Crystallization Tendency of Pharmaceutical Glasses: Relevance to Compound Properties, Impact of Formulation Process, and Implications for Design of Amorphous Solid Dispersions


Abstract
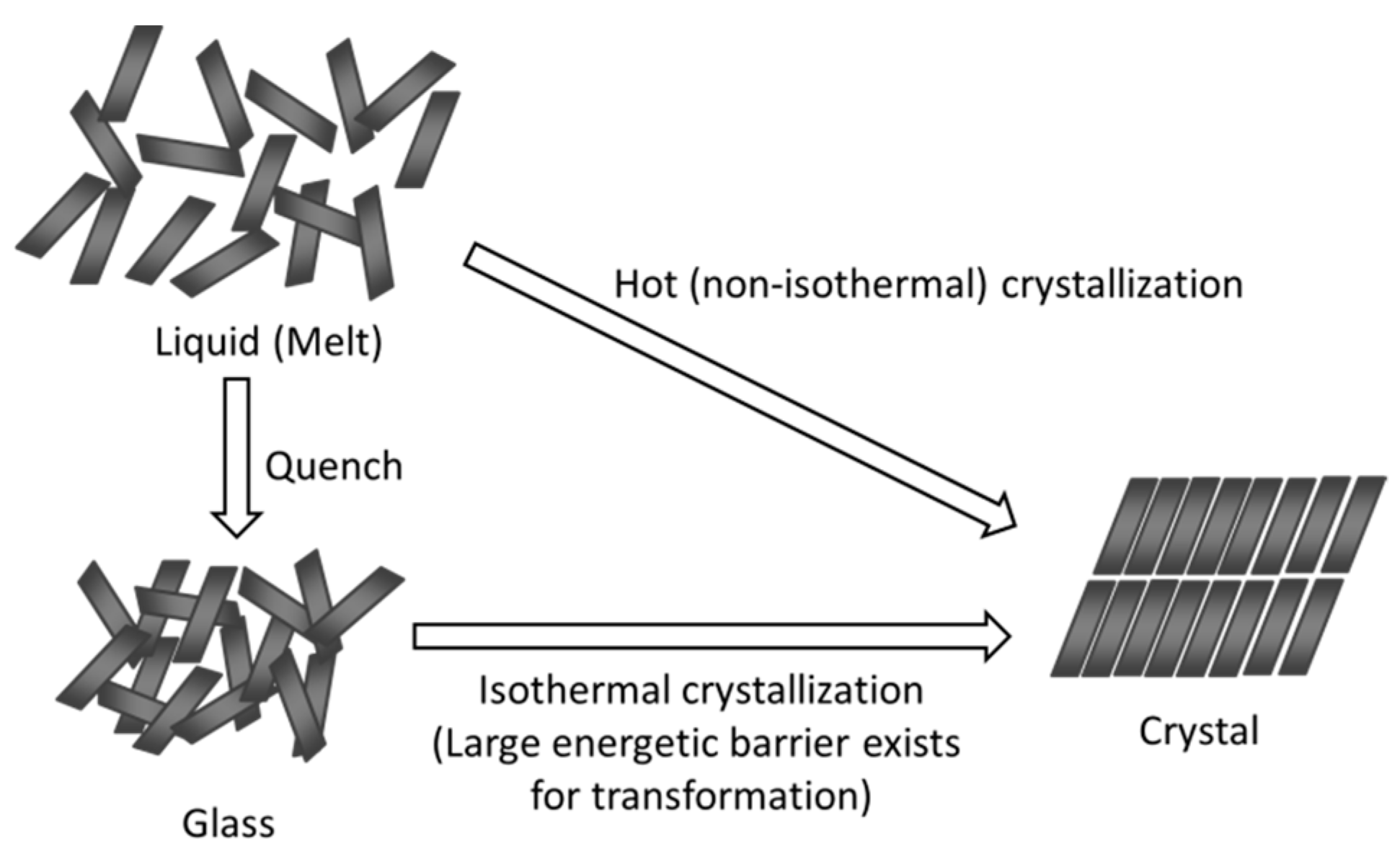
:1. Introduction
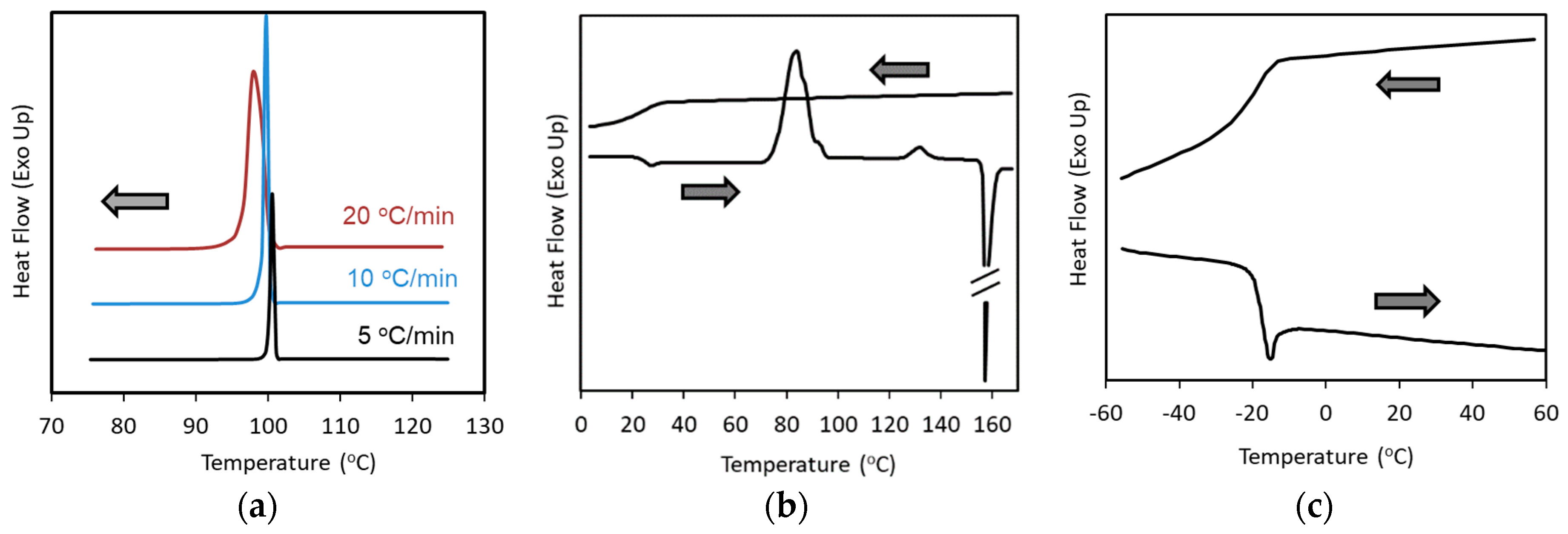
