In Vitro Immune Organs-on-Chip for Drug Development: A Review



Abstract
:1. Introduction
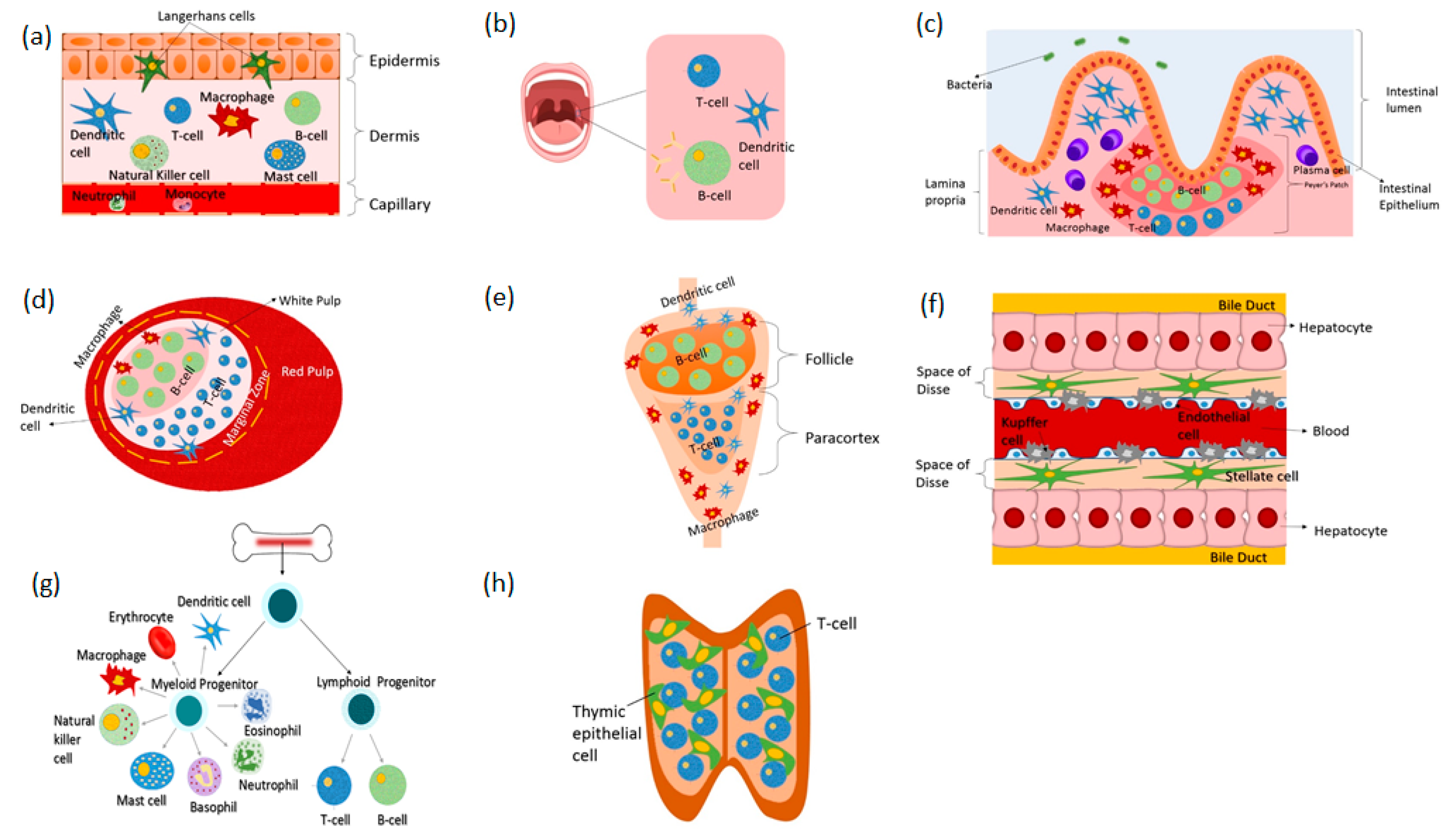
2. Immune Organs-on-Chip
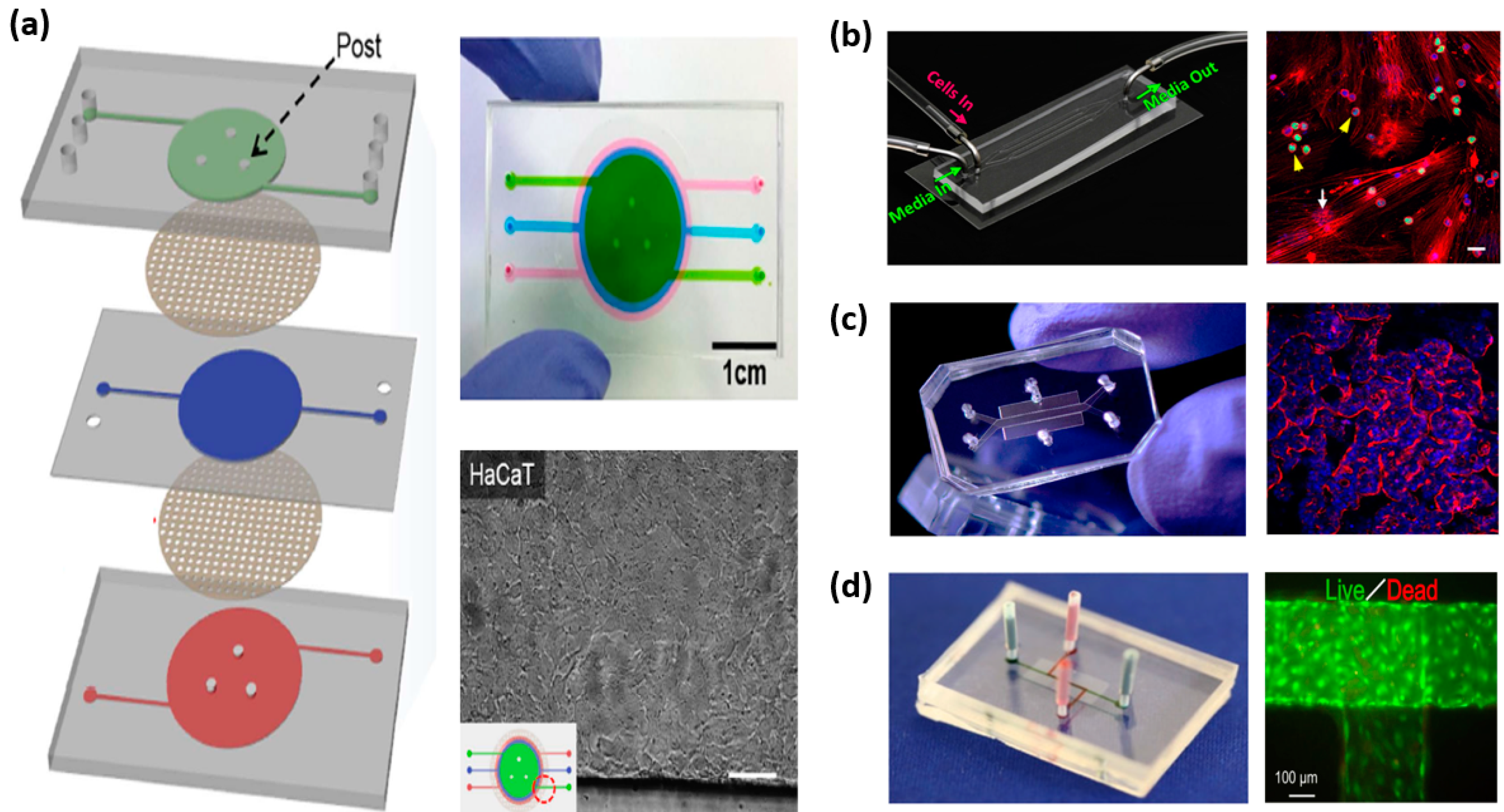
2.1. Skin-on-Chip
2.2. Gut-on-Chip
2.3. Liver-on-Chip
2.4. Lymph Node-on-Chip
2.5. Spleen-on-Chip
2.6. Bone Marrow-on-Chip
2.7. Lymphatic Vessel-on-Chip
2.8. Other Relevant Immune-on-Chip Platforms
3. Towards an Integrated Immune System on Chip
4. Challenges
Funding
Conflicts of Interest
References
- McKim, J.M. Building a Tiered Approach to In Vitro Predictive Toxicity Screening: A Focus on Assays with In Vivo Relevance. Comb. Chem. High Throughput Screen. 2010, 13, 188–206. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Mechanisms of Drug Toxicity and Relevance to Pharmaceutical Development. Drug Metab. Pharmacokinet. 2011, 26, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, R.A.; Kavanagh, S.L.; Mellor, H.R.; Pollard, C.E.; Robinson, S.; Platz, S.J. Reducing attrition in drug development: Smart loading preclinical safety assessment. Drug Discov. Today 2014, 19, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.-H.; Smith, A.S.T.; Lone, S.; Kwon, S.; Kim, D.-H. Biomimetic 3D Tissue Models for Advanced High-Throughput Drug Screening. J. Lab. Autom. 2015, 20, 201–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Gill, E.; Shery Huang, Y.Y. Microfluidic on-chip biomimicry for 3D cell culture: A fit-for-purpose investigation from the end user standpoint. Future Sci. OA 2017. [Google Scholar] [CrossRef] [PubMed]
- Ghaemmaghami, A.M.; Hancock, M.J.; Harrington, H.; Kaji, H.; Khademhosseini, A. Biomimetic tissues on a chip for drug discovery. Drug Discov. Today 2012, 17, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Shanks, N.; Greek, R.; Greek, J. Are animal models predictive for humans? Philos. Ethics Humanit. Med. 2009. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C.W. Of Mice and Not Men: Differences between Mouse and Human Immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Reese, T.A. Making Mouse Models That Reflect Human Immune Responses. Trends Immunol. 2017, 38, 181–193. [Google Scholar] [CrossRef]
- von Herrath, M.G.; Nepom, G.T. Lost in translation: Figure 1.: Barriers to implementing clinical immunotherapeutics for autoimmunity. J. Exp. Med. 2005, 202, 1159–1162. [Google Scholar] [CrossRef] [PubMed]
- Sackmann, E.K.; Fulton, A.L.; Beebe, D.J. The present and future role of microfluidics in biomedical research. Nature 2014, 507, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Hamilton, G.A.; Ingber, D.E. From Three-Dimensional Cell Culture to Organs-on-Chips. Trends Cell Biol. 2011, 21, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Lokuta, M.A.; Huttenlocher, A.; Beebe, D.J. Selective and tunable gradient device for cell culture and chemotaxis study. Lab Chip 2009, 9, 1797–1800. [Google Scholar] [CrossRef] [PubMed]
- Kothapalli, C.R.; van Veen, E.; de Valence, S.; Chung, S.; Zervantonakis, I.K.; Gertler, F.B.; Kamm, R.D. A high-throughput microfluidic assay to study neurite response to growth factor gradients. Lab Chip 2011, 11, 497–507. [Google Scholar] [CrossRef] [Green Version]
- Ataç, B.; Wagner, I.; Horland, R.; Lauster, R.; Marx, U.; Tonevitsky, A.G.; Azar, R.P.; Lindner, G. Skin and hair on-a-chip: In vitro skin models versus ex vivo tissue maintenance with dynamic perfusion. Lab Chip 2013, 13, 3555–3561. [Google Scholar] [CrossRef]
- Mota, C.; Danti, S.; D’Alessandro, D.; Trombi, L.; Ricci, C.; Puppi, D.; Dinucci, D.; Milazzo, M.; Stefanini, C.; Chiellini, F.; et al. Multiscale fabrication of biomimetic scaffolds for tympanic membrane tissue engineering. Biofabrication 2015, 7, 025005. [Google Scholar] [CrossRef]
- Spina, G.L.; Stefanini, C.; Menciassi, A.; Dario, P. A novel technological process for fabricating micro-tips for biomimetic adhesion. J. Micromech. Microeng. 2005, 15, 1576–1587. [Google Scholar] [CrossRef]
- Alhussein, G.; Shanti, A.; Farhat, I.A.H.; Timraz, S.B.H.; Alwahab, N.S.A.; Pearson, Y.E.; Martin, M.N.; Christoforou, N.; Teo, J.C.M. A spatiotemporal characterization method for the dynamic cytoskeleton: Characterization of the Dynamic Cytoskeleton. Cytoskeleton 2016, 73, 221–232. [Google Scholar] [CrossRef]
