Grinding as Solvent-Free Green Chemistry Approach for Cyclodextrin Inclusion Complex Preparation in the Solid State



Abstract
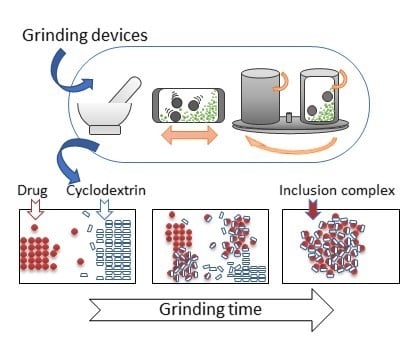
:
1. Introduction
- Methods in solution, where drug and CD are dissolved in water or organic solvent/water mixtures, with pH and temperature adjusted to achieve maximum interaction between the components. In the case of Bs-type complexes, mainly occurring with natural CDs, the product is isolated by crystallization [14], whereas in the case of A-type complexes, the solvent is removed by an adequate drying technique, such as coevaporation under reduced pressure (COE) [22,23], spray-drying (SPD) [24,25], or freeze-drying (FD) [26,27]. The drawbacks of such methods are in high consumption of time, energy and organic solvents (such as ethanol and methanol) whose complete removal from the final solid product could be quite costly and challenging, due to the ability of such solvents for the inclusion complex formation [28], that could lead to toxic effects. The use of supercritical fluid technology (SPF) could be also classified as a method of complex preparation in solution [29,30]. While, in this case, the use of organic solvents is in general avoided, this method requires the availability of specific and costly equipment.
- Methods in semisolid state, where the drug-CD physical mixture is kneaded with a small volume of water or ethanol/water mixture to obtain a homogeneous pasty product and continued until a back a powder is gotten and then dried to completely remove the solvent (KN) [31,32]. However, such a method often results in only partial drug/CD complexation [14].
- Methods in the solid state, where CD complexation is achieved by microwave irradiation (MWI) [33] or by gentle heating at temperatures below the fusion point of the compounds in a sealed container, eventually in the presence of a minimum water amount, according to the so-called “sealed-heating” method (SH) [34,35] or by mechanochemical activation through grinding (GR) with different types of mills [36,37] of the drug/cyclodextrin physical mixture. The drawback of both MW and SH technologies is in the possibility of drug degradation during microwave irradiation or heating [38]. Grinding, on the other hand, offers the advantages to be a simple, fast and highly effective method for the preparation of drug/CD inclusion complexes in the solid state, generally not requiring the use of organic solvents [39]. This provides additional advantages, avoiding problems and limitations of solution-based techniques, such as solubility issues, solvent complexation, or solvolysis. Therefore, grinding represents an economically and environmentally desirable technology and it will be the focus of this review.
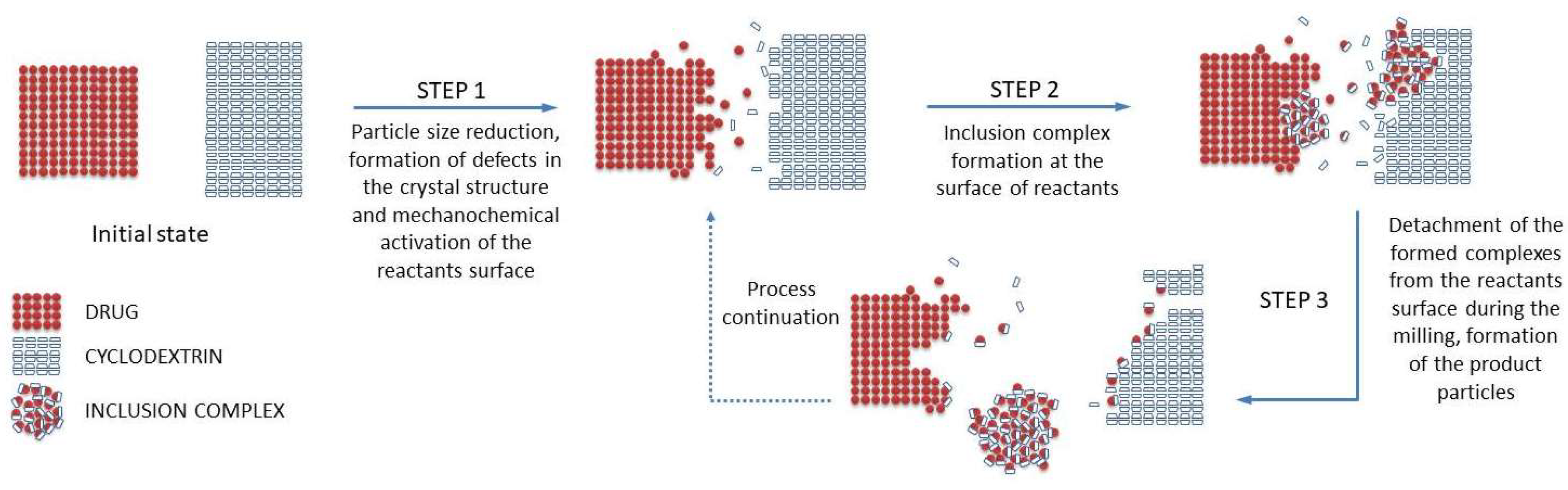
2. Mechanism of the Inclusion Complex Formation in the Solid State by Grinding
3. Challenges in the Analytical Characterization of Cyclodextrin Complexes in the Solid State
4. Process Variables of the Inclusion Complex Formation in the Solid State by Grinding
4.1. Inclusion Complex Preparation by Mortar and Pestle Grinding
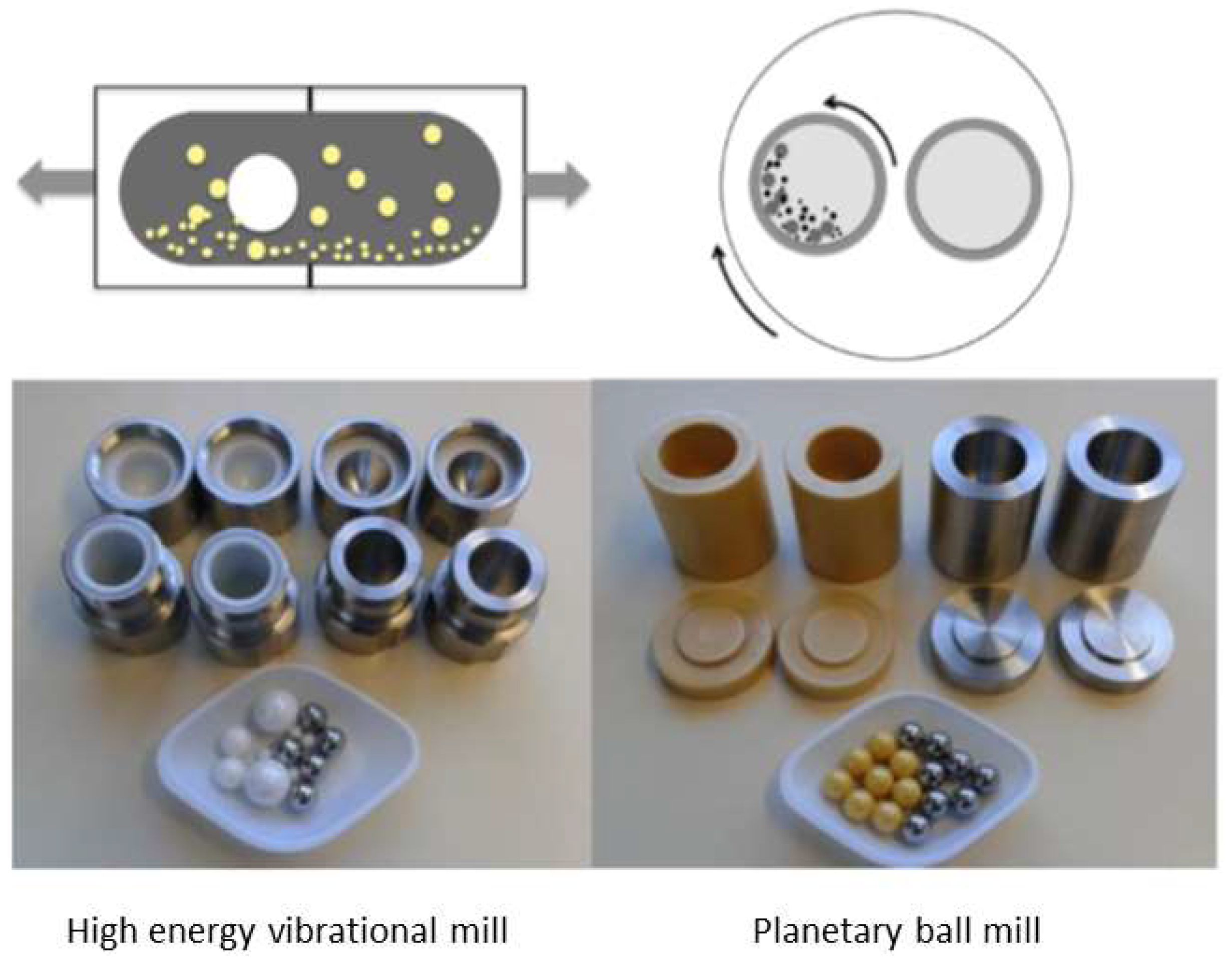
4.2. Inclusion Complex Preparation by Grinding in High Energy Vibrational Mills
4.3. Inclusion Complex Preparation by Grinding in Planetary Mills
4.4. Inclusion Complex Preparation by Grinding in Desktop Tumbling Ball Mills
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kurkov, S.V.; Loftsson, T. Cyclodextrins. Int. J. Pharm. 2013, 453, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Jansook, P.; Ogawa, N.; Loftsson, T. Cyclodextrins: Structure, physicochemical properties and pharmaceutical applications. Int. J. Pharm. 2018, 535, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Popielec, A.; Loftsson, T. Effects of cyclodextrins on the chemical stability of drugs. Int. J. Pharm. 2017, 531, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins: Effects on drug permeation through biological membranes. J. Pharm. Pharmacol. 2011, 63, 1119–1135. [Google Scholar] [CrossRef] [PubMed]
- Arima, H.; Higashi, T.; Motoyama, K. Improvement of the bitter taste of drugs by complexation with cyclodextrins: Applications, evaluations and mechanisms. Ther. Deliv. 2012, 3, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins: Basic science and product development. J. Pharm. Pharmacol. 2010, 62, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Marques, H.M.C. A review on cyclodextrin encapsulation of essential oils and volatiles. Flavour Fragr. J. 2010, 25, 313–326. [Google Scholar] [CrossRef]