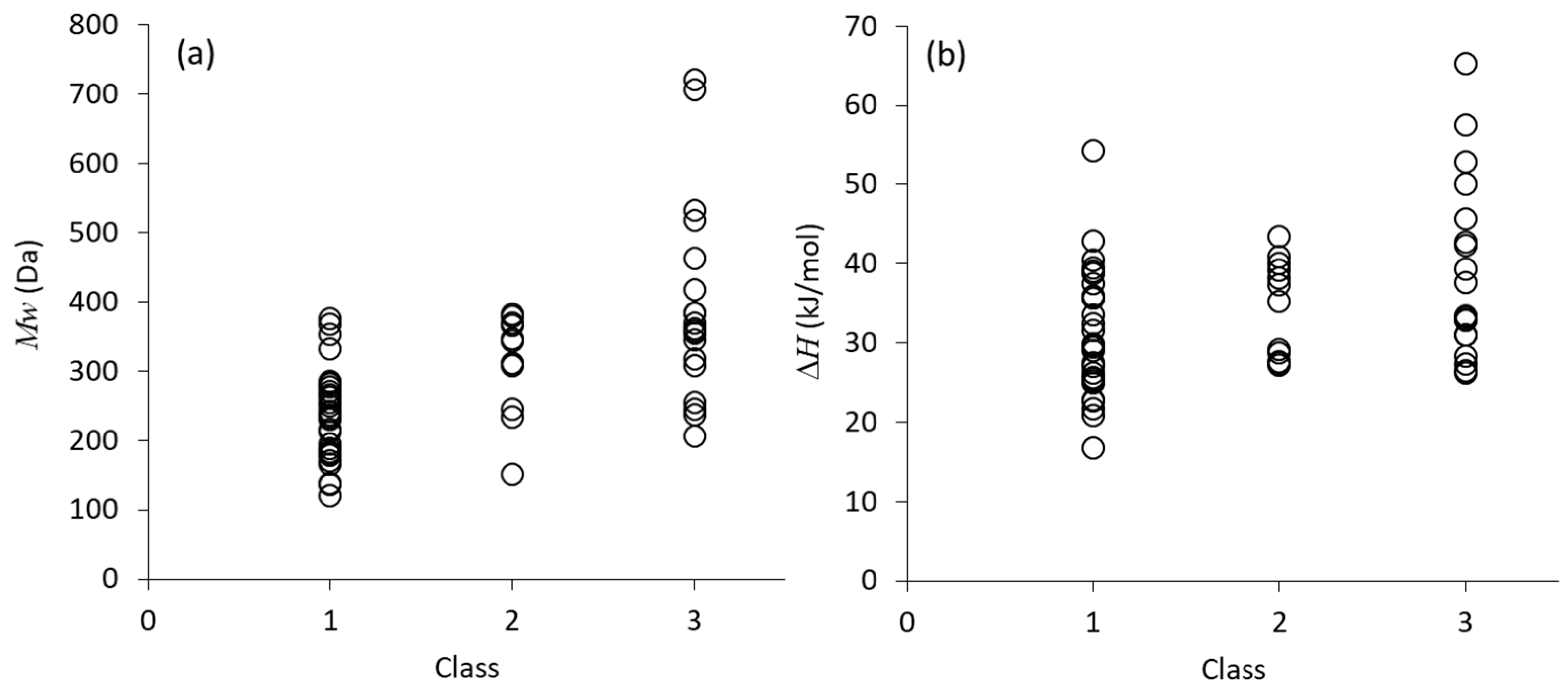
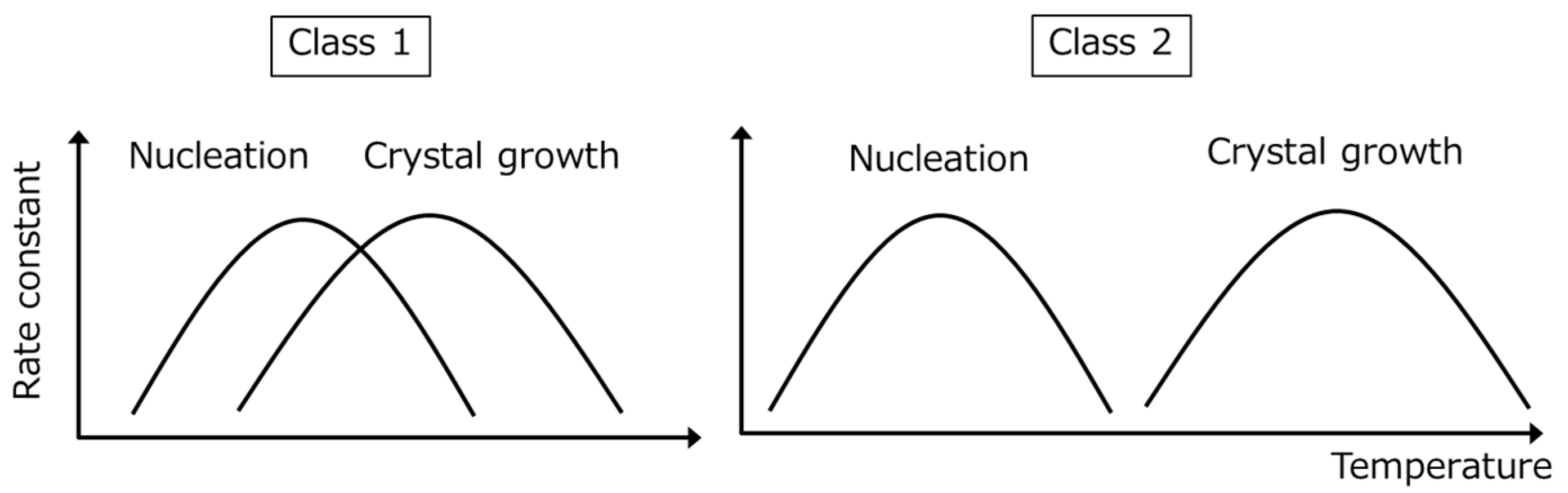
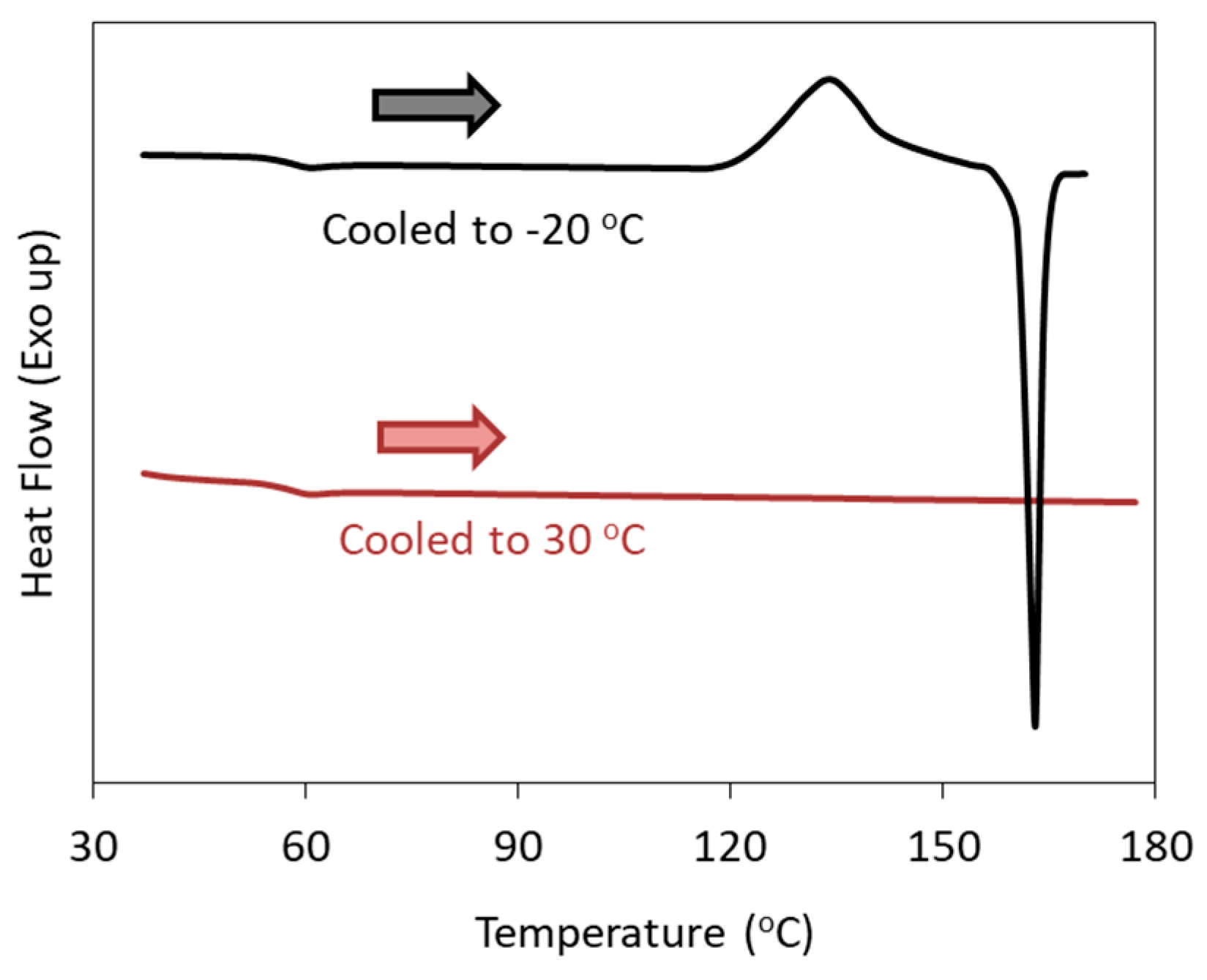
2. Classification of Crystallization Tendencies
- Class 1: Compounds that crystallize during cooling from the melt at 20 °C/min.