- Gater, D.L.; Widatalla, N.; Islam, K.; AlRaeesi, M.; Teo, J.C.M.; Pearson, Y.E. Quantification of sterol-specific response in human macrophages using automated imaged-based analysis. Lipids Health Dis. 2017. [Google Scholar] [CrossRef]
- Pearson, Y.E.; Lund, A.W.; Lin, A.W.H.; Ng, C.P.; Alsuwaidi, A.; Azzeh, S.; Gater, D.L.; Teo, J.C.M. Non-invasive single-cell biomechanical analysis using live-imaging datasets. J. Cell Sci. 2016, 129, 3351–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, D.R.; Singh, I.; Nguyen, M.N.; Kachurin, A.; Wittman, V.; Parkhill, R.; Kachurina, O.; Moser, J.M.; Burdin, N.; Moreau, M.; et al. In Vitro Biomimetic Model of the Human Immune System for Predictive Vaccine Assessments. Disrupt. Sci. Technol. 2012, 1, 28–40. [Google Scholar] [CrossRef]
- Mengus, C.; Muraro, M.G.; Mele, V.; Amicarella, F.; Manfredonia, C.; Foglietta, F.; Muenst, S.; Soysal, S.D.; Iezzi, G.; Spagnoli, G.C. In Vitro Modeling of Tumor-Immune System Interaction. ACS Biomater. Sci. Eng. 2018, 4, 314–323. [Google Scholar] [CrossRef]
- Gosselin, E.A.; Eppler, H.B.; Bromberg, J.S.; Jewell, C.M. Designing natural and synthetic immune tissues. Nat. Mater. 2018, 17, 484–498. [Google Scholar] [CrossRef] [PubMed]
- Vono, M.; Lin, A.; Norrby-Teglund, A.; Koup, R.A.; Liang, F.; Loré, K. Neutrophils acquire the capacity for antigen presentation to memory CD4+ T cells in vitro and ex vivo. Blood 2017, 129, 1991–2001. [Google Scholar] [CrossRef] [PubMed]
- Picker, L.J.; Michie, S.A.; Rott, L.S.; Butcher, E.C. A unique phenotype of skin-associated lymphocytes in humans. Preferential expression of the HECA-452 epitope by benign and malignant T cells at cutaneous sites. Am. J. Pathol. 1990, 136, 1053–1068. [Google Scholar]
- Craig, S.W.; Cebra, J.J. Peyer’s patches: An enriched source of precursors for IgA-producing immunocytes in the rabbit. J. Exp. Med. 1971, 134, 188–200. [Google Scholar] [CrossRef]
- Elson, C.O.; Heck, J.A.; Strober, W. T-cell regulation of murine IgA synthesis. J. Exp. Med. 1979, 149, 632–643. [Google Scholar] [CrossRef] [Green Version]
- Streilein, J.W. Skin-associated lymphoid tissues (SALT): Origins and functions. J. Investig. Dermatol. 1983, 80, 12s–16s. [Google Scholar] [CrossRef] [PubMed]
- Brodin, P.; Davis, M.M. Human immune system variation. Nat. Rev. Immunol. 2017, 17, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Racanelli, V.; Rehermann, B. The liver as an immunological organ. Hepatology 2006, 43, S54–S62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidd, B.A.; Wroblewska, A.; Boland, M.R.; Agudo, J.; Merad, M.; Tatonetti, N.P.; Brown, B.D.; Dudley, J.T. Mapping the effects of drugs on the immune system. Nat. Biotechnol. 2015, 34, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brake, K.; Gumireddy, A.; Tiwari, A.; Chauhan, H.; Kumari, D. In vivo Studies for Drug Development via Oral Delivery: Challenges, Animal Models and Techniques. Pharm. Anal. Acta 2017. [Google Scholar] [CrossRef]
- Li, X.; Wang, Z.; Lu, T.; Che, X. Modelling Immune System: Principles, Models, Analysis and Perspectives. J. Bionic Eng. 2009, 6, 77–85. [Google Scholar] [CrossRef]
- Kirschner, D.; Pienaar, E.; Marino, S.; Linderman, J.J. A review of computational and mathematical modeling contributions to our understanding of Mycobacterium tuberculosis within-host infection and treatment. Curr. Opin. Syst. Boil. 2017, 3, 170–185. [Google Scholar] [CrossRef]
- Fischbach, C.; Reagan, M. Editorial: Special Issue on Tissue Engineering and Biomaterials Approaches to Tumor Modeling. ACS Biomater. Sci. Eng. 2018, 4, 291–293. [Google Scholar] [CrossRef]
- Wufuer, M.; Lee, G.; Hur, W.; Jeon, B.; Kim, B.J.; Choi, T.H.; Lee, S. Skin-on-a-chip model simulating inflammation, edema and drug-based treatment. Sci. Rep. 2016. [Google Scholar] [CrossRef]
- Bruce, A.; Evans, R.; Mezan, R.; Shi, L.; Moses, B.S.; Martin, K.H.; Gibson, L.F.; Yang, Y. Three-Dimensional Microfluidic Tri-Culture Model of the Bone Marrow Microenvironment for Study of Acute Lymphoblastic Leukemia. PLoS ONE 2015, 10, e0140506. [Google Scholar] [CrossRef]
