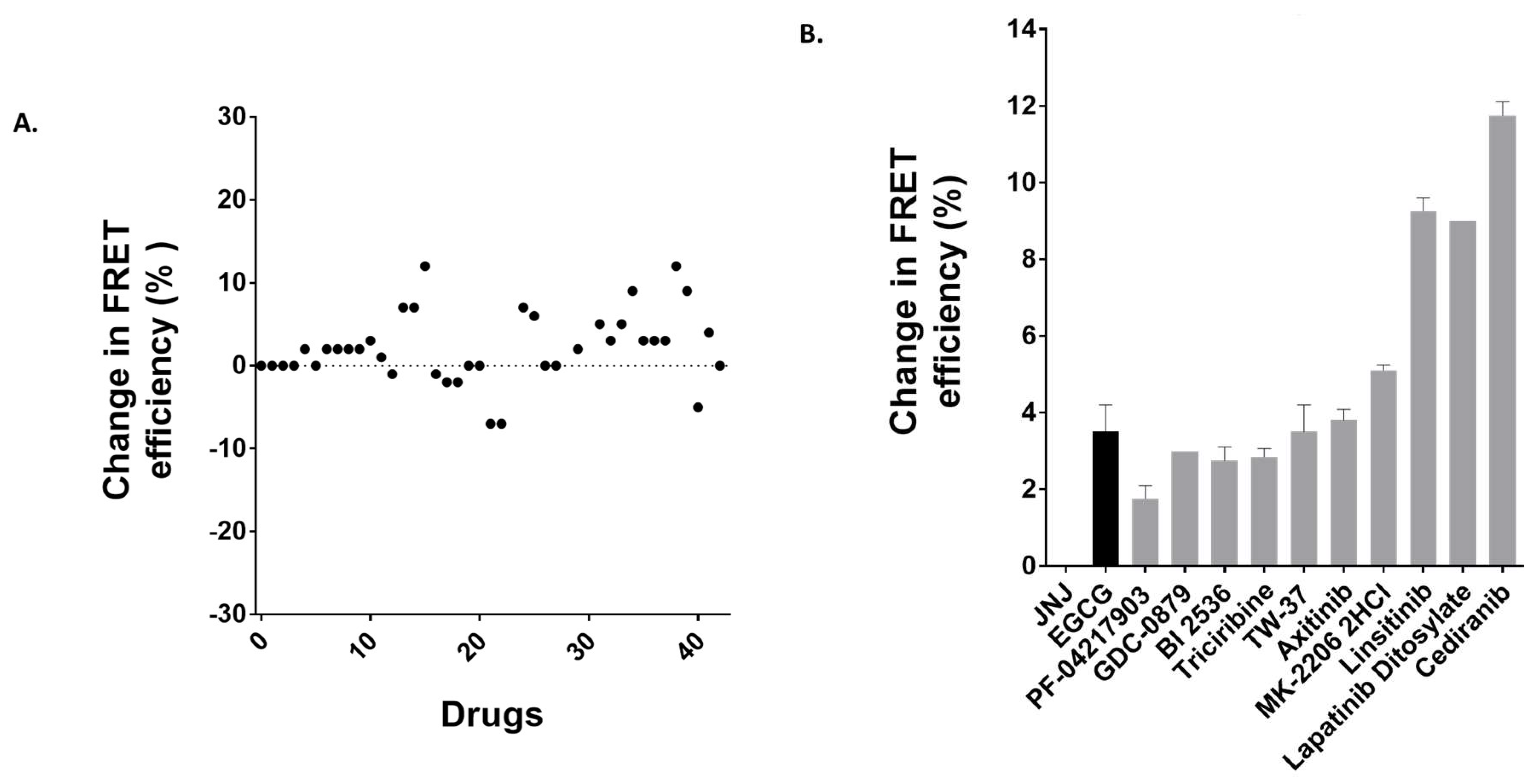
3.5. Identification of Anti-Cancer Drugs that Interact with MRP1
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. List of the 40 Anti-Cancer Drugs for FRET Screening with GR-881
| ABT-263 | Navitoclax |
| Afatinib | BIBW2992 |
| PD0325901 | |
| Trichostatin A | TSA |
| BMS-599626 | AC480 |
| AUY922 | NVP-AUY922 |
| Brivanib | BMS-540215 |
| PF-04217903 | |
| BI 2536 | |
| TW-37 | |
| Mocetinostat | MGCD0103 |
| SRT1720 | |
| YM155 | |
| MLN8237 | Alisertib |
| AT9283 | |
| Andarine | GTX-007 |
| AZD6244 | Selumetinib |
| CI-1040 | PD184352 |
| Motesanib Diphosphate | AMG-706 |
| Tandutinib | MLN518 |
| Entinostat | MS-275 SNDX-275 |
| SB 431542 | |
| SU11274 | |
| KU-55933 | |
| LY294002 | |
| XL147 | |
| Saracatinib | AZD0530 |
| Dovitinib | TKI-258 |
| Lenalidomide | Revlimid |
| Sunitinib Malate | Sutent |
| Elesclomol | |
| GDC-0941 | |
| MK-2206 2HCl | |
| Linsitinib | OSI-906 |
| GDC-0879 | |
| Triciribine | Triciribine phosphate |
| Axitinib | |
| Cediranib | AZD2171 |
| Lapatinib Ditosylate | Tykerb |
| STF-62247 |
Appendix B. Primers used in This Study
| Primer Names | Sequences (5’ → 3’) |
| MRP11–638 forward | GTA GAG CTC ATG GCG CTC CGG GGC TTC TGC AG |
| MRP11–638 reverse | CTA GTC GAC ATT CCT CAC GGT GAT GCT GTT CGT GCC C |
| MRP1639–1531 forward | GTA CCG CGG GCC ACA TTC ACC TGG GCC AGG AGC |
| MRP1639–1531 reverse | GTA ACC GGT CT CAC CAA GCC GGC GTC TTT GGC CAT G |
| MRP11–648 forward | GTA GAG CTC ATG GCG CTC CGG GGC TTC TGC AG |
| MRP11–648 reverse | CTA GTC GAC ATT CCT CAC GGT GAT GCT GTT CGT GCC C |
| MRP1649–1531 forward | GTA CCG CGG GCC ACA TTC ACC TGG GCC AGG AGC |
| MRP1649–1531 reverse | GTA ACC GGT CT CAC CAA GCC GGC GTC TTT GGC CAT G |
| MRP11–859 forward | GTA GAG CTC ATG GCG CTC CGG GGC TTC TGC AG |
| MRP11–859 reverse | CTA GTC GAC GTC TCG AGC CAG CAG CTC CTG GTA GG |
| MRP1860–1531 forward | GTA CCG CGG GGC GCC TTC GCT GAG TTC CTG |
| MRP1860–1531 reverse | GTA ACC GGT CT CAC CAA GCC GGC GTC TTT GGC CAT G |
| MRP11–881 forward | GTA GAG CTC ATG GCG CTC CGG GGC TTC TGC AG |
| MRP11–881 reverse | CTA GTC GAC GTT CTC CTC TGC ATC CTG CTC CTG C |
| MRP1882–1531 forward | GTA CCG CGG GGG GTC ACG GGC GTC AGC GGT |
| MRP1882–1531 reverse | GTA ACC GGT CT CAC CAA GCC GGC GTC TTT GGC CAT G |
| MRP11–888 forward | GTA GAG CTC ATG GCG CTC CGG GGC TTC TGC AG |
| MRP11–888 reverse | CTA GTC GAC ACC GCT GAC GCC CGT GAC CCC GT |
| MRP1889–1531 forward | GTA CCG CGG CCA GGG AAG GAA GCA AAG CAA ATG GAG |
| MRP1889–1531 reverse | GTA ACC GGT CT CAC CAA GCC GGC GTC TTT GGC CAT G |
| MRP11–905 forward | GTA GAG CTC ATG GCG CTC CGG GGC TTC TGC AG |
| MRP11–905 reverse | CTA GTC GAC ACT GTC CGT CAC CAG CAT GCC ATT CTC C |
| MRP1906–1531 forward | GTA CCG CGG GCA GGG AAG CAA CTG CAG AGA CAG C |
| MRP1906–1531 reverse | GTA ACC GGT CT CAC CAA GCC GGC GTC TTT GGC CAT G |
| GFP-forward | GTA GTC GAC ATG GTG AGC AAG GGC GAG GAG CTG |
| GFP-reverse | CTA CCG CGG CTT GTA CAG CTC GTC CAT GCC GAG AG |
References
- Cole, S.P. Multidrug resistance protein 1 (MRP1, ABCC1), a ‘multitasking’ ATP-binding cassette (ABC) transporter. J. Biol. Chem. 2014, 289, 30880–30888. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Annilo, T. Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annu. Rev. Genomics Hum. Genet. 2005, 6, 123–142. [Google Scholar] [CrossRef] [PubMed]