- Class 2: Compounds that do not crystallize during cooling from the melt, but crystallize during subsequent reheating at 10 °C/min.
- Class 3: Compounds that do not crystallize during the cooling/reheating cycle mentioned above.
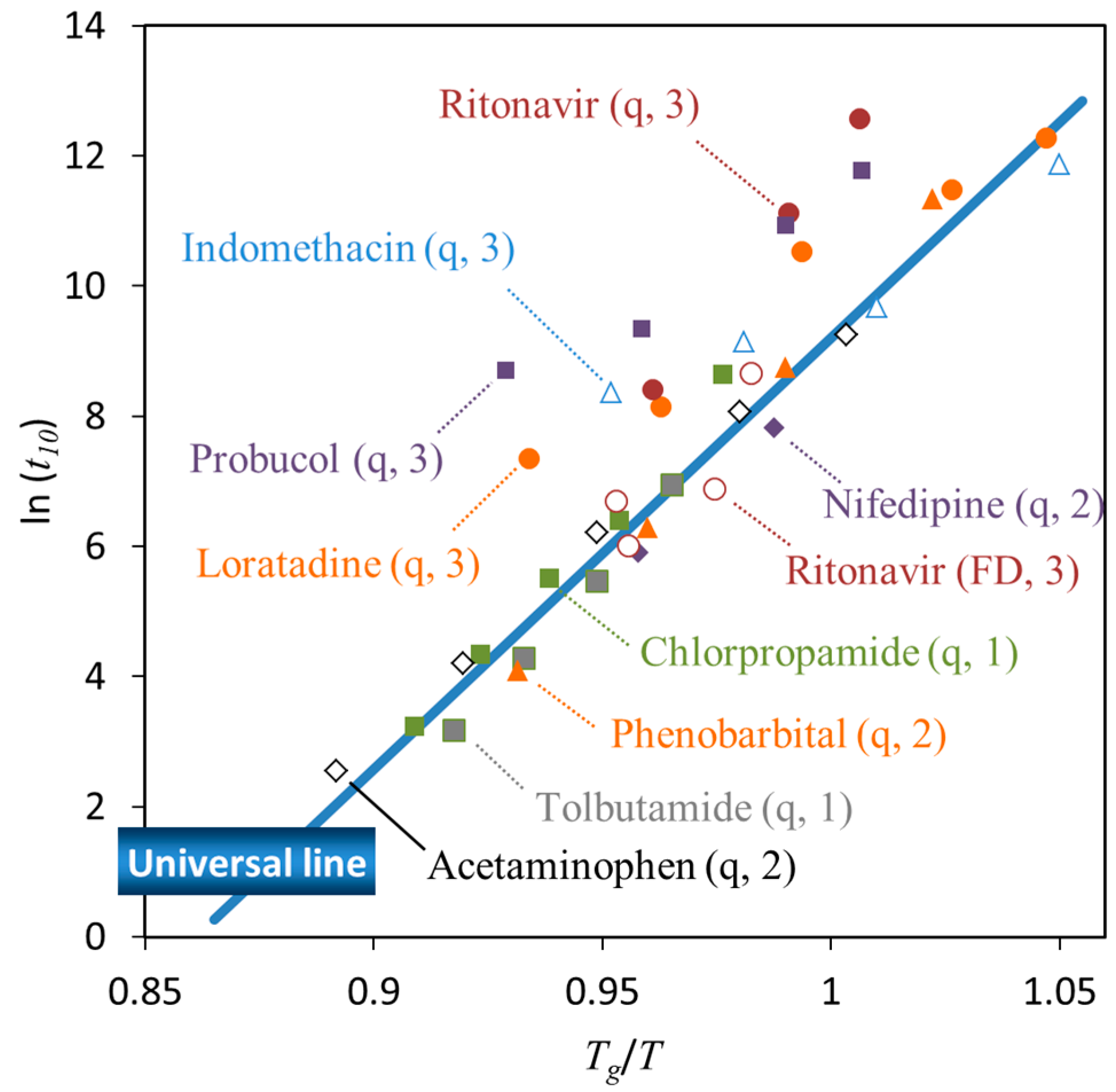
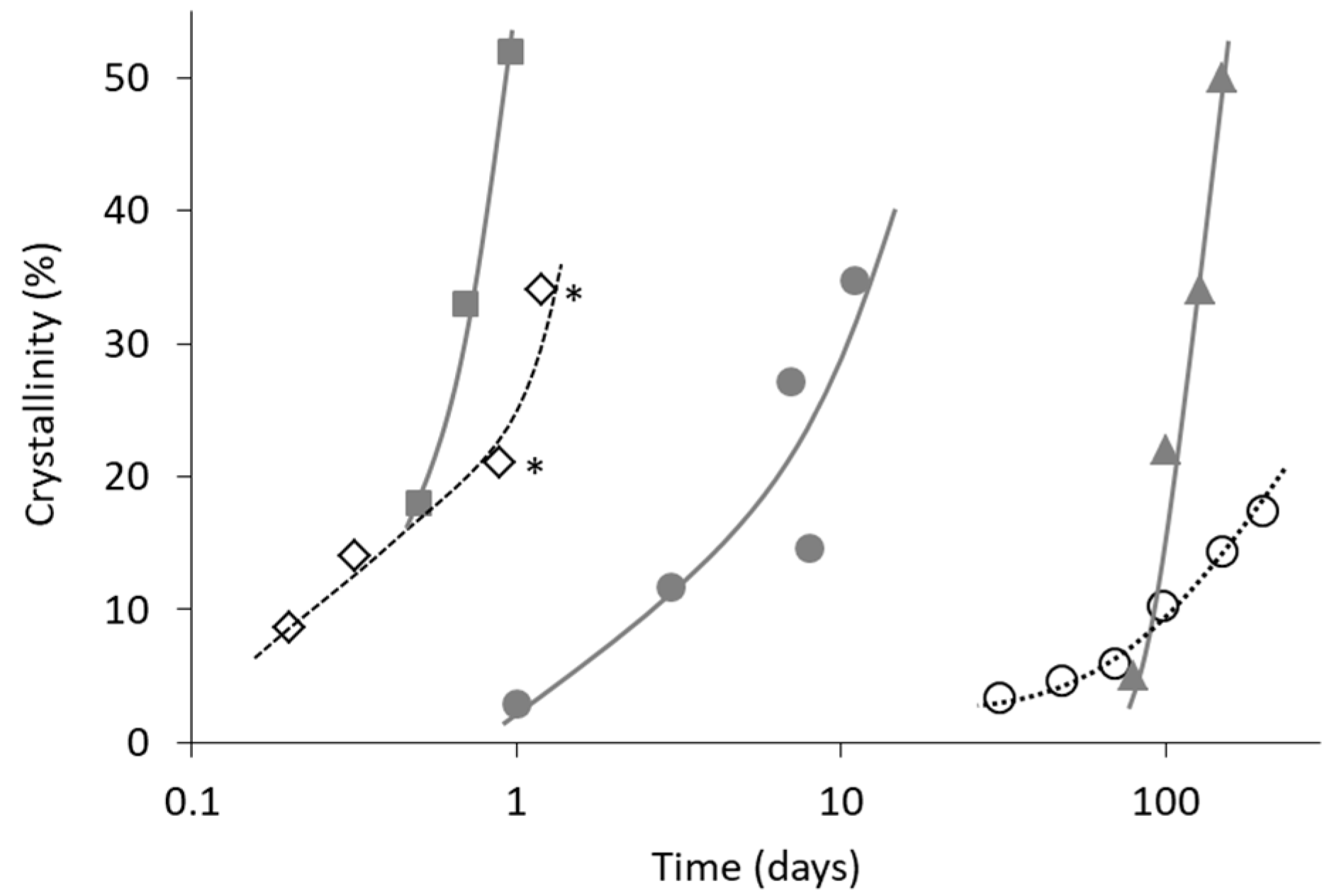
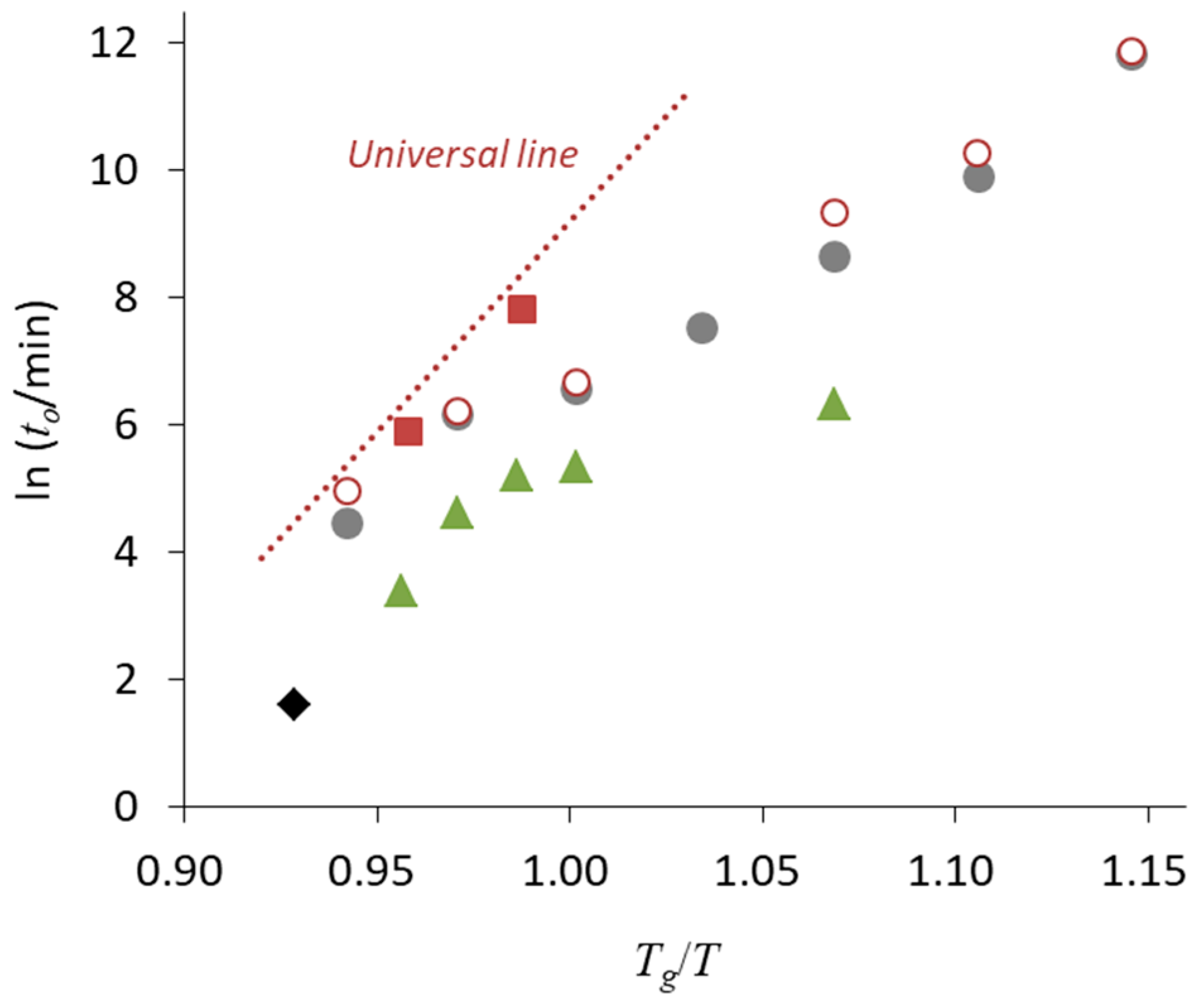
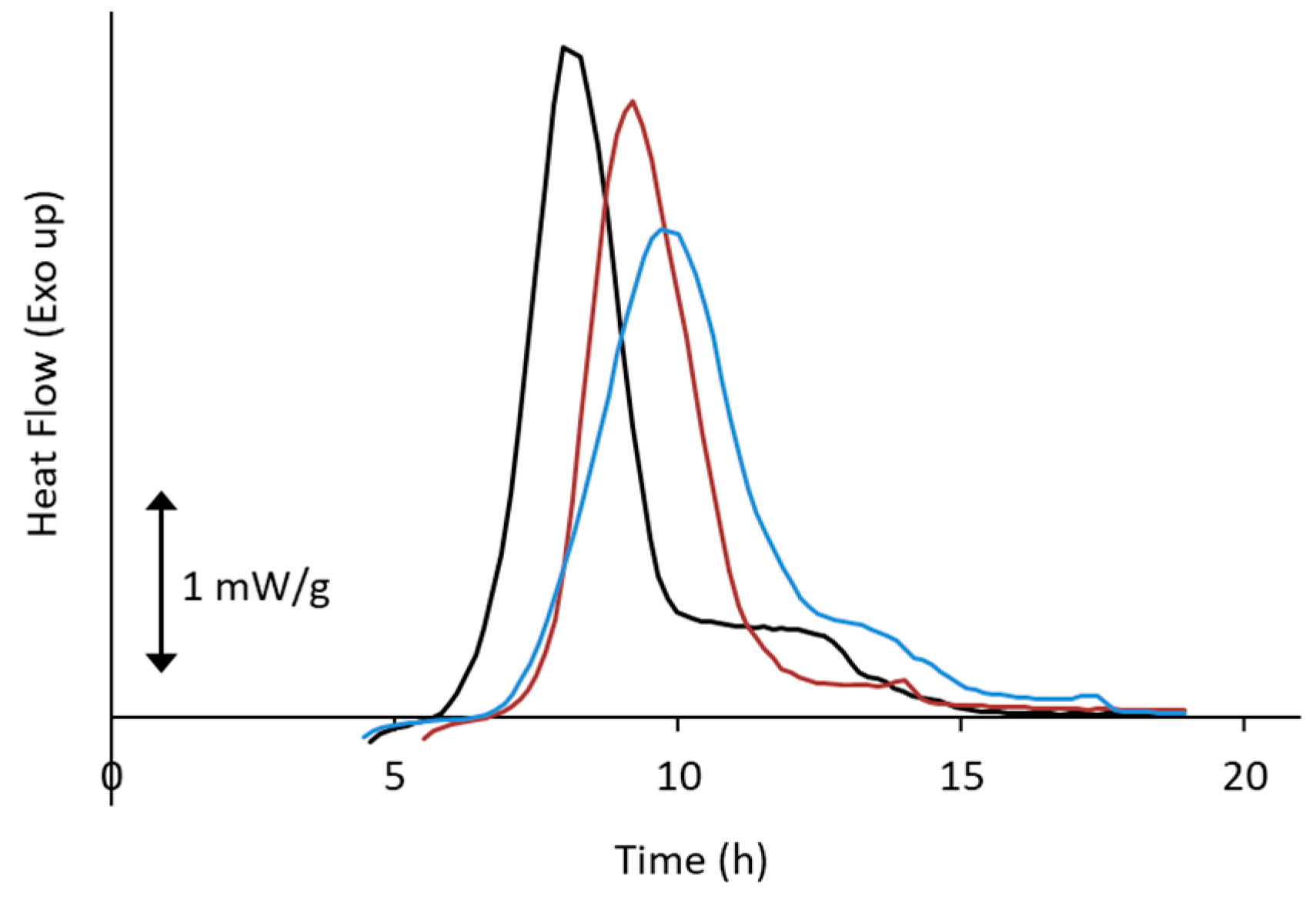
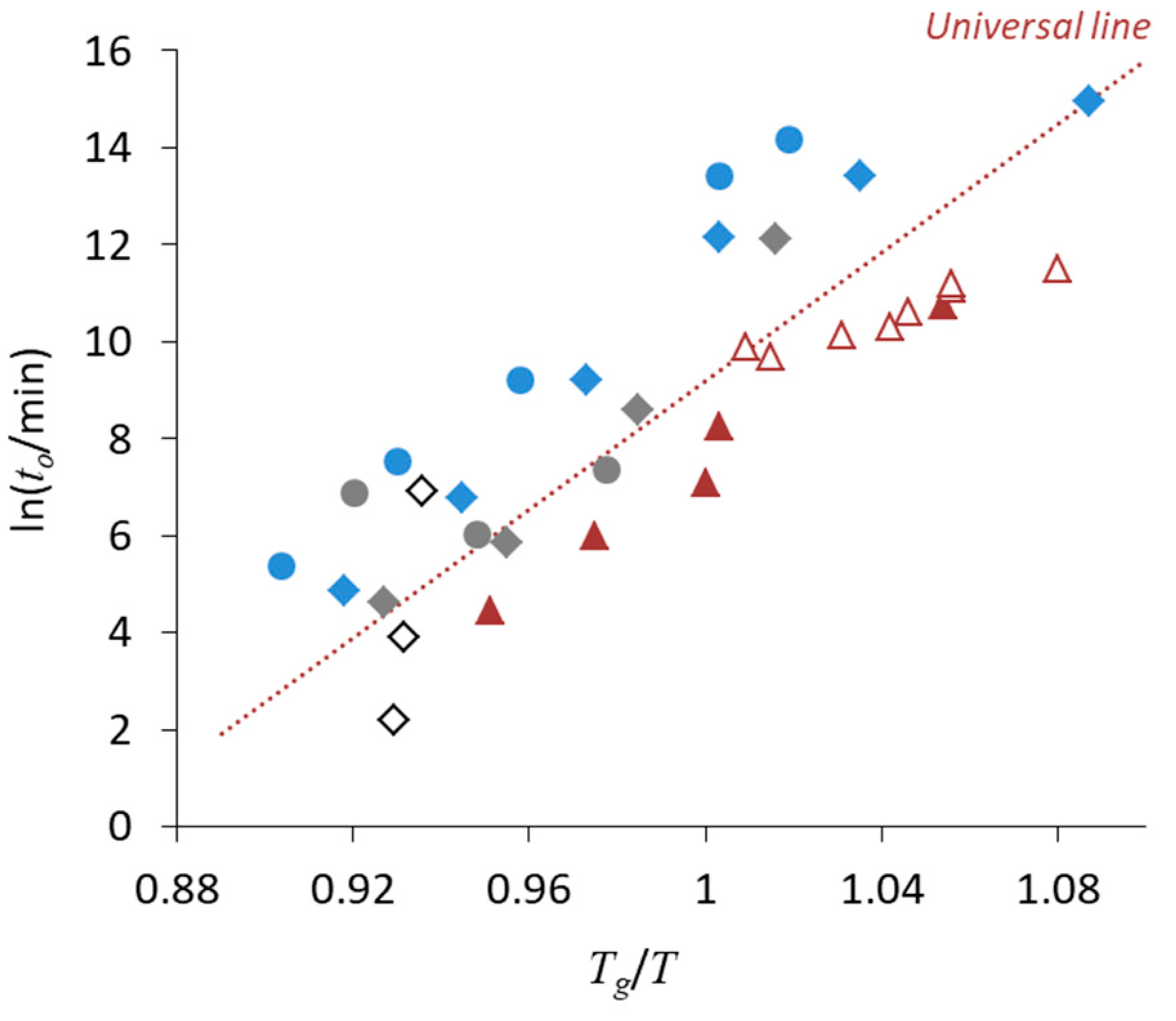
3. Relationship between Crystallization Tendency and Isothermal Crystallization
4. Non-Ideal Crystallization of Practical Glasses
5. Relevance to Formulation Research
6. Relevance to the Dissolution Benefits of ASDs
7. Summary
Funding
Conflicts of Interest
References
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating drug delivery systems: The answer to solubility-limited oral bioavailability? J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef]
- Kawakami, K. Modification of physicochemical characteristics of active pharmaceutical ingredients and application of supersaturatable dosage forms for improving bioavailability of poorly absorbed drugs. Adv. Drug Deliv. Rev. 2012, 64, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Paudel, A.; Worku, Z.A.; Meeus, J.; Guns, S.; Van den Mooter, G. Manufacturing of solid dispersions of poorly soluble drugs by spray drying: Formulation and process considerations. Int. J. Pharm. 2013, 453, 253–284. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K. Theory and practice of supersaturatable formulations for poorly soluble drugs. Ther. Deliv. 2015, 6, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Singh, A. Spray drying formulation of amorphous solid dispersions. Adv. Drug Deliv. Rev. 2016, 100, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Repka, M.A.; Bandari, S.; Kallakunta, V.R.; Vo, A.Q.; McFall, H.; Pimparade, M.B.; Bhagurkar, A.M. Melt extrusion with poorly solubloe drugs—An integrated review. Int. J. Pharm. 2018, 535, 68–85. [Google Scholar] [CrossRef]
- Jermain, S.V.; Brough, C.; Williams, R.O., III. Amorphous solid dispersions and nanocrystal technologies for poorly water-soluble drug delivery—An update. Int. J. Pharm. 2018, 535, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Twist, J.N.; Zatz, J.L. Influence of solvents on paraben permeation through idealized skin model membranes. J. Soc. Cosmet. Chem. 1986, 37, 429–444. [Google Scholar]
- Hens, B.; Brouwers, J.; Corsetti, M.; Augustijns, P. Gastrointestinal behavior of nano- and microsized fenofibrate: In vivo evaluation in man and in vitro simulation by assessment of the permeation potential. Eur. J. Pharm. Sci. 2015, 77, 40–47. [Google Scholar] [CrossRef]
- Porat, D.; Dahan, A. Active intestinal drug absorption and the solubility-permeability interplay. Int. J. Pharm. 2018, 537, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.S.; Zhang, G.G.Z. Physical chemistry of supersaturated solutions and implications for oral absorption. Adv. Drug Deliv. Rev. 2016, 101, 122–142. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K. Supersaturation and Crystallization: Non-equilibrium dynamics of amorphous solid dispersions for oral drug delivery. Exp. Opin. Drug Deliv. 2017, 14, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Sato, K.; Fukushima, M.; Miyazaki, A.; Yamamura, Y.; Sakuma, S. Phase separation of supersaturated solution created from amorphous solid dispersions: Relevance to oral absorption. Eur. J. Pharm. Biopharm. 2018, 132, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Maintaining supersaturation in aqueous drug solutions: Impact of different polymers on induction times. Cryst. Growth Des. 2013, 13, 740–751. [Google Scholar] [CrossRef]
- Jackson, M.J.; Toth, S.J.; Kestur, U.S.; Huang, J.; Qian, F.; Hussain, M.A.; Simpson, G.J.; Taylor, L.S. Impact of polymers on the precipitation behavior of highly supersaturated aqueous danazol solutions. Mol. Pharm. 2014, 11, 3027–3038. [Google Scholar] [CrossRef] [PubMed]