- Villenave, R.; Wales, S.Q.; Hamkins-Indik, T.; Papafragkou, E.; Weaver, J.C.; Ferrante, T.C.; Bahinski, A.; Elkins, C.A.; Kulka, M.; Ingber, D.E. Human Gut-On-A-Chip Supports Polarized Infection of Coxsackie B1 Virus In Vitro. PLoS ONE 2017. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Sasaki, N.; Ato, M.; Hirakawa, S.; Sato, K.; Sato, K. Microcirculation-on-a-Chip: A Microfluidic Platform for Assaying Blood- and Lymphatic-Vessel Permeability. PLoS ONE 2015, 10, e0137301. [Google Scholar] [CrossRef] [PubMed]
- Salmon, J.K.; Armstrong, C.A.; Ansel, J.C. The skin as an immune organ. West. J. Med. 1994, 160, 146–152. [Google Scholar] [PubMed]
- Matejuk, A. Skin Immunity. Arch. Immunol. Ther. Exp. 2018, 66, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.S.; Ovigne, J.-M.; Powles, A.V.; Corcoran, S.; Fry, L. Normal keratinocytes express Toll-like receptors (TLRs) 1, 2 and 5: Modulation of TLR expression in chronic plaque psoriasis. Br. J. Dermatol. 2003, 148, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Schauber, J.; Gallo, R.L. Antimicrobial peptides and the skin immune defense system. J. Allergy Clin. Immunol. 2008, 122, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Diamond, G.; Beckloff, N.; Weinberg, A.; Kisich, K.O. The roles of antimicrobial peptides in innate host defense. Curr. Pharm. Des. 2009, 15, 2377–2392. [Google Scholar] [CrossRef]
- O’Neill, A.T.; Monteiro-Riviere, N.A.; Walker, G.M. Characterization of microfluidic human epidermal keratinocyte culture. Cytotechnology 2008, 56, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Sriram, G.; Alberti, M.; Dancik, Y.; Wu, B.; Wu, R.; Feng, Z.; Ramasamy, S.; Bigliardi, P.L.; Bigliardi-Qi, M.; Wang, Z. Full-thickness human skin-on-chip with enhanced epidermal morphogenesis and barrier function. Mater. Today 2018, 21, 326–340. [Google Scholar] [CrossRef]
- Abaci, H.E.; Gledhill, K.; Guo, Z.; Christiano, A.M.; Shuler, M.L. Pumpless microfluidic platform for drug testing on human skin equivalents. Lab Chip 2015, 15, 882–888. [Google Scholar] [CrossRef] [Green Version]
- Abaci, H.E.; Guo, Z.; Coffman, A.; Gillette, B.; Lee, W.-H.; Sia, S.K.; Christiano, A.M. Human Skin Constructs with Spatially Controlled Vasculature Using Primary and iPSC-Derived Endothelial Cells. Adv. Healthc. Mater. 2016, 5, 1800–1807. [Google Scholar] [CrossRef] [Green Version]
- Mori, N.; Morimoto, Y.; Takeuchi, S. Skin integrated with perfusable vascular channels on a chip. Biomaterials 2017, 116, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, Q.; Ting, F.C.W. In vitro micro-physiological immune-competent model of the human skin. Lab Chip 2016, 16, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Mahler, G.J.; Esch, M.B.; Glahn, R.P.; Shuler, M.L. Characterization of a gastrointestinal tract microscale cell culture analog used to predict drug toxicity. Biotechnol. Bioeng. 2009, 104, 193–205. [Google Scholar] [CrossRef]
- McDermott, A.J.; Huffnagle, G.B. The microbiome and regulation of mucosal immunity. Immunology 2014, 142, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Kleinschek, M.A.; Boniface, K.; Sadekova, S.; Grein, J.; Murphy, E.E.; Turner, S.P.; Raskin, L.; Desai, B.; Faubion, W.A.; de Waal Malefyt, R.; et al. Circulating and gut-resident human Th17 cells express CD161 and promote intestinal inflammation. J. Exp. Med. 2009, 206, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Makita, S.; Kanai, T.; Nemoto, Y.; Totsuka, T.; Okamoto, R.; Tsuchiya, K.; Yamamoto, M.; Kiyono, H.; Watanabe, M. Intestinal Lamina Propria Retaining CD4+CD25+ Regulatory T Cells Is A Suppressive Site of Intestinal Inflammation. J. Immunol. 2007, 178, 4937–4946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.-J.; Wu, E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Bein, A.; Shin, W.; Jalili-Firoozinezhad, S.; Park, M.H.; Sontheimer-Phelps, A.; Tovaglieri, A.; Chalkiadaki, A.; Kim, H.J.; Ingber, D.E. Microfluidic Organ-on-a-Chip Models of Human Intestine. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 659–668. [Google Scholar] [CrossRef]
- Kimura, H.; Yamamoto, T.; Sakai, H.; Sakai, Y.; Fujii, T. An integrated microfluidic system for long-term perfusion culture and on-line monitoring of intestinal tissue models. Lab Chip 2008, 8, 741–746. [Google Scholar] [CrossRef]
- Kim, H.J.; Huh, D.; Hamilton, G.; Ingber, D.E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip 2012, 12, 2165–2174. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, S.M. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787. [Google Scholar] [CrossRef] [PubMed]
