Analysis of Vipadenant and Its In Vitro and In Vivo Metabolites via Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometry



Abstract
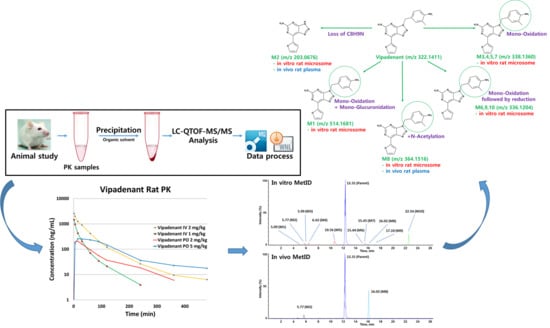
:
1. Introduction
2. Materials and Methods
2.1. Reagents & Chemicals
2.2. Preparation of Stock Solution, Calibration Standard (STD), Quality Control (QC) and Internal Standard (ISTD)
2.3. Sample Preparation—Plasma Samples (Method Qualification Samples and PK Samples)
2.4. Sample Preparation—In Vitro/Vivo Metabolite Identification
2.5. LC-QTOF-MS Condition
2.6. Method Qualification
2.7. Software
2.8. Application for Animal Study
3. Results
3.1. Method Development
3.1.1. Sample Preparation
3.1.2. Optimization of the LC-MS System
3.2. Method Qualification
3.2.1. Calibration Curve, Accuracy, Precision
3.2.2. Preliminary Stability
3.2.3. Species-Dependent Matrix Effect
3.3. Application
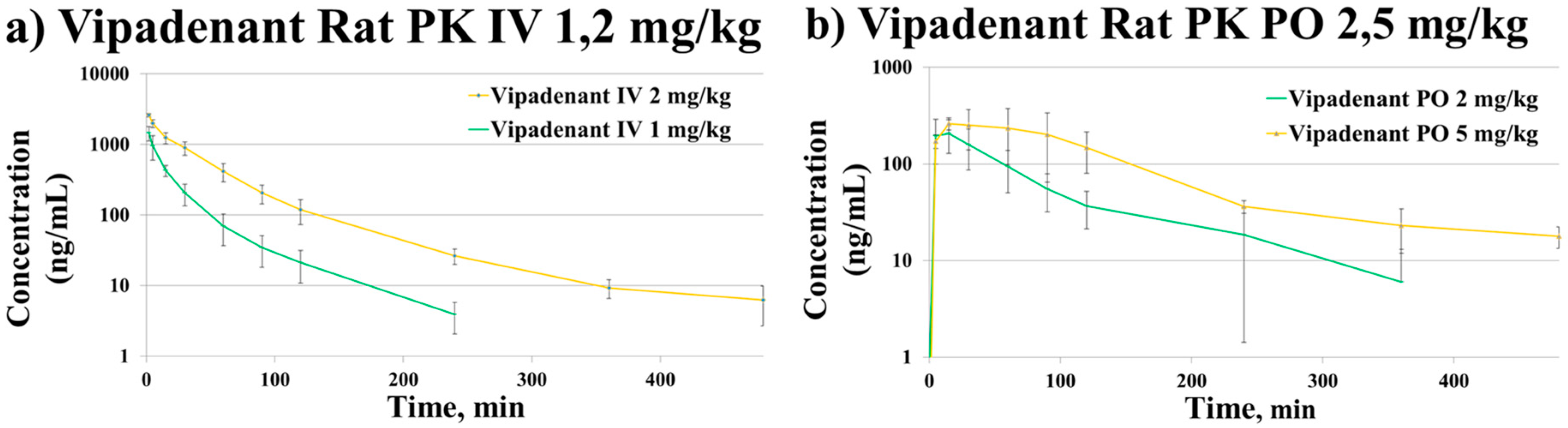
3.3.1. Pharmacokinetic Study
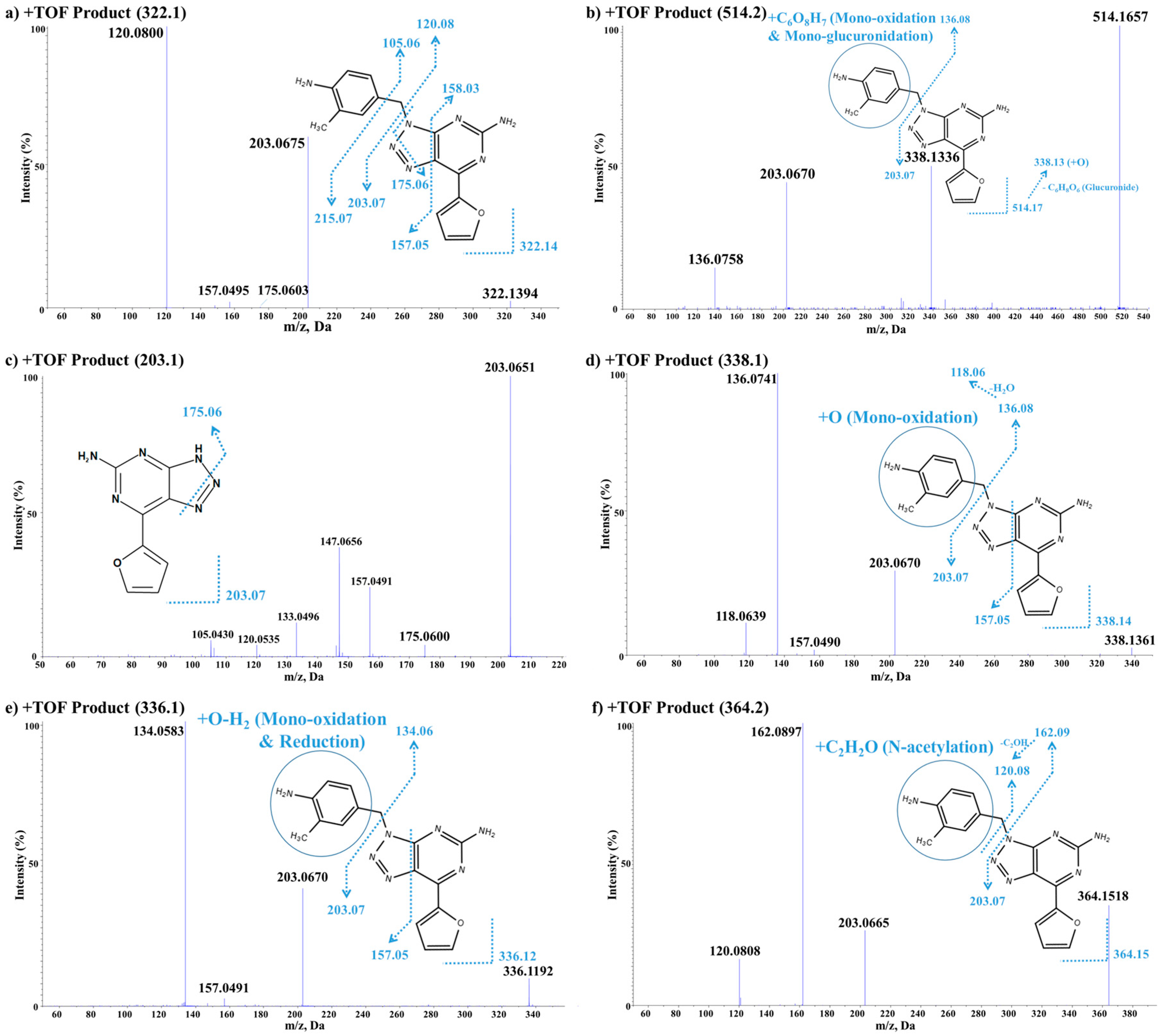
3.3.2. In Vitro/Vivo Metabolite Identification
Vipadenant
Metabolite M1
Metabolite M2
Metabolites M3, M4, M5 and M7
Metabolites M6, M9 and M10
Metabolite M8
4. Discussion & Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Jin, T.; Zhu, Y.; Dai, C. Immune checkpoint therapy in liver cancer. J. Exp. Clin. Cancer Res. 2018, 37, 110. [Google Scholar] [CrossRef]
- Dong, Y.; Sun, Q.; Zhang, X. PD-1 and its ligands are important immune checkpoints in cancer. Oncotarget 2017, 8, 2171–2186. [Google Scholar] [CrossRef] [PubMed]
- Dine, J.; Gordon, R.; Shames, Y.; Kasler, M.K.; Barton-Burke, M. Immune Checkpoint Inhibitors: An Innovation in Immunotherapy for the Treatment and Management of Patients with Cancer. Asia Pac. J. Oncol. Nurs. 2017, 4, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Afzal, M.Z.; Shirai, K. Immune checkpoint inhibitor (anti-CTLA-4, anti-PD-1) therapy alone versus immune checkpoint inhibitor (anti-CTLA-4, anti-PD-1) therapy in combination with anti-RANKL denosumuab in malignant melanoma: A retrospective analysis at a tertiary care center. Melanoma Res. 2018, 28, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Mediavilla-Varela, M.; Castro, J.; Chiappori, A.; Noyes, D.; Hernandez, D.C.; Allard, B.; Stagg, J.; Antonia, S.J. A Novel Antagonist of the Immune