- Salústio, P.J.; Pontes, P.; Conduto, C.; Sanches, I.; Carvalho, C.; Arrais, J.; Marques, H.M.C. Advanced Technologies for Oral Controlled Release: Cyclodextrins for Oral Controlled Release. AAPS PharmSciTech 2011, 12, 1276–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ma, P.X. Cyclodextrin-based supramolecular systems for drug delivery: Recent progress and future perspective. Adv. Drug Deliv. Rev. 2013, 65, 1215–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gref, R.; Duchêne, D. Cyclodextrins as “smart” components of polymer nanoparticles. J. Drug Deliv. Sci. Technol. 2012, 22, 223–233. [Google Scholar] [CrossRef]
- Adeoye, O.; Cabral-Marques, H. Cyclodextrin nanosystems in oral drug delivery: A mini review. Int. J. Pharm. 2017, 531, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Jung, H.; Li, X. Drug Delivery Approaches in Addressing Clinical Pharmacology-Related Issues: Opportunities and Challenges. AAPS J. 2015, 17, 1327–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchêne, D.; Bochot, A. Thirty years with cyclodextrins. Int. J. Pharm. 2016, 514, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.S.; Dressman, J.B.; Loftsson, T.; Moya-Ortega, M.D.; Alvarez-Lorenzo, C.; Concheiro, A. Pharmacokinetics of cyclodextrins and drugs after oral and parenteral administration of drug/cyclodextrin complexes. J. Pharm. Pharmacol. 2016, 68, 544–555. [Google Scholar] [CrossRef]
- Mura, P. Analytical techniques for characterization of cyclodextrin complexes in aqueous solution: A review. J. Pharm. Biomed. Anal. 2014, 101, 238–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mura, P. Analytical techniques for characterization of cyclodextrin complexes in the solid state: A review. J. Pharm. Biomed. Anal. 2015, 113, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, N.; Takahashi, C.; Yamamoto, H. Physicochemical characterization of cyclodextrin-drug interactions in the solid state and the effect of water on these interactions. J. Pharm. Sci. 2015, 104, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Szente, L.; Szemán, J.; Sohajda, T. Analytical characterization of cyclodextrins: History, official methods and recommended new techniques. J. Pharm. Biomed. Anal. 2016, 130, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Mura, P.; Faucci, M.; Manderioli, A.; Bramanti, G. Influence of the preparation method on the physicochemical properties of binary systems of econazole with cyclodextrins. Int. J. Pharm. 1999, 193, 85–95. [Google Scholar] [CrossRef]
- Mohit, V.; Harshal, G.; Neha, D.; Vilasrao, K.; Rajashree, H. A comparative study of complexation methods for cefdinir-hydroxypropyl-β-cyclodextrin system. J. Incl. Phenom. Macrocycl. Chem. 2011, 71, 57–66. [Google Scholar] [CrossRef]
- Miller, L.A.; Carrier, R.L.; Ahmed, I. Practical considerations in development of solid dosage forms that contain cyclodextrin. J. Pharm. Sci. 2007, 96, 1691–1707. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.S.; Bramhane, D.M.; Nagarsenker, M.S. Repaglinide-Cyclodextrin complexes: Preparation, Characterization and in vivo evaluation of antihyperglycemic activity. J. Incl. Phenom. Macrocycl. Chem. 2011, 70, 217–225. [Google Scholar] [CrossRef]
- Maestrelli, F.; Cirri, M.; Mennini, N.; Zerrouk, N.; Mura, P. Improvement of oxaprozin solubility and permeability by the combined use of cyclodextrin, chitosan, and bile components. Eur. J. Pharm. Biopharm. 2011, 78, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Jablan, J.; Szalontai, G.; Jug, M. Comparative analysis of zaleplon complexation with cyclodextrins and hydrophilic polymers in solution and in solid state. J. Pharm. Biomed. Anal. 2012, 71. [Google Scholar] [CrossRef] [PubMed]
- Bragagni, M.; Maestrelli, F.; Mura, P. Physical chemical characterization of binary systems of prilocaine hydrochloride with triacetyl-β-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2010, 68, 437–445. [Google Scholar] [CrossRef]
- Jug, M.; Bečirević-Laćan, M.; Bengez, S. Novel cyclodextrin-based film formulation intended for buccal delivery of atenolol Cyclodextrin-based film formulation for buccal delivery of atenolol. Drug Dev. Ind. Pharm. 2009, 35. [Google Scholar] [CrossRef] [PubMed]
- Promzeleva, M.; Volkova, T.; Proshin, A.; Siluykov, O.; Mazur, A.; Tolstoy, P.; Ivanov, S.; Kamilov, F.; Terekhova, I. Improved Biopharmaceutical Properties of Oral Formulations of 1,2,4-Thiadiazole Derivative with Cyclodextrins: In Vitro and in Vivo Evaluation. ACS Biomater. Sci. Eng. 2018, 4, 491–501. [Google Scholar] [CrossRef]
- Aree, T.; Chaichit, N. A new crystal form of β-cyclodextrin–ethanol inclusion complex: Channel-type structure without long guest molecules. Carbohydr. Res. 2003, 338, 1581–1589. [Google Scholar] [CrossRef]