- Raina, S.A.; Zhang, G.G.Z.; Alonzo, D.E.; Wu, J.; Zhu, D.; Catron, N.D.; Gao, Y.; Taylor, L.S. Impact of solubilizing additives on supersaturation and membrane transport of drugs. Pharm. Res. 2015, 32, 3350–3364. [Google Scholar] [CrossRef]
- Indulkar, A.S.; Gao, Y.; Raina, S.A.; Zhang, G.G.Z.; Taylor, L.S. Exploiting the phenomenon of liquid-liquid phase separation for enhanced and sustained membrane transport of a poorly soluble drug. Mol. Pharm. 2016, 13, 2059–2069. [Google Scholar] [CrossRef]
- Stewart, A.M.; Grass, M.E.; Brodeur, T.J.; Goodwin, A.K.; Morgen, M.M.; Friesen, D.T.; Vodak, D.T. Impact of drug-rich colloids of itraconazole and HPMCAS on membrane flux in vitro and oral bioavailability in rats. Mol. Pharm. 2017, 14, 2437–2449. [Google Scholar] [CrossRef]
- Liu, H.; Wang, P.; Zhang, X.; Shen, F.; Gogos, C.G. Effects of extrusion process parameters on the dissolution behavior of indomethacin in Eudragit E PO solid dispersions. Int. J. Pharm. 2010, 383, 161–169. [Google Scholar] [CrossRef]
- Zhang, S.; Kawakami, K.; Yamamoto, M.; Masaoka, Y.; Kataoka, M.; Yamashita, S.; Sakuma, S. Coaxial electrospray formulations for improving oral absorption of a poorly water-soluble drug. Mol. Pharm. 2011, 8, 807–813. [Google Scholar] [CrossRef]
- Xie, T.; Taylor, L.S. Effect of temperature and moisture on the physical stability of binary and ternary amorphous solid dispersion of celecoxib. J. Pharm. Sci. 2017, 106, 100–110. [Google Scholar] [CrossRef]
- Lehmkemper, K.; Kyeremateng, S.O.; Heinzerling, O.; Dagenhardt, M.; Sadowski, G. Impact of polymer type and relative humidity on the long-term physical stability of amorphous solid dispersions. Mol. Pharm. 2017, 14, 4374–4386. [Google Scholar] [CrossRef]
- Kawakami, K.; Bi, Y.; Yoshihashi, Y.; Sugano, K.; Terada, K. Time-dependent phase separation of amorphous solid dispersions: Implications for accelerated stability studies. J. Drug Deliv. Sci. Technol. 2018, 46, 197–206. [Google Scholar] [CrossRef]
- Mahlin, D.; Bergström, C.A.S. Early drug development predictions of glass-forming ability and physical stability of drugs. Eur. J. Pharm. Sci. 2013, 49, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, T.; Yoshioka, S.; Aso, Y.; Kawanishi, T. Crystallization rate of amorphous nifedipine analogues unrelated to the glass transition temperature. Int. J. Pharm. 2007, 336, 191–195. [Google Scholar] [CrossRef]
- Baird, J.A.; van Eerdenbrugh, B.; Taylor, L.S. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J. Pharm. Sci. 2010, 99, 3787–3806. [Google Scholar] [CrossRef]
- Mahlin, D.; Ponnambalam, S.; Hockerfeit, M.H.; Bergström, C.A.S. Toward in silico prediction of glass-forming ability from molecular structure alone: A screening tool in early drug development. Mol. Pharm. 2011, 8, 498–506. [Google Scholar] [CrossRef]
- Angell, C.A. Relaxation in liquids, polymers and plastic crystals—Strong/fragile patterns and problems. J. Non-Cryst. Solids 1991, 131–133, 13–31. [Google Scholar] [CrossRef]
- Crowley, K.J.; Zografi, G. The use of thermal methods for predicting glass-former ability. Thermochim. Acta 2001, 380, 79–93. [Google Scholar] [CrossRef]
- Senkov, O.N. Correlation between fragility and glass-forming ability of metallic alloys. Phys. Rev. B 2007, 76, 104202. [Google Scholar] [CrossRef]
- Kawakami, K.; Harada, T.; Yoshihashi, Y.; Yonemochi, E.; Terada, K.; Moriyama, H. Correlation between glass forming ability and fragility of pharmaceutical compounds. J. Phys. Chem. B 2015, 119, 4873–4780. [Google Scholar] [CrossRef]