Checkpoint Protein Adenosine A2a Receptor Restores Tumor-Infiltrating Lymphocyte Activity in the Context of the Tumor Microenvironment. Neoplasia 2017, 19, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Lo, Y.C.; Powell, J.D. A2aR antagonists: Next generation checkpoint blockade for cancer immunotherapy. Comput. Struct. Biotechnol. J. 2015, 13, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Available online: http://www.vernalis.com/nce-pipeline/oncology/v2006 (accessed on 30 November 2018).
- De Lera Ruiz, M.; Lim, Y.H.; Zheng, J. Adenosine A2A receptor as a drug discovery target. J. Med. Chem. 2014, 57, 3623–3650. [Google Scholar] [CrossRef] [PubMed]
- Pinna, A. Novel investigational adenosine A2A receptor antagonists for Parkinson’s disease. Expert Opin. Investig. Drugs 2009, 18, 1619–1631. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, R.J.; Bamford, S.J.; Botting, R.; Comer, M.; Denny, S.; Gaur, S.; Griffin, M.; Jordan, A.M.; Knight, A.R.; Lerpiniere, J.; et al. Antagonists of the human A(2A) adenosine receptor. 4. Design, synthesis, and preclinical evaluation of 7-aryltriazolo[4,5-d]pyrimidines. J. Med. Chem. 2009, 52, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.; Bleickardt, C.; Mullins, D.; Parker, E.; Hodgson, R. A2A receptor antagonists do not induce dyskinesias in drug-naive or L-dopa sensitized rats. Brain Res. Bull. 2013, 98, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Jones, G.B. Towards next generation adenosine A(2A) receptor antagonists. Curr. Med. Chem. 2014, 21, 3918–3935. [Google Scholar] [CrossRef] [PubMed]
- Shook, B.C. Adenosine A2a Receptor Antagonists. In Novel Therapeutic Approaches to the Treatment of Parkinson’s Disease; Hopkins, C.R., Ed.; Springer: Cham, Switzerland, 2014; Volume 18. [Google Scholar]
- Brooks, D.J.; Papapetropoulos, S.; Vandenhende, F.; Tomic, D.; He, P.; Coppell, A.; O’Neill, G. An open-label, positron emission tomography study to assess adenosine A2A brain receptor occupancy of vipadenant (BIIB014) at steady-state levels in healthy male volunteers. Clin. Neuropharmacol. 2010, 33, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Hop, C.E.; Wang, Z.; Chen, Q.; Kwei, G. Plasma-pooling methods to increase throughput for in vivo pharmacokinetic screening. J. Pharm. Sci. 1998, 87, 901–903. [Google Scholar] [CrossRef] [PubMed]