- Huang, Y.; Zu, Y.; Zhao, X.; Wu, M.; Feng, Z.; Deng, Y.; Zu, C.; Wang, L. Preparation of inclusion complex of apigenin-hydroxypropyl-β-cyclodextrin by using supercritical antisolvent process for dissolution and bioavailability enhancement. Int. J. Pharm. 2016, 511, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Rudrangi, S.R.S.; Kaialy, W.; Ghori, M.U.; Trivedi, V.; Snowden, M.J.; Alexander, B.D. Solid-state flurbiprofen and methyl-β-cyclodextrin inclusion complexes prepared using a single-step, organic solvent-free supercritical fluid process. Eur. J. Pharm. Biopharm. 2016, 104, 164–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.-L.; Lin, S.-Y.; Lin, C.-C.; Hsu, C.-H.; Wu, T.-K.; Huang, Y.-T. Mechanical grinding effect on thermodynamics and inclusion efficiency of loratadine-cyclodextrin inclusion complex formation. Carbohydr. Polym. 2011, 87, 512–517. [Google Scholar] [CrossRef]
- Mennini, N.; Bragagni, M.; Maestrelli, F.; Mura, P. Physico-chemical characterization in solution and in the solid state of clonazepam complexes with native and chemically-modified cyclodextrins. J. Pharm. Biomed. Anal. 2014, 89, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Cirri, M.; Maestrelli, F.; Mennini, N.; Mura, P. Influence of the preparation method on the physical-chemical properties of ketoprofen-cyclodextrin-phosphatidylcholine ternary systems. J. Pharm. Biomed. Anal. 2009, 50, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Maestrelli, F.; Cecchi, M.; Cirri, M.; Capasso, G.; Mennini, N.; Mura, P. Comparative study of oxaprozin complexation with natural and chemically-modified cyclodextrins in solution and in the solid state. J. Incl. Phenom. Macrocycl. Chem. 2009, 63, 17–25. [Google Scholar] [CrossRef]
- Higashi, K.; Tozuka, Y.; Moribe, K.; Yamamoto, K. Salicylic Acid/γ-Cydodextrin 2:1 and 4:1 Complex Formation by Sealed-Heating Method. J. Pharm. Sci. 2010, 99, 4192–4200. [Google Scholar] [CrossRef] [PubMed]
- Cugovčan, M.; Jablan, J.; Lovrić, J.; Cinčić, D.; Galić, N.; Jug, M. Biopharmaceutical characterization of praziquantel cocrystals and cyclodextrin complexes prepared by grinding. J. Pharm. Biomed. Anal. 2017, 137, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Brusnikina, M.; Silyukov, O.; Chislov, M.; Volkova, T.; Proshin, A.; Mazur, A.; Tolstoy, P.; Terekhova, I. Effect of cyclodextrin complexation on solubility of novel anti-Alzheimer 1,2,4-thiadiazole derivative. J. Therm. Anal. Calorim. 2017, 130, 443–450. [Google Scholar] [CrossRef]
- Prekodravac, B.; Damm, M.; Kappe, C.O. Microwave-assisted forced degradation using high-throughput microtiter platforms. J. Pharm. Biomed. Anal. 2011, 56, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Colombo, I.; Grassi, G.; Grassi, M. Drug mechanochemical actvation. J. Pharm. Sci. 2009, 398, 3961–3986. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.; Eddleston, M.D. Introductory Lecture: Mechanochemistry, a versatile synthesis strategy for new materials. Faraday Discuss. 2014, 170, 9–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández, J.G.; Bolm, C. Altering Product Selectivity by Mechanochemistry. J. Org. Chem. 2017, 82, 4007–4019. [Google Scholar] [CrossRef] [PubMed]
- Friščić, T. Supramolecular concepts and new techniques in mechanochemistry: Cocrystals, cages, rotaxanes, open metal–organic frameworks. Chem. Soc. Rev. 2012, 41, 3493–3510. [Google Scholar] [CrossRef] [PubMed]
- Bikiaris, D.N. Solid dispersions, Part I: Recent evolutions and future opportunities in manufacturing methods for dissolution rate enhancement of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2011, 8, 1501–1519. [Google Scholar] [CrossRef] [PubMed]
- Friščič, T.; Jones, W. Recent advances in understanding the mechanism of cocrystal formation via grinding. Cryst. Growth Des. 2009, 9, 1621–1637. [Google Scholar] [CrossRef]
- Aigner, Z.; Hassan, H.B.; Berkesi, O.; Kata, M.; Erős, I. Thermoanalytical, FTIR and X-ray studies of gemfibrozil-cyclodextrin complexes. J. Therm. Anal. Calorim. 2005, 81, 267–272. [Google Scholar] [CrossRef]
- Hasa, D.; Schneider Rauber, G.; Voinovich, D.; Jones, W. Cocrystal Formation through Mechanochemistry: From Neat and Liquid-Assisted Grinding to Polymer-Assisted Grinding. Angew. Chem. Int. Ed. 2015, 7371–7375. [Google Scholar] [CrossRef] [PubMed]
- Cirri, M.; Bragagni, M.; Mennini, N.; Mura, P. Development of a new delivery system consisting in “drug–In cyclodextrin–In nanostructured lipid carriers” for ketoprofen topical delivery. Eur. J. Pharm. Biopharm. 2012, 80, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Wongmekiat, A.; Tozuka, Y.; Oguchi, T.; Yamamoto, K. Formation of fine drug particles by cogrinding with cyclodextrins. I. The use of β-cyclodextrin anhydrate and hydrate. Pharm. Res. 2002, 19, 1867–1872. [Google Scholar] [CrossRef] [PubMed]
- Wongmekiat, A.; Tozuka, Y.; Oguchi, T.; Yamamoto, K. Formation of fine drug particle by cogrinding with cyclodextrins: Part II. The influence of moisture condition during cogrinding process on fine particle formation. Int. J. Pharm. 2003, 265, 85–93. [Google Scholar] [CrossRef]