- Bisgaard, H.; Li, N.; Bonnelykke, K.; Chawes, B.L.K.; Skov, T.; Paludan-Müller, G.; Stokholm, J.; Smith, B.; Krogfelt, K.A. Reduced diversity of the intestinal microbiota during infancy is associated with increased risk of allergic disease at school age. J. Allergy Clin. Immunol. 2011. [Google Scholar] [CrossRef]
- Ferreira, C.M.; Vieira, A.T.; Vinolo, M.A.R.; Oliveira, F.A.; Curi, R.; dos Martins, F.S. The Central Role of the Gut Microbiota in Chronic Inflammatory Diseases. J. Immunol. Res. 2014. [Google Scholar] [CrossRef]
- Kim, H.J.; Ingber, D.E. Gut-on-a-Chip microenvironment induces human intestinal cells to undergo villus differentiation. Integr. Boil. 2013, 5, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Li, H.; Collins, J.J.; Ingber, D.E. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc. Natl. Acad. Sci. USA 2016, 113, E7–E15. [Google Scholar] [CrossRef]
- Shim, K.-Y.; Lee, D.; Han, J.; Nguyen, N.-T.; Park, S.; Sung, J.H. Microfluidic gut-on-a-chip with three-dimensional villi structure. Biomed. Microdevices 2017. [Google Scholar] [CrossRef]
- Shah, P.; Fritz, J.V.; Glaab, E.; Desai, M.S.; Greenhalgh, K.; Frachet, A.; Niegowska, M.; Estes, M.; Jäger, C.; Seguin-Devaux, C.; et al. A microfluidics-based in vitro model of the gastrointestinal human–microbe interface. Nat. Commun. 2016, 7, 11535. [Google Scholar] [CrossRef] [Green Version]
- Gao, B. Basic liver immunology. Cell. Mol. Immunol. 2016, 13, 265–266. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Nguyen-Lefebvre, A.T.; Horuzsko, A. Kupffer Cell Metabolism and Function. J. Enzym. Metab. 2015, 1, 101. [Google Scholar]
- Bilzer, M.; Roggel, F.; Gerbes, A.L. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006, 26, 1175–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer Cells in the Liver. In Comprehensive Physiology; Terjung, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; ISBN 978-0-470-65071-4. [Google Scholar]
- Schütte, J.; Hagmeyer, B.; Holzner, F.; Kubon, M.; Werner, S.; Freudigmann, C.; Benz, K.; Böttger, J.; Gebhardt, R.; Becker, H.; et al. Artificial micro organs—A microfluidic device for dielectrophoretic assembly of liver sinusoids. Biomed. Microdevices 2011, 13, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Domansky, K.; Inman, W.; Serdy, J.; Dash, A.; Lim, M.H.M.; Griffith, L.G. Perfused multiwell plate for 3D liver tissue engineering. Lab Chip 2010, 10, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Midwoud, P.M.; Verpoorte, E.; Groothuis, G.M.M. Microfluidic devices for in vitro studies on liver drug metabolism and toxicity. Integr. Boil. 2011, 3, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Toh, Y.-C.; Lim, T.C.; Tai, D.; Xiao, G.; van Noort, D.; Yu, H. A microfluidic 3D hepatocyte chip for drug toxicity testing. Lab Chip 2009, 9, 2026–2035. [Google Scholar] [CrossRef] [PubMed]
- Ostrovidov, S.; Jiang, J.; Sakai, Y.; Fujii, T. Membrane-based PDMS microbioreactor for perfused 3D primary rat hepatocyte cultures. Biomed. Microdevices 2004, 6, 279–287. [Google Scholar] [CrossRef]
- Powers, M.J.; Domansky, K.; Kaazempur-Mofrad, M.R.; Kalezi, A.; Capitano, A.; Upadhyaya, A.; Kurzawski, P.; Wack, K.E.; Stolz, D.B.; Kamm, R.; et al. A microfabricated array bioreactor for perfused 3D liver culture. Biotechnol. Bioeng. 2002, 78, 257–269. [Google Scholar] [CrossRef]
- Lee, P.J.; Hung, P.J.; Lee, L.P. An artificial liver sinusoid with a microfluidic endothelial-like barrier for primary hepatocyte culture. Biotechnol. Bioeng. 2007, 97, 1340–1346. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.-T.; Lin, R.-Z.; Chen, R.-J.; Chin, C.-K.; Gong, S.-E.; Chang, H.-Y.; Peng, H.-L.; Hsu, L.; Yew, T.-R.; Chang, S.-F.; et al. Liver-cell patterning lab chip: Mimicking the morphology of liver lobule tissue. Lab Chip 2013, 13, 3578–3587. [Google Scholar] [CrossRef]
- Kane, B.J.; Zinner, M.J.; Yarmush, M.L.; Toner, M. Liver-specific functional studies in a microfluidic array of primary mammalian hepatocytes. Anal. Chem. 2006, 78, 4291–4298. [Google Scholar] [CrossRef] [PubMed]
- Goral, V.N.; Hsieh, Y.-C.; Petzold, O.N.; Clark, J.S.; Yuen, P.K.; Faris, R.A. Perfusion-based microfluidic device for three-dimensional dynamic primary human hepatocyte cell culture in the absence of biological or synthetic matrices or coagulants. Lab Chip 2010, 10, 3380–3386. [Google Scholar] [CrossRef]