- Annilo, T.; Tammur, J.; Hutchinson, A.; Rzhetsky, A.; Dean, M.; Allikmets, R. Human and mouse orthologs of a new ATP-binding cassette gene, ABCG4. Cytogenet. Cell Genet. 2001, 94, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Revalde, J.; Paxton, J.W. The effects of dietary and herbal phytochemicals on drug transporters. Adv. Drug Deliv. Rev. 2017, 116, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Allikmets, R. Evolution of ATP-binding cassette transporter genes. Curr. Opin. Genet. Dev. 1995, 5, 779–785. [Google Scholar] [CrossRef]
- Mamo, G.; Pandi, A. A Review on ATP Binding Cassette (ABC) Transporters. Int. J. Pharma. Res. Health Sci. 2017, 5, 1607–1615. [Google Scholar]
- Langmann, T.; Klucken, J.; Reil, M.; Liebisch, G.; Luciani, M.F.; Chimini, G.; Kaminski, W.E.; Schmitz, G. Molecular cloning of the human ATP-binding cassette transporter 1 (hABC1): Evidence for sterol-dependent regulation in macrophages. Biochem. Biophys. Res. Commun. 1999, 257, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.; Hunke, S. ATP-binding-cassette (ABC) transport systems: Functional and structural aspects of the ATP-hydrolyzing subunits/domains. FEMS Microbiol. Rev. 1998, 22, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome. Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Deeley, R.G.; Westlake, C.; Cole, S.P. Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol. Rev. 2006, 86, 849–899. [Google Scholar] [CrossRef] [PubMed]
- Conseil, G.; Deeley, R.G.; Cole, S.P. Functional importance of three basic residues clustered at the cytosolic interface of transmembrane helix 15 in the multidrug and organic anion transporter MRP1 (ABCC1). J. Biol. Chem. 2006, 281, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC transporters: The power to change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.L.; Dassa, E.; Orelle, C.; Chen, J. Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol. Mol. Biol. Rev. 2008, 72, 317–364. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Allikmets, R. Complete characterization of the human ABC gene family. J. Bioenerg. Biomembr. 2001, 33, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Iram, S.H.; Cole, S.P. Expression and function of human MRP1 (ABCC1) is dependent on amino acids in cytoplasmic loop 5 and its interface with nucleotide binding domain 2. J. Biol. Chem. 2011, 286, 7202–7213. [Google Scholar] [CrossRef] [PubMed]
- Locher, K.P. Review. Structure and mechanism of ATP-binding cassette transporters. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2009, 364, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Aittoniemi, J.; Fotinou, C.; Craig, T.J.; de Wet, H.; Proks, P.; Ashcroft, F.M. Review. SUR1: A unique ATP-binding cassette protein that functions as an ion channel regulator. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2009, 364, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Iram, S.H.; Cole, S.P. Differential functional rescue of Lys(513) and Lys(516) processing mutants of MRP1 (ABCC1) by chemical chaperones reveals different domain-domain interactions of the transporter. Biochim. Biophys. Acta 2014, 1838, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Biemans-Oldehinkel, E.; Doeven, M.K.; Poolman, B. ABC transporter architecture and regulatory roles of accessory domains. FEBS Lett. 2006, 580, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.J.; Hollenstein, K.; Locher, K.P. Uptake or extrusion: Crystal structures of full ABC transporters suggest a common mechanism. Mol. Microbiol. 2007, 65, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Iram, S.H.; Cole, S.P. Mutation of Glu521 or Glu535 in cytoplasmic loop 5 causes differential misfolding in multiple domains of multidrug and organic anion transporter MRP1 (ABCC1). J. Biol. Chem. 2012, 287, 7543–7555. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Z.N.; Yu, L.C.; Bao, Q.L.; Wu, J.R.; Shi, S.B.; Li, X.Q. Association of expression of MRP1, BCRP, LRP and ERCC1 with outcome of patients with locally advanced non-small cell lung cancer who received neoadjuvant chemotherapy. Lung Cancer 2010, 69, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Bagnoli, M.; Beretta, G.L.; Gatti, L.; Pilotti, S.; Alberti, P.; Tarantino, E.; Barbareschi, M.; Canevari, S.; Mezzanzanica, D.; Perego, P. Clinicopathological impact of ABCC1/MRP1 and ABCC4/MRP4 in epithelial ovarian carcinoma. Biomed. Res. Int. 2013, 2013, 143202. [Google Scholar] [CrossRef] [PubMed]
- Hlavac, V.; Brynychova, V.; Vaclavikova, R.; Ehrlichova, M.; Vrana, D.; Pecha, V.; Kozevnikovova, R.; Trnkova, M.; Gatek, J.; Kopperova, D.; et al. The expression profile of ATP-binding cassette transporter genes in breast carcinoma. Pharmacogenomics 2013, 14, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Peterson, B.G.; Tan, K.W.; Osa-Andrews, B.; Iram, S.H. High-content screening of clinically tested anticancer drugs identifies novel inhibitors of human MRP1 (ABCC1). Pharmacol. Res. 2017, 119, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Iram, S.H.; Gruber, S.J.; Raguimova, O.N.; Thomas, D.D.; Robia, S.L. ATP-Binding Cassette Transporter Structure Changes Detected by Intramolecular Fluorescence Energy Transfer for High-Throughput Screening. Mol. Pharmacol. 2015, 88, 84–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2012, 64, 138–153. [Google Scholar] [CrossRef]
- Cole, S.P. Targeting multidrug resistance protein 1 (MRP1, ABCC1): Past, present, and future. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Johnson, Z.L.; Chen, J. Structural Basis of Substrate Recognition by the Multidrug Resistance Protein MRP1. Cell 2017, 168, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, M.S.; Lee, J.H.; Kim, S.W.; Kang, R.H.; Choi, M.J.; Park, S.J.; Kim, S.J.; Lee, J.M.; Cole, S.P.; et al. MRP1 polymorphisms associated with citalopram response in patients with major depression. J. Clin. Psychopharmacol. 2010, 30, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Knauer, M.J.; Urquhart, B.L.; Meyer zu Schwabedissen, H.E.; Schwarz, U.I.; Lemke, C.J.; Leake, B.F.; Kim, R.B.; Tirona, R.G. Human skeletal muscle drug transporters determine local exposure and toxicity of statins. Circ. Res. 2010, 106, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Pasternak, G.W. The role of multidrug resistance-associated protein in the blood-brain barrier and opioid analgesia. Synapse 2013, 67, 609–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leslie, E.M.; Deeley, R.G.; Cole, S.P. Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharmacol. 2005, 204, 216–237. [Google Scholar] [CrossRef] [PubMed]