- Wongmekiat, A.; Yoshimatsu, S.; Tozuka, Y.; Moribe, K.; Yamamoto, K. Investigation of drug nanoparticle formation by co-grinding with cyclodextrins: Studies for indomethacin, furosemide and naproxen. J. Incl. Phenom. Macrocycl. Chem. 2006, 56, 29–32. [Google Scholar] [CrossRef]
- Sun, Y.; Du, L.; Liu, Y.; Li, X.; Li, M.; Jin, Y.; Qian, X. Transdermal delivery of the in situ hydrogels of curcumin and its inclusion complexes of hydroxypropyl-β-cyclodextrin for melanoma treatment. Int. J. Pharm. 2014, 469, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Li, L.; Liu, Y.; Wen, S.; Guo, Y.; Niu, X. Study on the Inclusion Complex of Rutin/Sulfobutylether-β-Cyclodextrin. Adv. Mater. Res. 2012, 456, 1177–1181. [Google Scholar] [CrossRef]
- Gu, F.G.; Wang, Y.; Meng, G.D.L.; Han, H.B.; Wu, C.Z. Investigation of a fenofibrate-hydroxypropyl-β-cyclodextrin system prepared by a co-grinding method. Pharmazie 2012, 67, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhai, Y.; Yan, J.; Wang, L.; Xu, K.; Li, H. Effect of preparation processes and structural insight into the supermolecular system: Bisacodyl and β-cyclodextrin inclusion complex. Mater. Sci. Eng. C 2016, 58, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Inoue, Y.; Tsunoda, Y.; Murata, I.; Isshiki, Y.; Kondo, S.; Kanamoto, I. Effect of γ-cyclodextrin derivative complexation on the physicochemical properties and antimicrobial activity of hinokitiol. J. Incl. Phenom. Macrocycl. Chem. 2015, 83, 177–186. [Google Scholar] [CrossRef]
- Zhang, J.; He, D.; Tan, Q.; Wu, M.; Yang, L.; Ren, Y.; Liu, J. Characterization, activity, and computer modeling of a molecular inclusion complex containing rifaldazine. Int. J. Nanomed. 2013, 477. [Google Scholar] [CrossRef] [PubMed]
- Aigner, Z.; Berkesi, O.; Farkas, G.; Szabó-Révész, P. DSC, X-ray and FTIR studies of a gemfibrozil/dimethyl-β-cyclodextrin inclusion complex produced by co-grinding. J. Pharm. Biomed. Anal. 2012, 57, 62–67. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Deng, P.; Yang, L.; Tan, Q.; Liu, J.; Yang, M.; Zhang, J. Molecular encapsulation of rifampicin as an inclusion complex of hydroxypropyl-β-cyclodextrin: Design; characterization and in vitro dissolution. Colloids Surf. B Biointerfaces 2013, 103, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, N.; Higashi, K.; Nagase, H.; Endo, T.; Moribe, K.; Loftsson, T.; Yamamoto, K.; Ueda, H. Effects of cogrinding with β-cyclodextrin on the solid state fentanyl. J. Pharm. Sci. 2010, 99, 5019–5029. [Google Scholar] [CrossRef] [PubMed]
- Anzai, K.; Mizoguchi, J.; Yanagi, T.; Hirayama, F.; Arima, H.; Uekama, K. Improvement of dissolution properties of a new Helicobacter pylori eradicating agent (TG44) by inclusion complexation with beta-cyclodextrin. Chem. Pharm. Bull. 2007, 55, 1466–1470. [Google Scholar] [CrossRef] [PubMed]
- Mura, P.; Furlanetto, S.; Cirri, M.; Maestrelli, F.; Corti, G.; Pinzauti, S. Interaction of naproxen with ionic cyclodextrins in aqueous solution and in the solid state. J. Pharm. Biomed. Anal. 2005, 37, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Mura, P.; Bettinetti, G.P.; Cirri, M.; Maestrelli, F.; Sorrenti, M.; Catenacci, L. Solid-state characterization and dissolution properties of Naproxen-Arginine-Hydroxypropyl-β-cyclodextrin ternary system. Eur. J. Pharm. Biopharm. 2005, 59, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Caira, M.R.; Dodds, D.R.; Nassimbeni, L.R. Polymorphism and Cyclodextrin Inclusion of Salbutamol Laurate. J. Therm. Anal. Calorim. 2002, 68, 647–655. [Google Scholar] [CrossRef]
- Bettinetti, G.; Mura, P.; Faucci, M.T.; Sorrenti, M.; Setti, M. Interaction of naproxen with noncrystalline acetyl beta- and acetyl gamma-cyclodextrins in the solid and liquid state. Eur. J. Pharm. Sci. 2002, 15, 21–29. [Google Scholar] [CrossRef]
- Mitrevej, A.; Sinchaipanid, N.; Junyaprasert, V.; Warintornuwat, L. Effect of grinding of beta-cyclodextrin and glibenclamide on tablet properties. 1. In vitro. Drug Dev. Ind. Pharm. 1996, 22, 1237–1241. [Google Scholar] [CrossRef]
- Fumić, B.; Jablan, J.; Cinčić, D.; Zovko Končić, M.; Jug, M. Cyclodextrin encapsulation of daidzein and genistein by grinding: Implication on the glycosaminoglycan accumulation in mucopolysaccharidosis type II and III fibroblasts. J. Microencapsul. 2017, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Majewska, K.; Skwierawska, A.; Kamińska, B.; Prześniak-Welenc, M. Improvement of opipramol base solubility by complexation with β-cyclodextrin. Supramol. Chem. 2018, 30, 20–31. [Google Scholar] [CrossRef]
- Ali, H.R.H.; Saleem, I.Y.; Tawfeek, H.M. Insight into inclusion complexation of indomethacin nicotinamide cocrystals. J. Incl. Phenom. Macrocycl. Chem. 2016, 84, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Borba, P.A.A.; Pinotti, M.; Andrade, G.R.S.; Da Costa, N.B.; Olchanheski, L.R.; Fernandes, D.; De Campos, C.E.M.; Stulzer, H.K. The effect of mechanical grinding on the formation, crystalline changes and dissolution behaviour of the inclusion complex of telmisartan and β-cyclodextrins. Carbohydr. Polym. 2015, 133, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.I.; Braga, T.M.; Silva, P.; Fernandes, J.A.; Ribeiro-Claro, P.; Lopes, M.D.F.S.; Paz, F.A.A.; Braga, S.S. Chloramphenicol·cyclodextrin inclusion compounds: Co-dissolution and mechanochemical preparations and antibacterial action. CrystEngComm 2013, 15, 2822–2834. [Google Scholar] [CrossRef]