- Jinno, N.; Ohashi, S.; Tagashira, M.; Kohira, T.; Yamada, S. A simple method to evaluate reactivity of acylglucuronides optimized for early stage drug discovery. Biol. Pharm. Bull. 2013, 36, 1509–1513. [Google Scholar] [CrossRef]
- Costi, M. Enzyme Inhibition in Drug Discovery and Development: The Good and the Bad; Lu, C., Li, A.P., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; Volume 5. [Google Scholar]
- Hill, H. Bioanalysis in drug discovery. Bioanalysis 2011, 3, 2155–2158. [Google Scholar] [CrossRef]
- Gupta, S.; Kesarla, R.; Chotai, N.; Omri, A. Development and validation of reversed-phase HPLC gradient method for the estimation of efavirenz in plasma. PLoS ONE 2017, 12, e0174777. [Google Scholar] [CrossRef]
- Hidau, M.K.; Kolluru, S.; Palakurthi, S. Development and validation of a high-performance liquid chromatography method for the quantification of talazoparib in rat plasma: Application to plasma protein binding studies. Biomed. Chromatogr. 2018, 32. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, J.B.; Han, Y.; Huang, W.; Wang, Q.J. The most convenient and general approach for plasma sample clean-up: Multifunction adsorption and supported liquid extraction. Bioanalysis 2012, 4, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.H.; Park, M.H.; Byeon, J.J.; Kim, Y.C.; Shin, Y.G. A Highly Sensitive Liquid Chromatography-Electrospray Ionization-Time of Flight/Mass Spectrometric Assay for the Quantitation of 4-β-Hydroxycholesterol and Its Application to in vivo Cytochrome P450 3a Induction by AGM-130. J. Liq. Chromatogr. Relat. Technol. 2015, 38, 1675–1680. [Google Scholar] [CrossRef]
- Park, Y.; Kim, N.; Choi, J.; Park, M.H.; Lee, B.I.; Shin, S.H.; Byeon, J.J.; Shin, Y.G. Qualification and Application of a Liquid Chromatography-Quadrupole Time-of-Flight Mass Spectrometric Method for the Determination of Adalimumab in Rat Plasma. Pharmaceutics 2018, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Whalley, P.M.; Bartels, M.; Bentley, K.S.; Corvaro, M.; Funk, D.; Himmelstein, M.W.; Neumann, B.; Strupp, C.; Zhang, F.; Mehta, J. An in vitro approach for comparative interspecies metabolism of agrochemicals. Regul. Toxicol. Pharmacol. 2017, 88, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Luo, G.; Ding, X.; Lu, C. Preclinical experimental models of drug metabolism and disposition in drug discovery and development. Acta Pharm. Sin. B 2012, 2, 549–561. [Google Scholar] [CrossRef]