- Khetani, S.R.; Bhatia, S.N. Microscale culture of human liver cells for drug development. Nat. Biotechnol. 2008, 26, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-A.; No, D.Y.; Kang, E.; Ju, J.; Kim, D.-S.; Lee, S.-H. Spheroid-based three-dimensional liver-on-a-chip to investigate hepatocyte-hepatic stellate cell interactions and flow effects. Lab Chip 2013, 13, 3529–3537. [Google Scholar] [CrossRef]
- Prodanov, L.; Jindal, R.; Bale, S.S.; Hegde, M.; McCarty, W.J.; Golberg, I.; Bhushan, A.; Yarmush, M.L.; Usta, O.B. Long-term maintenance of a microfluidic 3D human liver sinusoid: Maintenance of a Microfluidic 3D Human Liver Sinusoid. Biotechnol. Bioeng. 2016, 113, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.B.A.; Sodunke, T.R.; Lamontagne, J.; Cirillo, J.; Rajiv, C.; Bouchard, M.J.; Noh, M. Liver sinusoid on a chip: Long-term layered co-culture of primary rat hepatocytes and endothelial cells in microfluidic platforms. Biotechnol. Bioeng. 2015, 112, 2571–2582. [Google Scholar] [CrossRef] [PubMed]
- Ahadian, S.; Civitarese, R.; Bannerman, D.; Mohammadi, M.H.; Lu, R.; Wang, E.; Davenport-Huyer, L.; Lai, B.; Zhang, B.; Zhao, Y.; et al. Organ-On-A-Chip Platforms: A Convergence of Advanced Materials, Cells, and Microscale Technologies. Adv. Healthc. Mater. 2018, 7. [Google Scholar] [CrossRef]
- Chen, M.; Vijay, V.; Shi, Q.; Liu, Z.; Fang, H.; Tong, W. FDA-approved drug labeling for the study of drug-induced liver injury. Drug Discov. Today 2011, 16, 697–703. [Google Scholar] [CrossRef]
- Fullerton, J.N.; Gilroy, D.W. Resolution of inflammation: A new therapeutic frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef]
- Girard, J.-P.; Moussion, C.; Förster, R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat. Rev. Immunol. 2012, 12, 762–773. [Google Scholar] [CrossRef]
- Cupedo, T.; Stroock, A.; Coles, M. Application of tissue engineering to the immune system: Development of artificial lymph nodes. Front. Immunol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Moura Rosa, P.; Gopalakrishnan, N.; Ibrahim, H.; Haug, M.; Halaas, Ø. The intercell dynamics of T cells and dendritic cells in a lymph node-on-a-chip flow device. Lab Chip 2016, 16, 3728–3740. [Google Scholar] [CrossRef]
- Lian, J.; Luster, A.D. Chemokine-guided cell positioning in the lymph node orchestrates the generation of adaptive immune responses. Curr. Opin. Cell Boil. 2015. [Google Scholar] [CrossRef]
- Pape, K.A.; Catron, D.M.; Itano, A.A.; Jenkins, M.K. The Humoral Immune Response Is Initiated in Lymph Nodes by B Cells that Acquire Soluble Antigen Directly in the Follicles. Immunity 2007, 26, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Mitra, B.; Jindal, R.; Lee, S.; Dong, D.X.; Li, L.; Sharma, N.; Maguire, T.; Schloss, R.; Yarmush, M.L. Microdevice integrating innate and adaptive immune responses associated with antigen presentation by dendritic cells. RSC Adv. 2013, 3, 16002–16010. [Google Scholar] [CrossRef] [PubMed]
- Ricart, B.G.; John, B.; Lee, D.; Hunter, C.A.; Hammer, D.A. Dendritic Cells Distinguish Individual Chemokine Signals through CCR7 and CXCR4. J. Immunol. 2011, 186, 53–61. [Google Scholar] [CrossRef]
- Nandagopal, S.; Wu, D.; Lin, F. Combinatorial Guidance by CCR7 Ligands for T Lymphocytes Migration in Co-Existing Chemokine Fields. PLoS ONE 2011, 6, e18183. [Google Scholar] [CrossRef]
- Giese, C.; Lubitz, A.; Demmler, C.D.; Reuschel, J.; Bergner, K.; Marx, U. Immunological substance testing on human lymphatic micro-organoids in vitro. J. Biotechnol. 2010, 148, 38–45. [Google Scholar] [CrossRef]
- Giese, C.; Demmler, C.D.; Ammer, R.; Hartmann, S.; Lubitz, A.; Miller, L.; Müller, R.; Marx, U. A Human Lymph Node In Vitro? Challenges and Progress. Artif. Organs 2006, 30, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Pittet, M.J. The spleen in local and systemic regulation of immunity. Immunity 2013, 39, 806–818. [Google Scholar] [CrossRef]
- Timens, W. The human spleen and the immune system: Not just another lymphoid organ. Res. Immunol. 1991, 142, 316–320. [Google Scholar] [CrossRef]