- Zong, W.; Bi, S. The preparation and characterization of inclusion complex of ursolic acid with γ-cyclodextrin. In Proceedings of the 2011 7th International Conference on MEMS NANO, and Smart Systems (ICMENS 2011), Kuala Lumpur, Malaysia, 4–6 November 2011; Volume 403–408, pp. 712–716. [Google Scholar] [CrossRef]
- Corti, G.; Capasso, G.; Maestrelli, F.; Cirri, M.; Mura, P. Physical-chemical characterization of binary systems of metformin hydrochloride with triacetyl-β-cyclodextrin. J. Pharm. Biomed. Anal. 2007, 45, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Cirri, M.; Maestrelli, F.; Furlanetto, S.; Mura, P. Solid-state characterization of glyburide-cyclodextrin co-ground products. J. Therm. Anal. 2004, 77, 413–422. [Google Scholar] [CrossRef]
- Mura, P.; Faucci, M.T.; Maestrelli, F.; Furlanetto, S.; Pinzauti, S. Characterization of physicochemical properties of naproxen systems with amorphous β-cyclodextrin-epichlorohydrin polymers. J. Pharm. Biomed. Anal. 2002, 29, 1015–1024. [Google Scholar] [CrossRef]
- Mura, P.; Faucci, M.T.; Manderioli, A.; Bramanti, G. Multicomponent systems of econazole with hydroxyacids and cyclodextrins. J. Incl. Phenom. 2001, 39, 131–138. [Google Scholar] [CrossRef]
- Mura, P.; Faucci, M.T.; Parrini, P.L.; Furlanetto, S.; Pinzauti, S. Influence of the preparation method on the physicochemical properties of ketoprofen-cyclodextrin binary systems. Int. J. Pharm. 1999, 179, 117–128. [Google Scholar] [CrossRef]
- Jablan, J.; Bačić, I.; Kujundžić, N.; Jug, M. Zaleplon co-ground complexes with natural and polymeric β-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2013, 76. [Google Scholar] [CrossRef]
- Mura, P.; Faucci, M.T.; Bettinetti, G.P. The influence of polyvinylpyrrolidone on naproxen complexation with hydroxypropyl-β-cyclodextrin. Eur. J. Pharm. Sci. 2001, 13, 187–194. [Google Scholar] [CrossRef]
- Jug, M.; Kosalec, I.; Maestrelli, F.; Mura, P. Analysis of triclosan inclusion complexes with β-cyclodextrin and its water-soluble polymeric derivative. J. Pharm. Biomed. Anal. 2011, 54, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Jug, M.; Mennini, N.; Kövér, K.E.; Mura, P. Comparative analysis of binary and ternary cyclodextrin complexes with econazole nitrate in solution and in solid state. J. Pharm. Biomed. Anal. 2014, 91, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Jug, M.; Maestrelli, F.; Bragagni, M.; Mura, P. Preparation and solid-state characterization of bupivacaine hydrochloride cyclodextrin complexes aimed for buccal delivery. J. Pharm. Biomed. Anal. 2010, 52, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.; Kakumanu, V.K.; Bansal, A.K. Analytical techniques for quantification of amorphous/crystalline phases in pharmaceutical solids. J. Pharm. Sci. 2006, 95, 1641–1665. [Google Scholar] [CrossRef] [PubMed]
- Vogt, F.G.; Strohmeier, M. 2D Solid-State NMR Analysis of Inclusion in Drug–Cyclodextrin Complexes. Mol. Pharm. 2012, 9, 3357–3374. [Google Scholar] [CrossRef] [PubMed]
- Redenti, E.; Peveri, T.; Zanol, M.; Ventura, P.; Gnappi, G.; Montenero, A. A study on the differentiation between amorphous piroxicam:β-cyclodextrin complex and a mixture of the two amorphous components. Int. J. Pharm. 1996, 129, 289–294. [Google Scholar] [CrossRef]
- Anzai, K.; Kono, H.; Mizoguchi, J.; Yanagi, T.; Hirayama, F.; Arima, H.; Uekama, K. Two-dimensional 13C–1H heteronuclear correlation NMR spectroscopic studies for the inclusion complex of cyclomaltoheptaose (β-cyclodextrin) with a new Helicobacter pylori eradicating agent (TG44) in the amorphous state. Carbohydr. Res. 2006, 341, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Takacs, L. The historical development of mechanochemistry. Chem. Soc. Rev. 2013, 42, 7649–7659. [Google Scholar] [CrossRef] [PubMed]
- Bettinetti, G.; Sorrenti, M.; Negri, A.; Setti, M.; Mura, P.; Melani, F. Interaction of naproxen with alpha-cyclodextrin and its noncyclic analog maltohexaose. Pharm. Res. 1999, 16, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Veiga, M.D.; Ahsan, F.; Merino, M. Differential scanning calorimetry as an analytical tool in determining the interaction between drug and cyclodextrin. J. Therm. Anal. Calorim. 1999, 73, 635–646. [Google Scholar]
- Mixer Mill MM 200–RETSCH–High Performance and Great Flexibility. Available online: https://www.retsch.com/products/milling/ball-mills/mixer-mill-mm-200/function-features/ (accessed on 24 May 2018).