- Wijnholds, J.; Evers, R.; van Leusden, M.R.; Mol, C.A.; Zaman, G.J.; Mayer, U.; Beijnen, J.H.; van der Valk, M.; Krimpenfort, P.; Borst, P. Increased sensitivity to anticancer drugs and decreased inflammatory response in mice lacking the multidrug resistance-associated protein. Nat. Med. 1997, 3, 1275–1279. [Google Scholar] [CrossRef] [PubMed]
- Wijnholds, J.; deLange, E.C.; Scheffer, G.L.; van den Berg, D.J.; Mol, C.A.; van der Valk, M.; Schinkel, A.H.; Scheper, R.J.; Breimer, D.D.; Borst, P. Multidrug resistance protein 1 protects the choroid plexus epithelium and contributes to the blood-cerebrospinal fluid barrier. J. Clin. Invest. 2000, 105, 279–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umscheid, C.A.; Margolis, D.J.; Grossman, C.E. Key concepts of clinical trials: A narrative review. Postgrad. Med. 2011, 123, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.W.; Sampson, A.; Osa-Andrews, B.; Iram, S.H. Calcitriol and Calcipotriol Modulate Transport Activity of ABC Transporters and Exhibit Selective Cytotoxicity in MRP1 overexpressing Cells. Drug Metab. Dispos. 2018. [Google Scholar] [CrossRef] [PubMed]
- Verhalen, B.; Ernst, S.; Borsch, M.; Wilkens, S. Dynamic ligand-induced conformational rearrangements in P-glycoprotein as probed by fluorescence resonance energy transfer spectroscopy. J. Biol. Chem. 2012, 287, 1112–1127. [Google Scholar] [CrossRef] [PubMed]
- Remedios, C.G.D. Fluorescence Resonance Energy. In Encyclopedia of Life Sciences; Nature Publishing Group: London, UK, 2001. [Google Scholar]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The good, the bad and the ugly effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J.; Fulop, T.; Dezsi, L.; Metselaar, B.; Storm, G. Liposomal doxorubicin: The good, the bad and the not-so-ugly. J. Drug Target 2016, 24, 765–767. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.S.; Oldham, M.L.; Zhang, Q.; Chen, J. Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 2012, 490, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, V.; Pierik, A.J.; Lill, R. Crystal structures of nucleotide-free and glutathione-bound mitochondrial ABC transporter Atm1. Science 2014, 343, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Korkhov, V.M.; Mireku, S.A.; Locher, K.P. Structure of AMP-PNP-bound vitamin B12 transporter BtuCD-F. Nature 2012, 490, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Wang, Z.; James, N.R.; Voss, J.E.; Klimont, E.; Ohene-Agyei, T.; Venter, H.; Chiu, W.; Luisi, B.F. Structure of the AcrAB-TolC multidrug efflux pump. Nature 2014, 509, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, H.G.; Tong, Z.; Mathavan, I.; Li, Y.; Iwata, S.; Zirah, S.; Rebuffat, S.; van Veen, H.W.; Beis, K. Structure of an antibacterial peptide ATP-binding cassette transporter in a novel outward occluded state. Proc. Natl. Acad. Sci. USA 2014, 111, 9145–9150. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.J.; Hu, Z.; Blackwell, D.J.; Miller, T.D.; Thomas, D.D.; Robia, S.L. 2-Color Calcium Pump Reveals Closure of the Cytoplasmic Headpiece with Calcium Binding. PLoS ONE 2012, 7, e40369. [Google Scholar] [CrossRef] [PubMed]
