Development of a Ternary Solid Dispersion Formulation of LW6 to Improve the In Vivo Activity as a BCRP Inhibitor: Preparation and In Vitro/In Vivo Characterization




Abstract
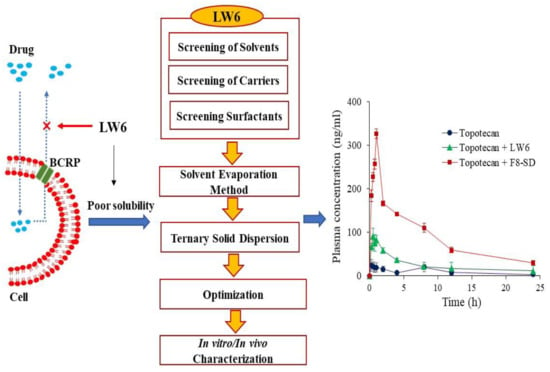
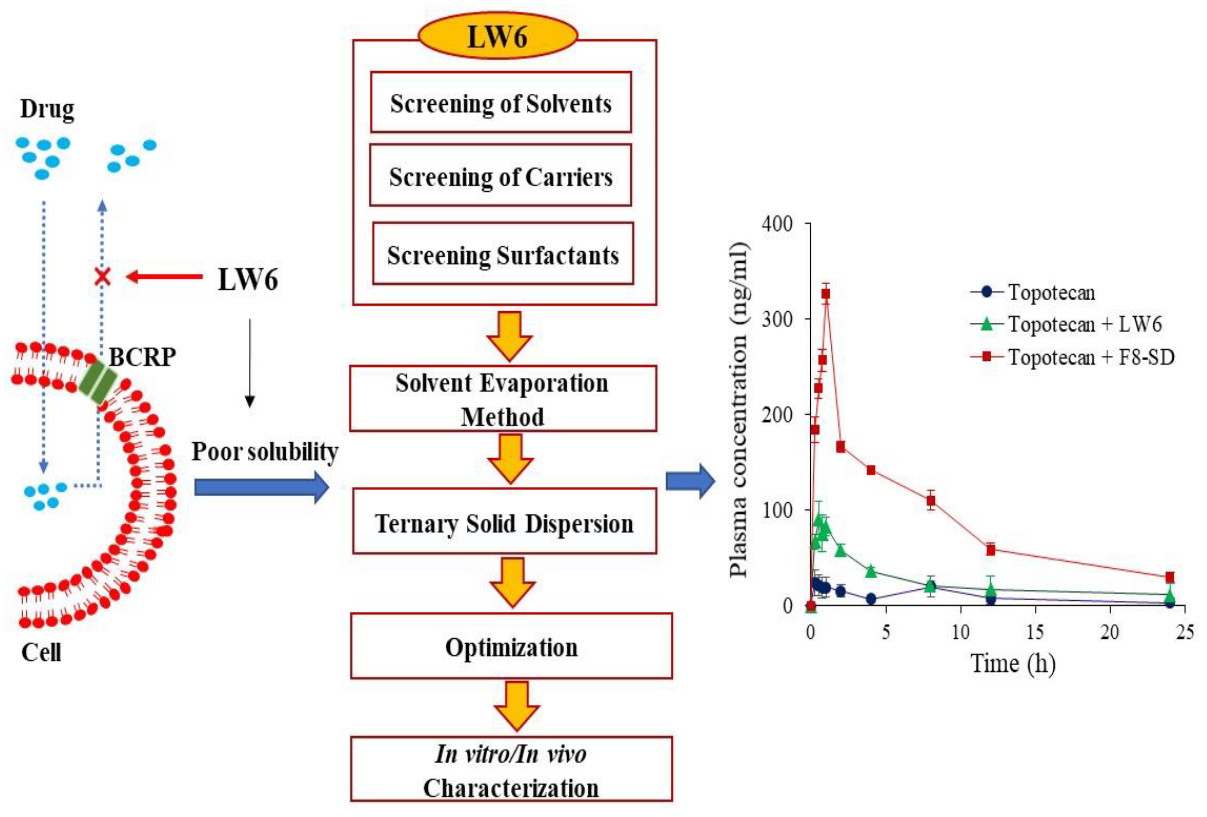
:
1. Introduction
2. Materials and Methods
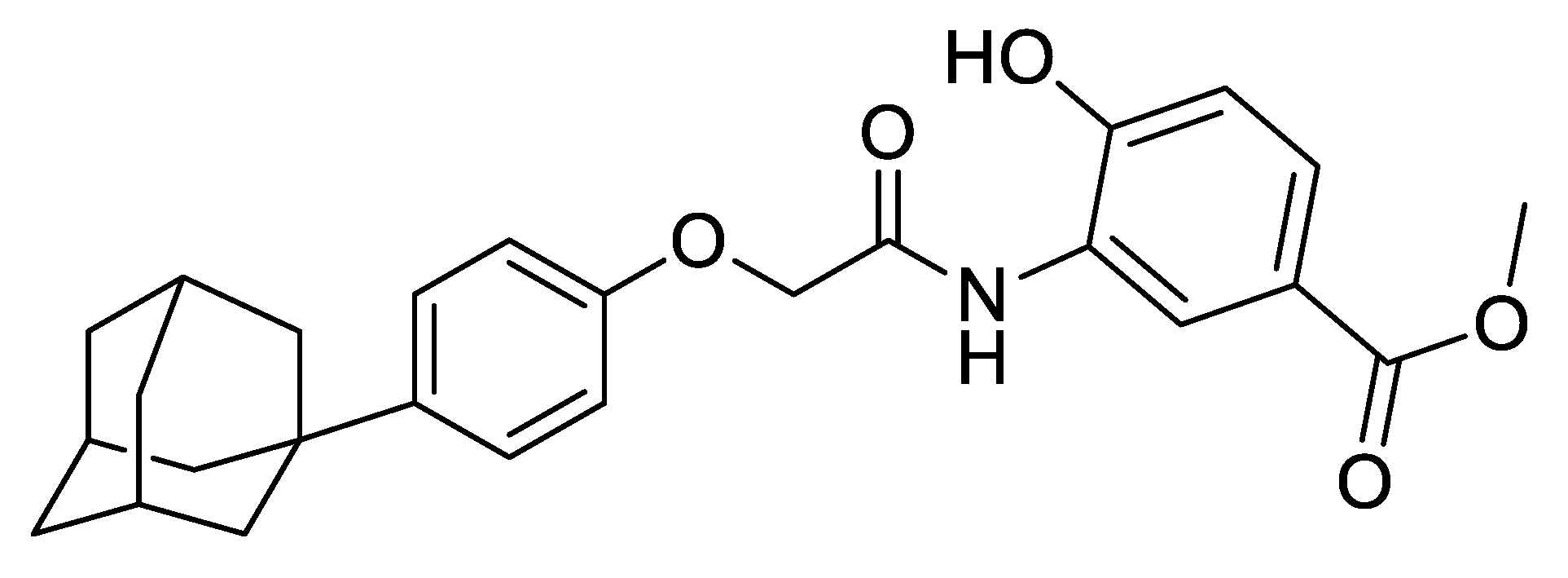