- 8000D Mixer/Mill—Dual Clamp, High-Energy Ball Mill, Mechanical Alloying. Grinds Hard Samples|SPEX SamplePrep. Available online: https://www.spexsampleprep.com/8000D-mixermill (accessed on 24 May 2018).
- Howard, J.L.; Cao, Q.; Browne, D.L. Mechanochemistry as an emerging tool for molecular synthesis: What can it offer? Chem. Sci. 2018, 9, 3080–3094. [Google Scholar] [CrossRef] [PubMed]
- Mura, P.; Faucci, M.T.; Parrini, P.L. Effects of grinding with microcrystalline cellulose and cyclodextrins on the ketoprofen physicochemical properties. Drug Dev. Ind. Pharm. 2001, 27, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Hsu, C.H.; Sheu, M.T. Curve-fitting FTIR studies of loratadine/hydroxypropyl-β-cyclodextrin inclusion complex induced by co-grinding process. J. Pharm. Biomed. Anal. 2010, 53, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Corvi Mora, P.; Cirri, M.; Allolio, B.; Carli, F.; Mura, P. Enhancement of Dehydroepiandrosterone Solubility and Bioavailability by Ternary Complexation with α-Cyclodextrin and Glycine. J. Pharm. Sci. 2003, 92, 2177–2184. [Google Scholar] [CrossRef] [PubMed]
- Mennini, N.; Maestrelli, F.; Cirri, M.; Mura, P. Analysis of physicochemical properties of ternary systems of oxaprozin with randomly methylated-ß-cyclodextrin and L-arginine aimed to improve the drug solubility. J. Pharm. Biomed. Anal. 2016, 129, 350–358. [Google Scholar] [CrossRef] [PubMed]
- James, S.L.; Adams, C.J.; Bolm, C.; Braga, D.; Collier, P.; Frisčc, T.; Grepioni, F.; Harris, K.D.M.; Hyett, G.; Jones, W.; et al. Mechanochemistry: Opportunities for new and cleaner synthesis. Chem. Soc. Rev. 2012, 41, 413–447. [Google Scholar] [CrossRef] [PubMed]
- Planetary Ball Mill PM 200–RETSCH–Short Grinding Times. Available online: https://www.retsch.com/products/milling/ball-mills/planetary-ball-mill-pm-200/function-features/ (accessed on 28 May 2018).
- Iwata, M.; Fukami, T.; Kawashima, D.; Sakai, M.; Furuishi, T.; Suzuki, T.; Tomono, K.; Ueda, H. Effectiveness of mechanochemical treatment with cyclodextrins on increasing solubility of glimepiride. Pharmazie 2009, 64, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Tang, W.; Kong, R.; Zhu, X. Inclusion Complex of α -Lipoic Acid Containing Alkalizer for Improving the Solubility and Stability Prepared by Co-grinding. Indian J. Pharm. Sci. 2017, 79, 544–552. [Google Scholar] [CrossRef]
- Li, S.; Lin, X.; Xu, K.; He, J.; Yang, H.; Li, H. Co-grinding Effect on Crystalline Zaltoprofen with β-cyclodextrin/Cucurbit[7]uril in Tablet Formulation. Sci. Rep. 2017, 7, 45984. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Drug | CD 1 | Drug/CD Ratio 2 | Grinding Conditions | Properties of the Obtained Product | Reference |
|---|---|---|---|---|---|
| Chloramphenicol (mp 155.23 °C) | β-CD | 1:1 | up to 120 min | Partial inclusion (ca. 32%) after 120 min grinding | [70] |
| Gemfibrozil (mp. 59.25 °C) | DIMEB | 1:1 | up to 35 min | Amorphous product after 35 min grinding | [57] |
| Naproxen (mp 153.4 °C) | βCDEPI βCDEPS | 50/50, 20/80, 15/85, 10/90 (w/w) | up to 40 min | Amorphous product with enhanced dissolution properties | [74] |
| Naproxen (mp. 153.4 °C) | αCD maltohexaose | 0.3–0.1 (w/w) | up to 30 min | Pseudo-inclusion complex formation with maltohexaose, partial interaction with αCD | [87] |
| Trimetoprim (mp 170 °C) Sulphadiazine (mp 260.6 °C) Sulphamethoxazole (mp 201 °C) | αCD βCD γCD RAMEB DIMEB | 1:1 | 15 min | Amorphous products with RAMEB | [88] |
| Rifaldazine (mp 259 °C) | βCD | 1:1 | 3 min trituration followed by 30 min grinding | Amorphous product, 4.4 times higher solubility; inclusion complexation confirmed by FTIR | [56] |
| Rifampicin (mp 259 °C) | HPβCD | 1:1 | 3 min trituration followed by 30 min grinding | Amorphous product with 2.5 times higher solubility | [58] |
| Drug | CD 1 | Drug/CD Ratio 2 | Grinding Conditions | Properties of the Obtained Product | Reference |
|---|---|---|---|---|---|
| Bupivacaine hydrochloride (mp 128.19 °C) | βCD βCD-EPI | 1:1 | 30–60 min at 24 Hz, ambient conditions | Amorphous products with enhanced dissolution properties by GR with βCD-EPI; partially crystalline by GR with βCD | [81] |
| Clonazepam (mp 239.1 °C) | αCD HPαCD βCD HPβCD RAMEB γCD HPγCD | 1:1 | 30 min at 24 Hz, ambient conditions | 61.1 and 16.4% RDC for αCD and HPαCD GR, respectively; amorphous with other CDs; the most efficient was RAMEB (dissolution rate rank GR > COE ≈ KN >PM) | [32] |
| Daidzein (mp 336.4 °C) Genistein (mp 308.5 °C) | HPβCD SBEβCD | 1:1 | 30 min at ambient conditions in 10 mL SS jars with two 7 mm SS balls | Partially crystalline products; SBEβCD more efficient as amorphizing agent | [66] |
| Econazole (mp 89.0 °C) | αCD | 1:1 | 60 min at 24 Hz, ambient conditions, batch size 1 g | Partially amorphous system obtained by GR; completely amorphous complex by FD. | [75] |
| Econazole nitrate (mp 163.78 °C) | HEβCD HPβCD SBEβCD EPIβCD | 1:1 | 15–60 min at 24 Hz, ambient conditions | RDC always decreased as a function of grinding time; amorphous product with HPβCD and SBEβCD after 60 min GR | [80] |