- Pajula, K.; Taskinen, M.; Lehto, V.P.; Ketolainen, J.; Korhonen, O. Predicting the formation and stability of amorphous small molecule binary mixtures from computationally determined Flory-Huggins interaction parameter and phase diagram. Mol. Pharm. 2010, 7, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Usui, T.; Hattori, M. Understanding the glass-forming ability of active pharmaceutical ingredients for designing supersaturating dosage forms. J. Pharm. Sci. 2012, 101, 3239–3248. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.A.; Thomas, L.C.; Aubuchon, S.R.; Taylor, L.S. Evaluating the non-isothermal crystallization behavior of organic molecules from the undercooled melt state during rapid heat/cool calorimetry. CrystEngComm 2013, 15, 111–119. [Google Scholar] [CrossRef]
- Wyttenback, N.; Kirchmeyer, W.; Alsenz, J.; Kuentz, M. Theoretical considerations of the prigogine-defay ratio with regard to the glass-forming ability of drugs from undercooled melts. Mol. Pharm. 2016, 13, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Li, X.; Chen, Z.; Liu, Y.D.; Labardi, M.; Capaccioli, S.; Paluch, M.; Wang, L.M. Glass formability in medium-sized molecular system/pharmaceuticals. I. Thermodynamics vs. kinetics. J. Chem. Phys. 2016, 144, 174502. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Chamarthy, S.P.; Byrn, S.R.; Pinal, R. A calorimetric method to estimate molecular mobility of amorphous solids at relatively low temperatures. Pharm. Res. 2006, 23, 2269–2276. [Google Scholar] [CrossRef] [PubMed]
- Blaabjerg, L.I.; Lindenberg, E.; Löbmann, K.; Grohganz, H.; Rades, T. Glass forming ability of amorphous drugs investigated by continuous cooling and isothermal transformation. Mol. Pharm. 2016, 13, 3318–3325. [Google Scholar] [CrossRef]
- Kawakami, K.; Ohba, C. Crystallization of probucol from solution and the glassy state. Int. J. Pharm. 2017, 517, 322–328. [Google Scholar] [CrossRef]
- Kawakami, K. Dynamics of ribavirin glass in sub-Tg temperature region. J. Phys. Chem. B 2011, 115, 11375–11381. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Gutzow, I. Structural order parameters, the Prigogine-Defay ratio and the behavior of the entropy in vitrification. J. Non-Cryst. Solids 2009, 355, 653–662. [Google Scholar] [CrossRef]
- Blaabjerg, L.I.; Lindenberg, E.; Rades, T.; Grohganz, H.; Löbmann, K. Influence of preparation pathway on the glass forming ability. Int. J. Pharm. 2017, 521, 232–238. [Google Scholar] [CrossRef]
- Kawakami, K. Nucleation and crystallization of celecoxib glass: Impact of experience of low temperature on physical stability. Thermochim. Acta 2019, 671, 43–47. [Google Scholar] [CrossRef]
- Kawakami, K.; Harada, T.; Miura, K.; Yoshihashi, Y.; Yonemochi, E.; Terada, K.; Moriyama, H. Relationship between crystallization tendencies during cooling from melt and isothermal storage: Toward a general understanding of physical stability of pharmaceutical glasses. Mol. Pharm. 2014, 11, 1835–1843. [Google Scholar] [CrossRef]
- Wu, T.; Yu, L. Surface crystallization of indomethacin below Tg. Pharm. Res. 2006, 23, 2350–2355. [Google Scholar] [CrossRef]
- Wu, T.; Sun, Y.; Li, N.; de Villiers, M.M.; Yu, L. Inhibiting surface crystallization of amorphous indomethacin by nanocoating. Langmuir 2007, 23, 5148–5153. [Google Scholar] [CrossRef]
- Kawakami, K. Surface effects on the crystallization of ritonavir glass. J. Pharm. Sci. 2015, 104, 276–279. [Google Scholar] [CrossRef]
- Bell, R.C.; Wang, H.; Iedema, M.J.; Cowin, J.P. Nanometer-resolved interfacial fluidity. J. Am. Chem. Soc. 2003, 125, 5176–5185. [Google Scholar] [CrossRef]
- Mirigian, S.; Schweizer, K.S. Slow relaxation, spatial mobility gradients, and vitrification in confined films. J. Chem. Phys. 2014, 141, 161103. [Google Scholar] [CrossRef]
- Tominaka, S.; Kawakami, K.; Fukushima, M.; Miyazaki, A. Physical stabilization of pharmaceutical glasses based on hydrogen bond reorganization under sub-Tg temperature. Mol. Pharm. 2017, 14, 264–273. [Google Scholar] [CrossRef]