2.1. Materials
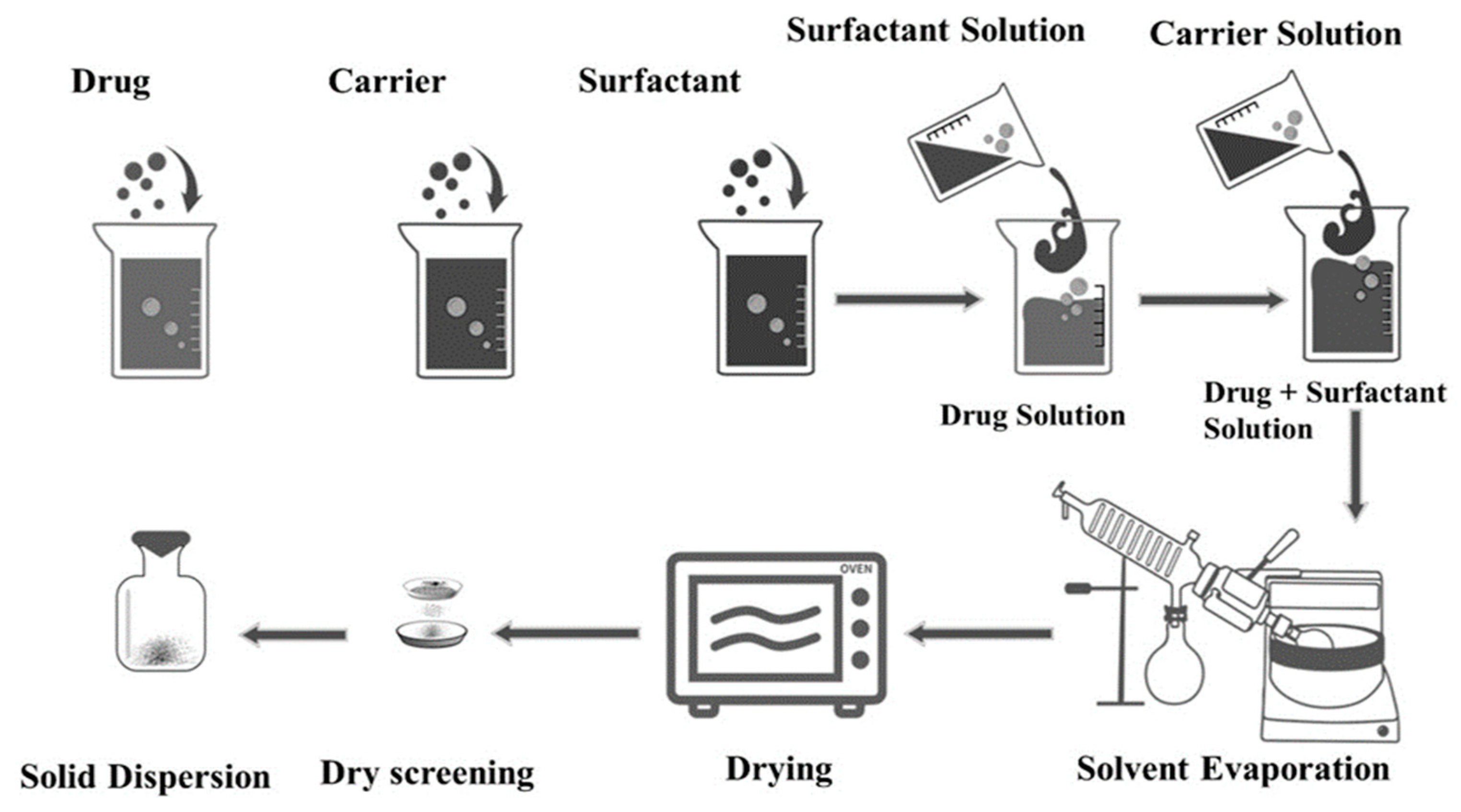
2.2. Screening of Excipients and Preparation of SDs
2.3. Solubility Studies