- Rigat-Brugarolas, L.G.; Elizalde-Torrent, A.; Bernabeu, M.; De Niz, M.; Martin-Jaular, L.; Fernandez-Becerra, C.; Homs-Corbera, A.; Samitier, J.; del Portillo, H.A. A functional microengineered model of the human splenon-on-a-chip. Lab Chip 2014, 14, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Mohandas, N.; Gallagher, P.G. Red cell membrane: Past, present, and future. Blood 2008, 112, 3939–3948. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Super, M.; Yung, C.W.; Cooper, R.M.; Domansky, K.; Graveline, A.R.; Mammoto, T.; Berthet, J.B.; Tobin, H.; Cartwright, M.J.; et al. An extracorporeal blood-cleansing device for sepsis therapy. Nat. Med. 2014, 20, 1211–1216. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, T.; Nagasawa, T. Bone Marrow Niches for Hematopoietic Stem Cells and Immune Cells. Inflamm. Allergy-Drug Targets 2012, 11, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Yu, V.W.C.; Scadden, D.T. Hematopoietic Stem Cell and Its Bone Marrow Niche. Curr. Top. Dev. Biol. 2016, 118, 21–44. [Google Scholar] [CrossRef]
- Torisawa, Y.; Spina, C.S.; Mammoto, T.; Mammoto, A.; Weaver, J.C.; Tat, T.; Collins, J.J.; Ingber, D.E. Bone marrow-on-a-chip replicates hematopoietic niche physiology in vitro. Nat. Methods 2014, 11, 663–669. [Google Scholar] [CrossRef]
- Torisawa, Y.; Mammoto, T.; Jiang, E.; Jiang, A.; Mammoto, A.; Watters, A.L.; Bahinski, A.; Ingber, D.E. Modeling Hematopoiesis and Responses to Radiation Countermeasures in a Bone Marrow-on-a-Chip. Tissue Eng. Part C Methods 2016, 22, 509–515. [Google Scholar] [CrossRef]
- Sieber, S.; Wirth, L.; Cavak, N.; Koenigsmark, M.; Marx, U.; Lauster, R.; Rosowski, M. Bone marrow-on-a-chip: Long-term culture of human haematopoietic stem cells in a three-dimensional microfluidic environment. J. Tissue Eng. Regen. Med. 2018, 12, 479–489. [Google Scholar] [CrossRef]
- Petrova, T.V.; Koh, G.Y. Organ-specific lymphatic vasculature: From development to pathophysiology. J. Exp. Med. 2018, 215, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Clasper, S.; Holt, A.P.; Lalor, P.F.; Baban, D.; Jackson, D.G. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J. Exp. Med. 2006, 203, 2763–2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irimia, D.; Wang, X. Inflammation-on-a-Chip: Probing the Immune System Ex Vivo. Trends Biotechnol. 2018, 36, 923–937. [Google Scholar] [CrossRef] [PubMed]
- Biselli, E.; Agliari, E.; Barra, A.; Bertani, F.R.; Gerardino, A.; De Ninno, A.; Mencattini, A.; Di Giuseppe, D.; Mattei, F.; Schiavoni, G.; et al. Organs on chip approach: A tool to evaluate cancer-immune cells interactions. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Parlato, S.; De Ninno, A.; Molfetta, R.; Toschi, E.; Salerno, D.; Mencattini, A.; Romagnoli, G.; Fragale, A.; Roccazzello, L.; Buoncervello, M.; et al. 3D Microfluidic model for evaluating immunotherapy efficacy by tracking dendritic cell behaviour toward tumor cells. Sci. Rep. 2017, 7, 1093. [Google Scholar] [CrossRef] [PubMed]
- Hamza, B.; Irimia, D. Whole blood human neutrophil trafficking in a microfluidic model of infection and inflammation. Lab Chip 2015, 15, 2625–2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.N.; Dalli, J.; Dimisko, L.; Wong, E.; Serhan, C.N.; Irimia, D. Microfluidic chambers for monitoring leukocyte trafficking and humanized nano-proresolving medicines interactions. Proc. Natl. Acad. Sci. USA 2012, 109, 20560–20565. [Google Scholar] [CrossRef] [Green Version]
- Amor, S.; Puentes, F.; Baker, D.; van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef] [Green Version]
- Wevers, N.R.; Kasi, D.G.; Gray, T.; Wilschut, K.J.; Smith, B.; van Vught, R.; Shimizu, F.; Sano, Y.; Kanda, T.; Marsh, G.; et al. A perfused human blood-brain barrier on-a-chip for high-throughput assessment of barrier function and antibody transport. Fluids Barriers CNS 2018. [Google Scholar] [CrossRef]
- Caballero, D.; Kaushik, S.; Correlo, V.M.; Oliveira, J.M.; Reis, R.L.; Kundu, S.C. Organ-on-chip models of cancer metastasis for future personalized medicine: From chip to the patient. Biomaterials 2017, 149, 98–115. [Google Scholar] [CrossRef]
- Tsai, H.-F.; Trubelja, A.; Shen, A.Q.; Bao, G. Tumour-on-a-chip: Microfluidic models of tumour morphology, growth and microenvironment. J. R. Soc. Interface 2017. [Google Scholar] [CrossRef] [PubMed]