- Crowley, K.J.; Zografi, G. Cryogenic grinding of indomethacin polymorphs and solvates: Assessment of amorphous phase formation and amorphous phase physical stability. J. Pharm. Sci. 2002, 91, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Bhugra, C.; Shmeis, R.; Pikal, M.J. Role of mechanical stress in crystallization and relaxation behavior of amorphous indomethacin. J. Pharm. Sci. 2008, 97, 4446–4458. [Google Scholar] [CrossRef] [PubMed]
- Descamps, M.; Dudognon, E. Crystallization from the amorphous state: Nucleation-growth decoupling, polymorphism interplay, and the role of interfaces. J. Pharm. Sci. 2014, 103, 2615–2628. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Kaneniwa, N. A kinetic study of the crystallization process of noncrystalline indomethacin under isothermal conditions. Chem. Pharm. Bull. 1988, 36, 4026–4032. [Google Scholar] [CrossRef]
- Andronis, V.; Zografi, G. Crystal nucleation and growth of indomethacin polymorphs from the amorphous state. J. Non-cryst. Solids 2000, 271, 236–248. [Google Scholar] [CrossRef]
- Bhugra, C.; Shmeis, R.; Krill, S.L.; Pikal, M.J. Prediction of onset of crystallization from experimental relaxation times. II. Comparison between predicted and experimental onset times. J. Pharm. Sci. 2008, 97, 455–472. [Google Scholar] [CrossRef]
- Aso, Y.; Yoshioka, S.; Kojima, S. Relationship between the crystallization rates of amorphous nifedipine, phenobarbital, and flopropione, and their molecular mobility as measured by their enthalpy relaxation and 1H NMR relaxation times. J. Pharm. Sci. 2000, 89, 408–416. [Google Scholar] [CrossRef]
- Kothari, K.; Ragoonanan, V.; Suryanarayanan, R. The role of polymer concentration on the molecular mobility and physical stability of nifedipine solid dispersions. Mol. Pharm. 2015, 12, 1477–1484. [Google Scholar] [CrossRef]
- Kawakami, K.; Miyoshi, K.; Tamura, N.; Yamaguchi, T.; Ida, Y. Crystallization of sucrose glass under ambient conditions: Evaluation of crystallization rate and unusual melting behavior of resultant crystals. J. Pharm. Sci. 2006, 95, 1354–1363. [Google Scholar] [CrossRef]
- Ayenew, Z.; Paudel, A.; Rombaut, P.; Van den Mooter, G. Effect of compression on non-isothermal crystallization behaviour of amorphous indomethacin. Pharm. Res. 2012, 29, 2489–2498. [Google Scholar] [CrossRef]
- Rams-Baron, M.; Pacult, J.; Jedrzejowska, A.; Knapik-Kowalczuk, J.; Paluch, M. Changes in physical stability of supercooled etoricoxib after compression. Mol. Pharm. 2018, 15, 2969–3978. [Google Scholar] [CrossRef] [PubMed]
- van Eerdenbrugh, B.; Baird, J.A.; Taylor, L.S. Crystallization tendency of active pharmaceutical ingredients following rapid solvent evaporation—Classification and comparison with crystallization tendency from undercooled melts. J. Pharm. Sci. 2010, 99, 3826–3838. [Google Scholar] [CrossRef]
- Kawakami, K. Miscibility analysis of particulate solid dispersions prepared by electrospray deposition. Int. J. Pharm. 2012, 433, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Vehring, R. Phamaceutical particle engineering via spray drying. Pharm. Res. 2008, 25, 999–1022. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Hasegawa, Y.; Deguchi, K.; Ohki, S.; Shimizu, T.; Yoshihashi, Y.; Yonemochi, E.; Terada, K. Competition of thermodynamic and dynamic factors during formation of multicomponent particles via spray drying. J. Pharm. Sci. 2013, 102, 518–529. [Google Scholar] [CrossRef]
- Calahan, J.L.; Azali, S.C.; Munson, E.J.; Nagapudi, K. Investigation of phase mixing in amorphous solid dispersions of AMG 517 in HPMC-AS using DSC, solid-state NMR, and solution calorimetry. Mol. Pharm. 2015, 12, 4115–4123. [Google Scholar] [CrossRef] [PubMed]
- Bank, M.; Leffingwell, J.; Thies, C. The influence of solvent upon the compatibility of polystyrene and poly(vinyl methyl ether). Macromolecules 1971, 4, 43–46. [Google Scholar] [CrossRef]