- Ufer, M.; Juif, P.E.; Boof, M.L.; Muehlan, C.; Dingemanse, J. Metabolite profiling in early clinical drug development: Current status and future prospects. Expert Opin. Drug Met. 2017, 13, 803–806. [Google Scholar] [CrossRef]
- Uusitalo, J.; Turpeinen, M.; Tolonen, A.; Koskimies, P.; Lammintausta, R.; Pelkonen, O. Metabolism and metabolite profiles in vitro and in vivo of ospemifene in humans and preclinical species. Drug Metab. Pers. Ther. 2016, 31, 35–40. [Google Scholar] [CrossRef]
- Hewitt, N.J.; Buhring, K.U.; Dasenbrock, J.; Haunschild, J.; Ladstetter, B.; Utesch, D. Studies comparing in vivo:in vitro metabolism of three pharmaceutical compounds in rat, dog, monkey, and human using cryopreserved hepatocytes, microsomes, and collagen gel immobilized hepatocyte cultures. Drug Metab. Dispos. 2001, 29, 1042–1050. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1. LC Gradient for Quantification | |
| Time (min) | Mobile Phase B (%) |
| 0 | 10 |
| 0.5 | 10 |
| 0.9 | 95 |
| 1.5 | 95 |
| 1.6 | 10 |
| 3.0 | 10 |
| 2. LC Gradient for MetID | |
| Time (min) | Mobile Phase B (%) |
| 0 | 5 |
| 1.5 | 5 |
| 18 | 25 |
| 23 | 50 |
| 23.5 | 95 |
| 27 | 95 |
| 27.5 | 5 |
| 32 | 5 |
| Intra-Run Assay | |||||
| Run | Nominal QC Concentration (ng/mL) | Calculated Concentration (ng/mL) | Mean Accuracy (%) | Precision (% CV) | n |
| Day 1 | 15 | 16.9 | 112.8 | 12.1 | 3 |
| 165 | 175.2 | 106.2 | 7.9 | ||
| 1820 | 1793.4 | 98.5 | 3.2 | ||
| Day 2 | 15 | 17.3 | 114.8 | 9.6 | 3 |
| 165 | 164.2 | 99.2 | 6.3 | ||
| 1820 | 1727.6 | 94.9 | 5.3 | ||
| Day 3 | 15 | 18.5 | 123.3 | 4.9 | 3 |
| 165 | 187.1 | 113.4 | 6.9 | ||
| 1820 | 1923.1 | 105.7 | 5.3 | ||
| Inter-Run Assay (Day 1~3) | |||||
| Nominal QC Concentration (ng/mL) | Calculated Concentration (ng/mL) | Mean Accuracy (%) | Precision (% CV) | n | |
| 15 | 16.2 | 108.1 | 11.3 | 9 | |
| 165 | 178.1 | 107.8 | 7.4 | ||
| 1820 | 1843.7 | 101.3 | 4.6 | ||
| Freeze-Thaw, Long-Term and Post-Preparative Stability Assessment | ||||
|---|---|---|---|---|