- Pavesi, A.; Tan, A.T.; Koh, S.; Chia, A.; Colombo, M.; Antonecchia, E.; Miccolis, C.; Ceccarello, E.; Adriani, G.; Raimondi, M.T.; et al. A 3D microfluidic model for preclinical evaluation of TCR-engineered T cells against solid tumors. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loskill, P.; Sezhian, T.; Tharp, K.M.; Lee-Montiel, F.T.; Jeeawoody, S.; Reese, W.M.; Zushin, P.-J.H.; Stahl, A.; Healy, K.E. WAT-on-a-chip: A physiologically relevant microfluidic system incorporating white adipose tissue. Lab Chip 2017, 17, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; He, J.; Verano, M.; Brimmo, A.T.; Glia, A.; Qasaimeh, M.A.; Chen, P.; Aleman, J.O.; Chen, W. An integrated adipose-tissue-on-chip nanoplasmonic biosensing platform for investigating obesity-associated inflammation. Lab Chip 2018. [Google Scholar] [CrossRef] [PubMed]
- Bennet, D.; Estlack, Z.; Reid, T.; Kim, J. A microengineered human corneal epithelium-on-a-chip for eye drops mass transport evaluation. Lab Chip 2018, 18, 1539–1551. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Romero, R.; Han, Y.M.; Kim, H.C.; Kim, C.J.; Hong, J.-S.; Huh, D. Placenta-on-a-chip: A novel platform to study the biology of the human placenta. J. Matern. Fetal Neonatal Med. 2016, 29, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.W.; Dixit, V.D. Adipose tissue as an immunological organ. Obesity 2015, 23, 512–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrante, A.W. The immune cells in adipose tissue. Diabetes Obes. Metab. 2013, 15 (Suppl. 3), 34–38. [Google Scholar] [CrossRef] [Green Version]
- Nissen, M.H.; Röpke, C. Innate and Adaptive Immunity of the Eye. In Advances in Organ Biology; Elsevier: Amsterdam, The Netherlands, 2005; Volume 10, pp. 291–305. ISBN 978-0-444-50925-3. [Google Scholar]
- Mor, G.; Cardenas, I. The Immune System in Pregnancy: A Unique Complexity: Immune System in Pregnancy. Am. J. Reprod. Immunol. 2010, 63, 425–433. [Google Scholar] [CrossRef]
- Zhou, R.; Caspi, R.R. Ocular immune privilege. F1000 Biol. Rep. 2010. [Google Scholar] [CrossRef]
- Niederkorn, J.Y.; Wang, S. Immune privilege of the eye and fetus: Parallel universes? Transplantation 2005, 80, 1139–1144. [Google Scholar] [CrossRef]
- Abaci, H.E.; Shuler, M.L. Human-on-a-chip design strategies and principles for physiologically based pharmacokinetics/pharmacodynamics modeling. Integr. Biol. 2015, 7, 383–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esch, M.B.; Smith, A.S.T.; Prot, J.-M.; Oleaga, C.; Hickman, J.J.; Shuler, M.L. How multi-organ microdevices can help foster drug development. Adv. Drug Deliv. Rev. 2014, 69–70, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Imura, Y.; Sato, K.; Yoshimura, E. Micro Total Bioassay System for Ingested Substances: Assessment of Intestinal Absorption, Hepatic Metabolism, and Bioactivity. Anal. Chem. 2010, 82, 9983–9988. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Kam, C.; Shuler, M.L. A microfluidic device for a pharmacokinetic–pharmacodynamic (PK–PD) model on a chip. Lab Chip 2010, 10, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhao, Z.; Abdul Rahim, N.A.; van Noort, D.; Yu, H. Towards a human-on-chip: Culturing multiple cell types on a chip with compartmentalized microenvironments. Lab Chip 2009, 9, 3185–3192. [Google Scholar] [CrossRef]
- Maschmeyer, I.; Hasenberg, T.; Jaenicke, A.; Lindner, M.; Lorenz, A.K.; Zech, J.; Garbe, L.-A.; Sonntag, F.; Hayden, P.; Ayehunie, S.; et al. Chip-based human liver-intestine and liver-skin co-cultures—A first step toward systemic repeated dose substance testing in vitro. Eur. J. Pharm. Biopharm. 2015, 95, 77–87. [Google Scholar] [CrossRef]
- Maschmeyer, I.; Lorenz, A.K.; Schimek, K.; Hasenberg, T.; Ramme, A.P.; Hübner, J.; Lindner, M.; Drewell, C.; Bauer, S.; Thomas, A.; et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip 2015, 15, 2688–2699. [Google Scholar] [CrossRef] [Green Version]
- Low, L.A.; Tagle, D.A. Organs-on-chips: Progress, challenges, and future directions. Exp. Biol. Med. 2017, 242, 1573–1578. [Google Scholar] [CrossRef] [Green Version]
- Shirure, V.S.; George, S.C. Design considerations to minimize the impact of drug absorption in polymer-based organ-on-a-chip platforms. Lab Chip 2017, 17, 681–690. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Organ | Simulated Feature/s | Relevance to Drug development | Reference |