- Aso, Y.; Yoshioka, S.; Kojima, S. Molecular mobility-based estimation of the crystallization rates of amorphous nidedipine and phenobarbital in poly(vinylpyrrolidone) solid dispersions. J. Pharm. Sci. 2004, 93, 384–391. [Google Scholar] [CrossRef]
- Caron, V.; Bhugra, C.; Pikal, M.J. Prediction of onset of crystallization in amorphous pharmaceutical systems: Phenobarbital, nifedipine/PVP, and phenobarbital/PVP. J. Pharm. Sci. 2010, 99, 3887–3900. [Google Scholar] [CrossRef]
- Greco, S.; Authelin, J.R.; Leveder, C.; Segalini, A. A practical method to predict physical stability of amorphous solid dispersions. Mol. Pharm. 2012, 29, 2792–2805. [Google Scholar] [CrossRef] [PubMed]
- Greco, K.; Bogner, R. Solution-mediated phase transformation: Significance during dissolution and implications for bioavailability. J. Pharm. Sci. 2012, 101, 2996–3018. [Google Scholar] [CrossRef] [PubMed]
- Blaabjerg, L.I.; Lindenberg, E.; Löbmann, K.; Grohganz, H.; Rades, T. Is there a correlation between the glass forming ability of a drug and its supersaturation propensity? Int. J. Pharm. 2018, 538, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Alhalaweh, A.; Alzghoul, A.; Bergström, C.A.S. Molecular divers of crystallization kinetics for drugs in supersaturated aqueous solutions. J. Pharm. Sci. 2019, 108, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, N.; Kuentz, M. Glass-forming ability of compounds in marketed amorphous drug products. Eur. J. Pharm. Biopharm. 2017, 112, 204–208. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical-Structural Features | Physicochemical Features |
|---|---|
| Large molecular weight | Large melting enthalpy/entropy |
| Low number of benzene rings | High melting temperature |
| Low symmetry | Large crystal/amorphous energy difference |
| Large number of rotatable bonds | Large fragility |
| High branching degree | Large Tg/Tm |
| Large number of electronegative atoms | Large viscosity above Tg |
| Compounds | Mw (Da) | Tm (°C) | Tg (°C) | Tg/Tm | ΔH (kJ/mol) | m | Reference |
|---|---|---|---|---|---|---|---|
| Antipyrin | 188 | 111 | −25 | 0.65 | 25.2 | 81 | [31] |
| Anthranilic acid | 137 | 147 | 5 | 0.66 | 22.8 | - | [26] |
| Atenolol | 266 | 153 | 22 | 0.69 | 37.5 | - | [26] |
| Atovaquone | 367 | 219 | - | - | 33.5 | - | [35] |
| Benzamide | 121 | 127 | −10 | 0.66 | 21.7 | - | [26] |
| Benzocaine | 165 | 89 | −31 | 0.67 | 22.6 | - | [26] |
| Caffeine | 194 | 237 | 72 | 0.68 | 20.8 | - | [26] |
| Carbamazepine | 236 | 192 | 61 | 0.72 | 25.5 | - | [26] |
| Chlorpropamide | 277 | 118 | 17 | 0.74 | 27.4 | 219 | [31] |
| Chlorzoxazone | 170 | 191 | 38 | 0.67 | 25.6 | - | [26] |
| Clofibric acid | 215 | 121 | - | - | 29.0 | - | [35] |
| Diflunisal | 250 | 213 | - | - | 35.6 | - | [35] |
| Felbinac | 212 | 164 | 24 | 0.68 | 29.8 | - | [26] |
| Flufenamic acid | 281 | 135 | 17 | 0.71 | 27.1 | 78 | [26,36] |
| Griseofulvin | 353 | 218 | 89 | 0.74 | 39.1 | 74 | [26,37] |
| Haloperidol | 376 | 152 | 33 | 0.72 | 54.3 | - | [26] |
| Indoprofen | 281 | 212 | 50 | 0.67 | 36.0 | - | [26] |
| Lidocaine | 234 | 68 | −39 | 0.69 | 16.7 | - | [26] |
| Mefenamic acid | 241 | 231 | - | - | 39.4 | - | [35] |
| Naproxen | 230 | 157 | 56 | 0.77 | 32.4 | - | [35,38] |
| Nepafenac | 254 | 183 | - | - | 42.8 | - | [35] |
| Phenacetin | 179 | 136 | 2 | 0.67 | 31.5 | - | [26] |
| Piroxicam | 331 | 201 | - | - | 35.6 | - | [35] |
| Probenecid | 285 | 199 | - | - | 40.4 | - | [35] |
| Saccharin | 183 | 228 | - | - | 29.5 | - | [35] |
| Salicylic acid | 138 | 159 | - | - | 24.9 | - | [35] |
| Theophylline | 180 | 272 | 94 | 0.67 | 29.6 | - | [26] |
| Tolbutamide | 270 | 128 | 5 | 0.69 | 26.2 | 122 | [31] |
| Tolfenamic acid | 262 | 213 | 63 | 0.69 | 38.8 | - | [26] |
| Average | 237 | 172 | 27 | 0.69 | 31.1 | 115 | - |
| Compounds | Mw (Da) | Tm (°C) | Tg (°C) | Tg/Tm | ΔH (kJ/mol) | m | Reference |
|---|---|---|---|---|---|---|---|
| Acetaminophen | 151 | 169 | 23 | 0.67 | 27.2 | 77 | [31] |
| Bifonazole | 310 | 149 | 16 | 0.68 | 39.2 | 76 | [31] |