| Stability Test | Nominal QC Concentration (ng/mL) | Calculated Concentration (ng/mL) | Mean Accuracy (%) | Precision (% CV) |
| Short term (4 h, RT, n = 3) | 15 | 14.7 | 97.8 | 11.6 |
| 165 | 149.4 | 90.5 | 10.9 | |
| 1820 | 1685.6 | 92.6 | 3.9 | |
| Freeze-thaw (3 cycles, −80 °C, n = 3) | 15 | 17.8 | 118.8 | 5.2 |
| 165 | 145.8 | 88.5 | 0.4 | |
| 1820 | 1671.6 | 91.1 | 1.7 | |
| Long-term (2 weeks, −80 °C, n = 3) | 15 | 16.8 | 111.8 | 3.3 |
| 165 | 171.8 | 104.1 | 2.7 | |
| 1820 | 1887.7 | 103.7 | 4.8 | |
| Post-preparative (24 h, 4 °C, n = 6) | 15 | 17.3 | 114.8 | 9.6 |
| 165 | 164.2 | 99.2 | 6.3 | |
| 1820 | 1727.6 | 94.9 | 5.3 | |
| Stock storage (6 months, −80°C, n = 3) | 165 | 169.1 | 102.5 | 3.3 |
| 1820 | 1739.2 | 95.6 | 6.1 | |
| Species-Dependent Matrix Effect Assessment (5 Species) | ||||||
|---|---|---|---|---|---|---|
| Species | QC Medium (165 ng/mL, n = 3) | QC High (1820 ng/mL, n = 3) | ||||
| Mean Concentration (ng/mL) | Mean Accuracy (%) | Precision (% CV) | Mean Concentration (ng/mL) | Mean Accuracy (%) | Precision (% CV) | |
| Control (Rat) | 153.2 | 92.7 | 2.9 | 1788.0 | 98.3 | 5.9 |
| Mouse | 159.7 | 96.6 | 6.1 | 1881.6 | 103.5 | 9.6 |
| Dog | 159.8 | 96.7 | 4.8 | 1879.4 | 103.4 | 7.9 |
| Monkey | 163.7 | 99.1 | 3.3 | 1740.3 | 95.7 | 3.1 |
| Human | 174.0 | 105.3 | 6.6 | 1857.9 | 102.2 | 3.4 |
| PK Parameters of Vipadenant | ||||||||
|---|---|---|---|---|---|---|---|---|
| PK Study | Dose (mg/kg) | T1/2 (min) | Tmax (min) | C0 or Cmax (ng/mL) | AUClast (min ng/mL) | CL (mL/min/kg) | Vss (mL/kg) | BA (%) |
| PO | 2 | 65.2 ± 20.2 | 7.5 ± 5.0 | 229.7 ± 88.0 | 16716.4 ± 7245.0 | - | - | 30.4 ± 8.9 |
| 5 | 115.6 ± 59.0 | 30.0 ± 26.0 | 296.0 ± 87.3 | 53196.4 ± 4067.6 | - | - | ||
| IV | 1 | 48.0 ± 5.8 | 2.6 ± 1.3 | 2213.7 ± 1155.3 | 27060.9 ± 5826.5 | 37.8 ± 7.2 | 1082.9 ± 222.5 | |
| 2 | 71.2 ± 8.5 | 2.0 ± 0.0 | 3091.0 ± 221.6 | 90694.2 ± 18814.6 | 22.5 ± 4.3 | 1209.3 ± 174.4 | ||
| In Vitro MetID Result of Vipadenant | ||||||
| Peak ID | Name | Formula | R.T (min) | m/z | Nominal Mass Change (Da) | Error ppm |
| Parent | Vipadenant [M + H]+ | C16H15N7O | 12.3 | 322.1410 | - | −0.2 |
| M1 | Mono-oxidation + mono-glucuronidation [M + H]+ | C22H23N7O8 | 5.1 | 514.1656 | +192 | −4.8 |
| M2 | Loss of C8H9N [M + H]+ | C8H6N6O | 5.8 | 203.0651 | −120 | −0.9 |