| Glyburide (mp 175.3 °C) | αCD βCD HPβCD RAMEB γCD | 1:1 | 0–60 min at 24 Hz, ambient conditions | Amorphous products obtained in all cases after 60 min, but with different sensitivity to mechanochemical activation. No drug recrystallization during storage. | [73] |
| Indomethacin nicotinamide cocrystals (mp 128.5 °C) | βCD HPβCD | 1:1 | 15 min | Partial complexation for GR products with βCD and HPβCD. Amorphous system with enhanced dissolution rate by COE with HPβCD | [68] |
| Ketoprofen (mp 94.6 °C) | βCD-EPI CMβCD-EPI | 10:90 (w/w) | 10–120 min at 24 Hz ambient temperature with or without 10% water | Complete amorphization by dry GR with βCD-EPI (60 min) and CMβCD-EPI (30 min). In the moist conditions complete amorphization after 30 min GR with βCD-EPI, 120 min with CMβCD-EPI. | [47] |
| Ketoprofen (mp 94.6 °C) | βCD RAMEB | 1:1 | 10–60 min at frequency from 15 to 24 Hz and ambient temperature | Complete amorphization after 60 min GR at 15 Hz or 30 min at 25 Hz with RAMEB; partial crystalline systems with βCD regardless applied frequency. | [92] |
| Loratadine (mp 136.1 °C) | HPβCD | 1:1 or 1:2 | 25 mL SS jar with two 15 mm SS balls, at 15 Hz up to 30 min. Batch size 0.2 g | Complete amorphization after 7 min GR with HPβCD at 1:1 ratio and after 15 min at 1:2 ratio. Inclusion complex formation verified by FTIR. | [93] |
| Loratadine (mp 136.1 °C) | βCD HPβCD | 1:1 | 25 mL SS jar with two 15 mm SS balls at 15 Hz up to 30 min. Batch size 0.2 g | Complete amorphization after 7 and 15 min GR with HPβCD and βCD, respectively. Amorphization process followed zero-order kinetics. | [31] |
| Metformin hydrochloride (mp 231.0 °C) | TAβCD | 1:1 | 30 min at 24 Hz and ambient conditions | Partially crystalline product by GR, completely amorphous by SPD and characterized with the most pronounced sustained release profile | [72] |
| Naproxen (mp 155.9 °C) | SBEβCD TMAβCD | 1:1 | 30 min at 24 Hz and ambient conditions | Amorphous products by GR and FD; FD complex presented almost double dissolution efficiency. SBEβCD was the best carrier. | [61] |
| Naproxen (mp 155.9 °C) | TAβCD TAγCD | 1:1 | 30 min at 24 Hz and ambient conditions | Amorphous products by GR and FD. FD complexes showed faster initial dissolution followed by decline due to supersaturation, not observed for GR products | [64] |
| Oxaprozin (mp 161.3 °C) | βCD DIMEB RAMEB | 1:1 | 30 min at 24 Hz and ambient conditions | Partially crystalline product with βCD, amorphous with DIMEB and RAMEB. Drug dissolution rate increased 7.2, 4.4 and 1.9 times with RAMEB, DIMEB and βCD complexes, respectively. | [34] |
| Telmisartan (mp 265.3 °C) | βCD | 1:2 or 1:3 | 65 mL SS jars with 3 SS ball (two of 6.4 and one of 12.8 mm) at 3.7 ball-to-powder ratio for 7, 15 and 30 min Grinding frequency not stated | Formation of new solid phases in all samples after 30 min GR; 19-fold increase of drug dissolution and rapid and effective antihypertensive effect in rat model | [69] |
| Triclosan (mp 59.48 °C) | βCD βCD-EPI | 1:1 | 10–80 min at 24 Hz and ambient temperature | Complete amorphization after 60 and 80 min GR with βCD-EPI and βCD, respectively. Complexation with βCD-EPI enhanced drug dissolution and antimicrobial activity | [79] |
| Pranlukast hemihydrate (mp 192.1 °C) | βCD βCD 10.5 H2O | 1:2, 1:1, or 2:1 | 10 min Grinding frequency not stated | βCD hydrated GR systems appeared as amorphous stiff mass; those with anhydrous βCD as fine crystalline powder. βCD hydrated GR systems dispersed in water formed a fine suspension (particle size 0.04–0.06 μm) | [48] |
| Pranlukast hemihydrate (mp 192.1 °C) | αCD βCD γCD | 1:2 | 10 min with 0.75–20% of water Grinding frequency not stated | GR with βCD prepared with 13% of water almost completely transferred into fine drug particles after dispersion in water; similar behavior for αCD and γCD GR with 10 and 20% of water. | [49] |
| Praziquantel (mp 142.28 °C) | βCD HPβCD RAMEB SBEβCD | 1:1 | 30 min at 25 HZ in 10 mL SS jars with two 7 mm SS balls; ambient conditions; batch size 200 mg | Partially crystalline product with βCD (RDC 61.63%), amorphous products with other CDs. GR with HPβCD showed 10 fold dissolution rate increase and acceptable chemical stability during storage; substantial drug degradation in other products. | [36] |
| Prilocaine hydrochloride (mp 169.9 °C) | TAβCD | 1:1 | 30 min at 24 Hz and ambient temperature | Partially crystalline product by GR (RDC 28%), amorphous by SPD, with more pronounced sustained release. | [25] |
| Zaleplon (mp 185.27 °C) | βCD βCD-EPI | 1:1 | 10–90 min at 24 Hz and ambient conditions | Superior performance of βCD-EPI vs. βCD (RDC 12.05 and 51.10%, respectively) and 25% faster dissolution rate. Formation of actual inclusion complexes after dissolution in water proved by 1 H-NMR | [77] |
| Drug | CD/Ternary Compound 1 | Drug/CD/Ternary Compound Ratio 2 | Grinding Conditions | Properties of the Obtained Product | Reference |
|---|---|---|---|---|---|
| Dehydroepiandrosterone (mp 150.9 °C) | αCD + glycine | 1:1:2 or 1:2:3 | 60 min at 24 Hz, ambient conditions | Partially crystalline product at 1:1:2 and amorphous at 1:2:3 molar ratio. Superior performance of 1:2:3 complex confirmed in vivo | [94] |