- Pallikkuth, S.; Blackwell, D.J.; Hu, Z.; Hou, Z.; Zieman, D.T.; Svensson, B.; Thomas, D.D.; Robia, S.L. Phosphorylated phospholamban stabilizes a compact conformation of the cardiac calcium-ATPase. Biophys. J. 2013, 105, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.J.; St-Pierre, F.; Gong, Y.; Marshall, J.D.; Cranfill, P.J.; Baird, M.A.; McKeown, M.R.; Wiedenmann, J.; Davidson, M.W.; Schnitzer, M.J.; et al. Improving FRET Dynamic Range with Bright Green and Red Fluorescent Proteins. Biophys J. 2013, 104, 683a. [Google Scholar] [CrossRef] [Green Version]
- Beedholm-Ebsen, R.; van de Wetering, K.; Hardlei, T.; Nexo, E.; Borst, P.; Moestrup, S.K. Identification of multidrug resistance protein 1 (MRP1/ABCC1) as a molecular gate for cellular export of cobalamin. Blood 2010, 115, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- Leier, I.; Jedlitschky, G.; Buchholz, U.; Center, M.; Cole, S.P.; Deeley, R.G.; Keppler, D. ATP-dependent glutathione disulphide transport mediated by the MRP gene-encoded conjugate export pump. Biochem. J. 1996, 314, 433–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jedlitschky, G.; Leier, I.; Buchholz, U.; Barnouin, K.; Kurz, G.; Keppler, D. Transport of glutathione, glucuronate, and sulfate conjugates by the MRP gene-encoded conjugate export pump. Cancer Res. 1996, 56, 988–994. [Google Scholar] [PubMed]
- Senior, A.E.; Al-Shawi, M.K.; Urbatsch, I.L. The catalytic cycle of P-glycoprotein. FEBS Lett. 1995, 377, 285–289. [Google Scholar] [PubMed] [Green Version]
- Myette, R.L.; Conseil, G.; Ebert, S.P.; Wetzel, B.; Detty, M.R.; Cole, S.P. Chalcogenopyrylium dyes as differential modulators of organic anion transport by multidrug resistance protein 1 (MRP1), MRP2, and MRP4. Drug Metab. Dispos. 2013, 41, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Slot, A.J.; Wise, D.D.; Deeley, R.G.; Monks, T.J.; Cole, S.P. Modulation of human multidrug resistance protein (MRP) 1 (ABCC1) and MRP2 (ABCC2) transport activities by endogenous and exogenous glutathione-conjugated catechol metabolites. Drug Metab. Dispos. 2008, 36, 552–560. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osa-Andrews, B.; Tan, K.W.; Sampson, A.; Iram, S.H. Development of Novel Intramolecular FRET-Based ABC Transporter Biosensors to Identify New Substrates and Modulators. Pharmaceutics 2018, 10, 186. https://doi.org/10.3390/pharmaceutics10040186
Osa-Andrews B, Tan KW, Sampson A, Iram SH. Development of Novel Intramolecular FRET-Based ABC Transporter Biosensors to Identify New Substrates and Modulators. Pharmaceutics. 2018; 10(4):186. https://doi.org/10.3390/pharmaceutics10040186
Chicago/Turabian StyleOsa-Andrews, Bremansu, Kee W. Tan, Angelina Sampson, and Surtaj H. Iram. 2018. "Development of Novel Intramolecular FRET-Based ABC Transporter Biosensors to Identify New Substrates and Modulators" Pharmaceutics 10, no. 4: 186. https://doi.org/10.3390/pharmaceutics10040186
APA StyleOsa-Andrews, B., Tan, K. W., Sampson, A., & Iram, S. H. (2018). Development of Novel Intramolecular FRET-Based ABC Transporter Biosensors to Identify New Substrates and Modulators. Pharmaceutics, 10(4), 186. https://doi.org/10.3390/pharmaceutics10040186