| M3 | Mono-oxidation [M + H]+ | C16H15N7O2 | 6.0 | 338.1363 | +16 | 0.9 |
| M4 | Mono-oxidation [M + H]+ | C16H15N7O2 | 6.4 | 338.1338 | +16 | −6.5 |
| M5 | Mono-oxidation [M + H]+ | C16H15N7O2 | 10.6 | 338.1362 | +16 | 0.6 |
| M6 | Mono-oxidation followed by reduction [M + H]+ | C16H13N7O2 | 15.4 | 336.1193 | +14 | −3.1 |
| M7 | Mono-oxidation [M + H]+ | C16H15N7O2 | 15.5 | 338.1338 | +16 | 5.3 |
| M8 | N-Acetylation [M + H]+ | C18H17N7O | 16.0 | 364.1527 | +42 | 2.9 |
| M9 | Mono-oxidation followed by reduction [M + H]+ | C16H13N7O2 | 17.1 | 336.1192 | +14 | −3.4 |
| M10 | Mono-oxidation followed by reduction [M + H]+ | C16H13N7O2 | 22.5 | 336.1187 | +14 | −4.9 |
| In Vivo MetID Result of Vipadenant | ||||||
| Peak ID | Name | Formula | R.T (min) | m/z | Nominal Mass Change (Da) | Error ppm |
| Parent | Vipadenant [M + H]+ | C16H15N7O | 12.3 | 322.1417 | - | 1.9 |
| M2 | Loss of C8H9N [M + H]+ | C8H6N6O | 5.8 | 203.0673 | −120 | −1.4 |
| M8 | N-Acetylation [M + H]+ | C18H17N7O | 16.0 | 364.1526 | +42 | 2.6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, S.-H.; Park, M.-H.; Byeon, J.-J.; Lee, B.i.; Park, Y.; Kim, N.; Choi, J.; Shin, Y.G. Analysis of Vipadenant and Its In Vitro and In Vivo Metabolites via Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometry. Pharmaceutics 2018, 10, 260. https://doi.org/10.3390/pharmaceutics10040260
Shin S-H, Park M-H, Byeon J-J, Lee Bi, Park Y, Kim N, Choi J, Shin YG. Analysis of Vipadenant and Its In Vitro and In Vivo Metabolites via Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometry. Pharmaceutics. 2018; 10(4):260. https://doi.org/10.3390/pharmaceutics10040260
Chicago/Turabian StyleShin, Seok-Ho, Min-Ho Park, Jin-Ju Byeon, Byeong ill Lee, Yuri Park, Nahye Kim, Jangmi Choi, and Young G. Shin. 2018. "Analysis of Vipadenant and Its In Vitro and In Vivo Metabolites via Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometry" Pharmaceutics 10, no. 4: 260. https://doi.org/10.3390/pharmaceutics10040260
APA StyleShin, S.-H., Park, M.-H., Byeon, J.-J., Lee, B. i., Park, Y., Kim, N., Choi, J., & Shin, Y. G. (2018). Analysis of Vipadenant and Its In Vitro and In Vivo Metabolites via Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometry. Pharmaceutics, 10(4), 260. https://doi.org/10.3390/pharmaceutics10040260