| Econazol (mp 89.0 °C) | αCD + malic acid | 1:1:1 | 60 min at 24 Hz ambient conditions, batch size 1 g | Partially crystalline ternary product prepared by GR; FD was amorphous with superior dissolution properties | [75] |
| Econazol nitrate (mp 123.12 °C) | SBEβCD + citric acid | 1:1:1 | 60 min at 24 Hz and ambient conditions | Amorphous product with superior dissolution profile, without supersaturation phenomenon, as instead observed for binary GR complex | [80] |
| Ketoprofen (mp 96.5 °C) | βCD or RAMEB + EPC | 20:76:4 (w/w) | 15 or 30 min at 24 Hz and ambient conditions | Partially crystalline ternary complexes prepared by GR, amorphous when prepared by MWI that showed superior dissolution profile | [33] |
| Naproxen (155.9 °C) | HPβCD + L-arginine | 1:1:1 | 60 min at 24 Hz and ambient conditions; batch size 500 mg | Enhanced dissolution of ternary complex prepared by GR, further dissolution increase of ternary complex prepared by COE | [62] |
| Naproxen (155.9 °C) | HPβCD + PVP | 1:1 + 15% (w/w) | 60 min at 24 Hz and ambient conditions | Amorphous binary and ternary GR systems; ternary showed 100% higher drug dissolution efficiency than the binary ones | [78] |
| Oxaprozin (mp 161.3 °C) | RAMEB + bile acids/salts + chitosan | 1:1:1 + 0.0625% (w/w) | 30 min at 24 Hz and ambient conditions | Amorphous ternary and quaternary products with enhanced dissolution and permeability. GR products showed superior performance than those prepared by COE and SH | [23] |
| Oxaprozin (mp 161.3 °C) | RAMEB + L-arginin | 1:1:1 | 30 min at 24 Hz and ambient conditions | Amorphous products both in binary and ternary complexes; ternary with 10 times higher relative dissolution rate than binary complexes | [95] |
| Praziquantel (mp 142.28 °C) | HPβCD or RAMEB + malic acid | 1:1:1 | 10 mL SS jars containing two 7 mm SS balls for 30 min at 25 HZ and ambient conditions; batch size 200 mg | Amorphous ternary complexes with solubility lower than corresponding binary ones. Ternary complex formation led to pronounced chemical degradation of the drug | [36] |
| Drug | CD 1 | Drug/CD Ratio 2 | Grinding Conditions | Properties of the Obtained Product | Reference |
|---|---|---|---|---|---|
| 1,2,4-thiadiazole anti-Alzheimer drug candidate (mp not given) | βCD HPβCD | 1:1 | 12 mL agate jar with 5 mm agate balls for 60 min at 600 rpm with pauses to prevent sample overheating | Inclusion complex formation confirmed by 13C MAS CP/TSOO NMR; FD complex presented higher bioavailability | [27] |
| 1,2,4-thiadiazole anti-Alzheimer drug candidate (mp 50 and 102 °C) | βCD HPβCD | 1:1 | 12 mL agate jar with 5 mm agate balls for 60 min at 600 rpm with pauses to prevent sample overheating | Inclusion complex formation confirmed by 13C MAS CP/TSOO NMR; FD complex showed higher solubility | [37] |
| Bisacodyl (mp 136 °C) | βCD | 1:1 | 50 mL agate jar; agate balls (mixture of 15 × 10 mm, 55 × 5 mm, and 40 × 2 mm) for 5 h at 400 rpm alternating milling (5 min) and pause (1 min) periods to prevent sample overheating | Amorphous product with higher solubility than complexes prepared by FD and COE. Inclusion complexation confirmed by 13C CP/MAS NMR | [54] |
| Lipoic acid (mp 62.9 °C) | HPβCD + Na2CO3 | 1:1 and 1:1:1.2 | 25 mL jar and 12 mm balls (1/3 of the tank volume) for 120 min at 150 rpm | Partially crystalline binary system; amorphization achieved by addition of Na2CO3 that enhanced complexation by GR, increasing the compound solubility and chemical stability | [99] |
| Opipramol base (mp 100 °C) | βCD | 1:1 | 12 mL jar for 10 min at 400 rpm | Partial amorphization with GR, complete by FD. Both products showed comparable increase of drug dissolution rate. | [67] |
| Telmisartan (mp 265.3 °C) | βCD | 1:2 and 1:3 | 65 mL steel jar and 3 steel balls (two of 6.4 and one 12.8 mm) at 3.7 ball/powder ratio; milling time 7, 15 and 30 min | Amorphous systems at 1:2 and 1:3 ratio at 30 min GR | [69] |
| Zaltoprofen (mp 138 °C) | βCD cucurbit [7] uryl | 1:1 | 50 mL agate jar with mixture of 10 × 10 mm–20 × 5 mm agate balls for 5 h at room temperature and 400 rpm, with alternate grinding (5 min) and pause (1 min) periods | Amorphous products in both cases with increased dissolution rate. GR of drug alone failed to improve its solubility | [100] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jug, M.; Mura, P.A. Grinding as Solvent-Free Green Chemistry Approach for Cyclodextrin Inclusion Complex Preparation in the Solid State. Pharmaceutics 2018, 10, 189. https://doi.org/10.3390/pharmaceutics10040189
Jug M, Mura PA. Grinding as Solvent-Free Green Chemistry Approach for Cyclodextrin Inclusion Complex Preparation in the Solid State. Pharmaceutics. 2018; 10(4):189. https://doi.org/10.3390/pharmaceutics10040189
Chicago/Turabian StyleJug, Mario, and Paola Angela Mura. 2018. "Grinding as Solvent-Free Green Chemistry Approach for Cyclodextrin Inclusion Complex Preparation in the Solid State" Pharmaceutics 10, no. 4: 189. https://doi.org/10.3390/pharmaceutics10040189
APA StyleJug, M., & Mura, P. A. (2018). Grinding as Solvent-Free Green Chemistry Approach for Cyclodextrin Inclusion Complex Preparation in the Solid State. Pharmaceutics, 10(4), 189. https://doi.org/10.3390/pharmaceutics10040189