| Celecoxib | 381 | 163 | 58 | 0.76 | 37.4 | 85 | [26] |
| Cinnarizine | 369 | 120 | 7 | 0.71 | 40.9 | 84 | [31] |
| Clofoctol | 365 | 88 | −4 | 0.75 | 35.2 | 70 | [26] |
| Dibucaine | 343 | 65 | −35 | 0.70 | 29.2 | 132 | [26] |
| Droperidol | 379 | 143 | 29 | 0.73 | 40.0 | 108 | [26] |
| Flurbiprofen | 244 | 115 | −5 | 0.69 | 27.4 | 88 | [31] |
| Nifedipine | 346 | 172 | 46 | 0.72 | 38.2 | 112 | [31] |
| Phenobarbital | 233 | 174 | 42 | 0.70 | 28.7 | 96 | [31] |
| Phenylbutazone | 308 | 106 | −6 | 0.70 | 27.6 | 79 | [36] |
| Tolazamide | 311 | 172 | 18 | 0.65 | 43.4 | 18 | [26] |
| Average | 312 | 136 | 16 | 0.71 | 34.5 | 85 | - |
| Compounds | Mw (Da) | Tm (°C) | Tg (°C) | Tg/Tm | ΔH (kJ/mol) | m | Reference |
|---|---|---|---|---|---|---|---|
| Aceclofenac | 354 | 153 | 10 | 0.66 | 42.3 | 25 | [26] |
| Clotrimazole | 345 | 141 | 28 | 0.73 | 33.3 | 63 | [31] |
| Curcumin | 368 | 182 | 62 | 0.74 | 50.1 | 87 | - |
| Felodipine | 384 | 147 | 45 | 0.76 | 31.0 | 66 | [26] |
| Fenofibrate | 361 | 80 | −19 | 0.72 | 33.0 | 82 | [31] |
| Ibuprofen | 206 | 76 | −44 | 0.66 | 26.5 | 75 | [31] |
| Indomethacin | 358 | 161 | 45 | 0.73 | 37.6 | 85 | [31] |
| Itraconazole | 706 | 168 | 58 | 0.75 | 57.6 | 731 | [26] |
| Ketoconazole | 531 | 147 | 44 | 0.75 | 52.9 | 97 | [31] |
| Ketoprofen | 254 | 95 | −3 | 0.73 | 28.3 | 67 | [31] |
| Loratadine | 383 | 134 | 35 | 0.76 | 27.3 | 72 | [31] |
| Miconazole | 417 | 86 | 1 | 0.76 | 32.8 | 61 | [26] |
| Nilutamide | 317 | 155 | 33 | 0.72 | 31.0 | 106 | [26] |
| Nimesulide | 308 | 150 | 21 | 0.70 | 33.4 | 103 | [26] |
| Pimozide | 462 | 219 | 54 | 0.66 | 42.7 | 170 | [26] |
| Probucol | 517 | 126 | 27 | 0.75 | 39.3 | 138 | [39] |
| Procaine | 236 | 61 | −39 | 0.70 | 26.2 | 90 | [31] |
| Ribavirin | 244 | 168 | 56 | 0.75 | 45.7 | 70 | [40] |
| Ritonavir | 721 | 122 | 47 | 0.81 | 65.3 | 86 | [31] |
| Average | 393 | 135 | 24 | 0.73 | 38.8 | 120 | - |
| Classification | Compound | Avrami Exponent |
|---|---|---|
| Class 1 | Tolbutamide | 3.7–4.6 |
| Chlorpropamide | 3.0–4.2 | |
| Class 2 | Acetaminophen | 2.1–3.0 |
| Nifedipine | 2.0 | |
| Class 3 | Ritonavir | 2.2–3.1 |
| Indomethacin | 1.0–2.6 | |
| Loratadine | 1.1–1.5 | |
| Probucol | 1.2–1.3 |
| With increasing classification number: |
| • Nucleation becomes more heterogeneous |
| • The nucleation barrier becomes larger |
| • Surface effects become more important |
| • Vitrification during the formulation processes may become easier |
| • The supersaturation ability may increase |
| • The universal line in Figure 6 is applicable for Class 1 and 2 compounds, whereas the stability of Class 3 compounds may be better |
| • Stabilization may be achieved via thermal treatment |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawakami, K. Crystallization Tendency of Pharmaceutical Glasses: Relevance to Compound Properties, Impact of Formulation Process, and Implications for Design of Amorphous Solid Dispersions. Pharmaceutics 2019, 11, 202. https://doi.org/10.3390/pharmaceutics11050202
Kawakami K. Crystallization Tendency of Pharmaceutical Glasses: Relevance to Compound Properties, Impact of Formulation Process, and Implications for Design of Amorphous Solid Dispersions. Pharmaceutics. 2019; 11(5):202. https://doi.org/10.3390/pharmaceutics11050202
Chicago/Turabian StyleKawakami, Kohsaku. 2019. "Crystallization Tendency of Pharmaceutical Glasses: Relevance to Compound Properties, Impact of Formulation Process, and Implications for Design of Amorphous Solid Dispersions" Pharmaceutics 11, no. 5: 202. https://doi.org/10.3390/pharmaceutics11050202
APA StyleKawakami, K. (2019). Crystallization Tendency of Pharmaceutical Glasses: Relevance to Compound Properties, Impact of Formulation Process, and Implications for Design of Amorphous Solid Dispersions. Pharmaceutics, 11(5), 202. https://doi.org/10.3390/pharmaceutics11050202