|---|---|---|---|
| Skin | Barrier Function | The skin is a site of administration of some drugs Skin is an active immune organ that reacts to pathogens and foreign substances including drugs Immune rejection to newly developed drugs can be initiated in skin, thus, models assessing drug-skin immune interaction are desirable | [48,49] |
| Vascularization | [39,51,52] | ||
| Air-to-liquid interface | [49,50] | ||
| Absorption | [39] | ||
| Inflammation and Edema | [39] | ||
| Immune Competence | [53] | ||
| Gut | Epithelial Barrier function | The Gut is a site of administration of some drugs (oral administration) The Gut contains various immune components that react to pathogens and foreign substances including drugs Immune rejection to newly developed drugs can be initiated in gut, thus, models assessing drug-gut immune interaction are desirable | [60] |
| Peristalsis motion | [61,65,66] | ||
| Microbial interface | [61,66,68] | ||
| Substance Transport | [61] | ||
| Gut viral infection | [41] | ||
| Villi | [61,65,66,67] | ||
| Immune Competence | [66] | ||
| Liver | Liver sinusoid | The liver has been shown to play key roles in immunity containing the body’s largest population of macrophages (Kupffer cells) Drug toxicity to the liver is the most common cause for the discontinuation of clinical trials on a drug, as well as the most common reason for an approved withdrawal of a drug from the marketplace [89] Thus, interactions between liver immune components and drugs should be assessed | [74,80,86,87] |
| Substance Transport | [75,79,80,83,84] | ||
| Metabolism | [77,80,84] | ||
| Bile Canaliculi | [83] | ||
| Secretion of liver specific products (urea and albumin) | [82,84,86] | ||
| Hepatocyte-endothelial interface | [74,78,81,87] | ||
| Hepatocyte-fibroblast interaction | [82] | ||
| Hepatocyte-stellate cell interaction | [85] | ||
| Hepatitis B virus Replication | [87] | ||
| Drug toxicity testing | [77,80,84] | ||
| Lymph node | Chemotaxis | Lymph node is the site of immune coordination in the body Adaptive immune responses against newly developed drugs are initiated in the lymph node thus, lymph node models can be utilized to modulate response of immune cells to drugs | [97,98] |
| Immune cell interactions | [93,96] | ||
| Response to vaccines and drugs | [99,100] | ||
| Spleen | Blood filtration | Spleen filters the blood and removes pathogens and other foreign substances including pharmaceutical drugs Models assessing the cleansing process of the spleen which involves immune components such as macrophages are highly desirable especially for assessing how drugs are eliminated | [103,105] |
| Blood cleansing | [105] | ||
| Bone Marrow | Hematopoietic Niche Formation | The bone marrow is the site of production of all immune cells as well as the site of selection and maturation of B lymphocytes Models examining the development of lymphocytes within bone marrow and investigating the priming process of B-cells as well as the specific cellular mechanism of recognizing and responding to an antigen can aid in developing drugs that are less toxic to the immune system | [40,110] |
| Blood and immune cell production | [109,110] | ||
| Responsiveness to drugs | [109,111] | ||
| Lymphatic vessels | Microcirculation | Exchange of substances between circulatory and lymphatic system occur through lymphatic vessels therefore, models examining how pharmaceutical drugs leave bloodstream and enter into nearby afferent lymphatics are highly desirable | [42] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shanti, A.; Teo, J.; Stefanini, C. In Vitro Immune Organs-on-Chip for Drug Development: A Review. Pharmaceutics 2018, 10, 278. https://doi.org/10.3390/pharmaceutics10040278
Shanti A, Teo J, Stefanini C. In Vitro Immune Organs-on-Chip for Drug Development: A Review. Pharmaceutics. 2018; 10(4):278. https://doi.org/10.3390/pharmaceutics10040278
Chicago/Turabian StyleShanti, Aya, Jeremy Teo, and Cesare Stefanini. 2018. "In Vitro Immune Organs-on-Chip for Drug Development: A Review" Pharmaceutics 10, no. 4: 278. https://doi.org/10.3390/pharmaceutics10040278
APA StyleShanti, A., Teo, J., & Stefanini, C. (2018). In Vitro Immune Organs-on-Chip for Drug Development: A Review. Pharmaceutics, 10(4), 278. https://doi.org/10.3390/pharmaceutics10040278