2.4. Structural and Morphological Characterizations of SDs
2.5. In Vitro Drug Release Studies
2.6. Pharmacokinetic Studies in Rats
2.7. Analytical Methods
2.8. Pharmacokinetic and Statistical Analysis
3. Results and Discussions
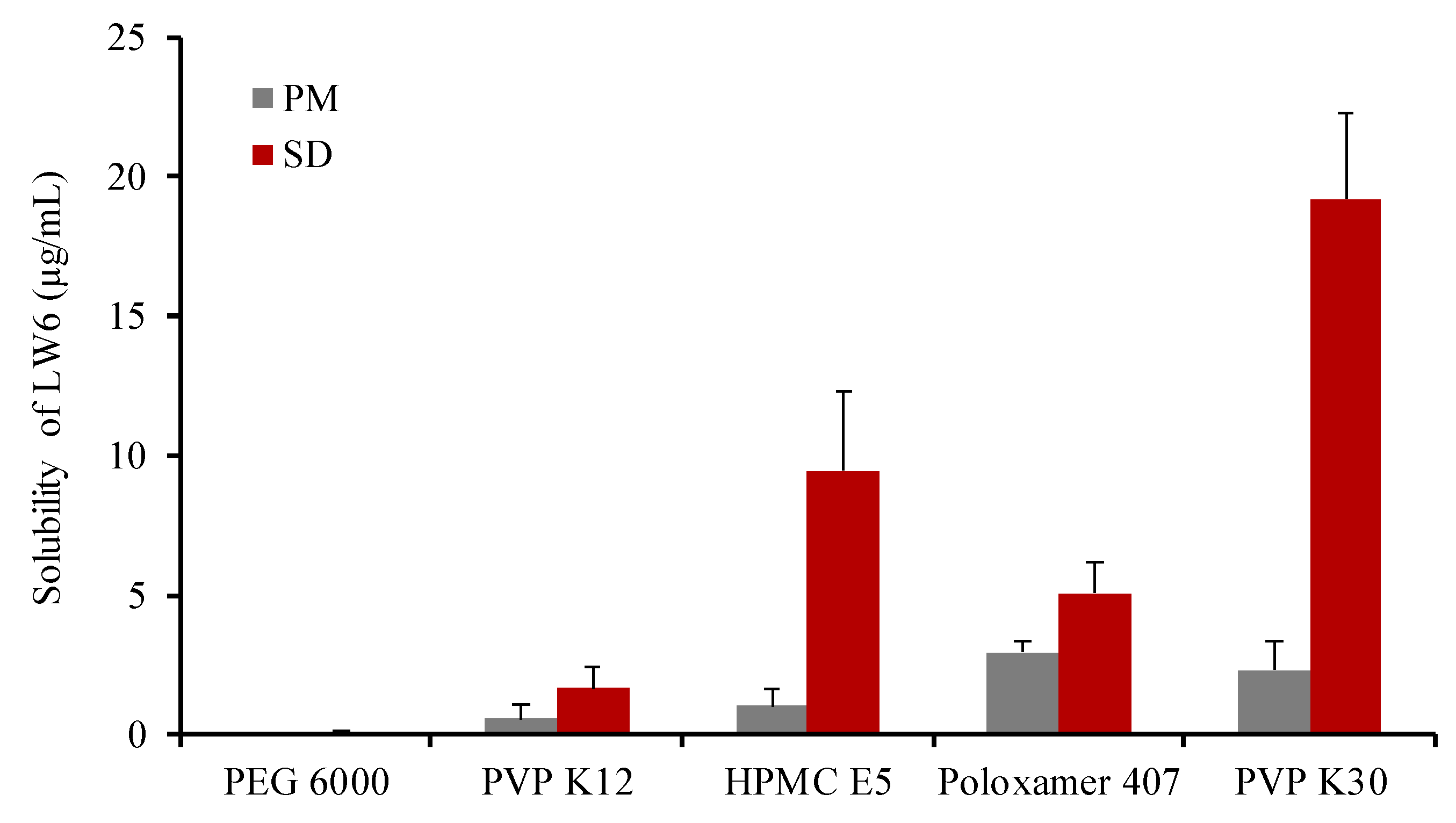
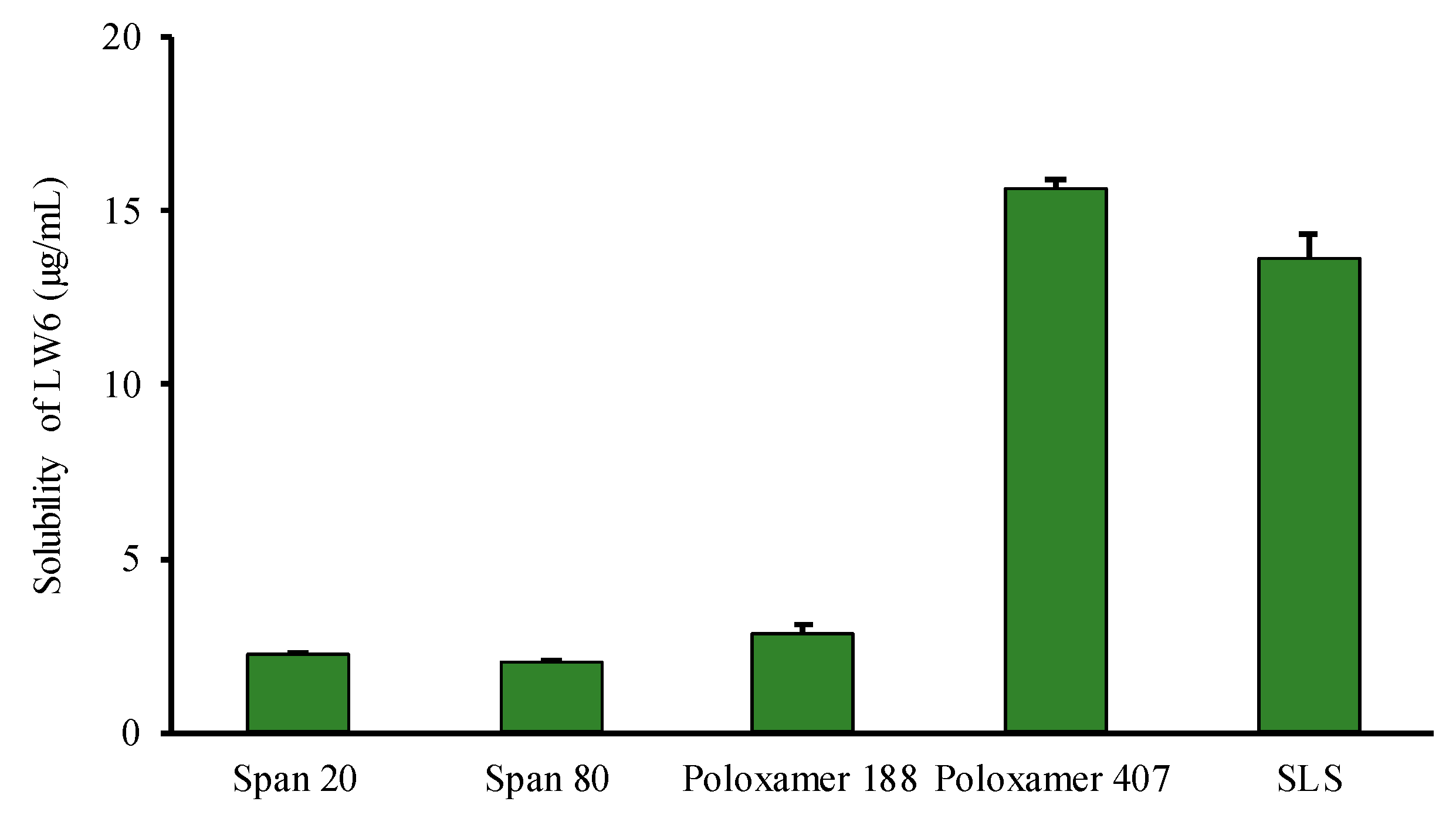
3.1. Selection of Excipients
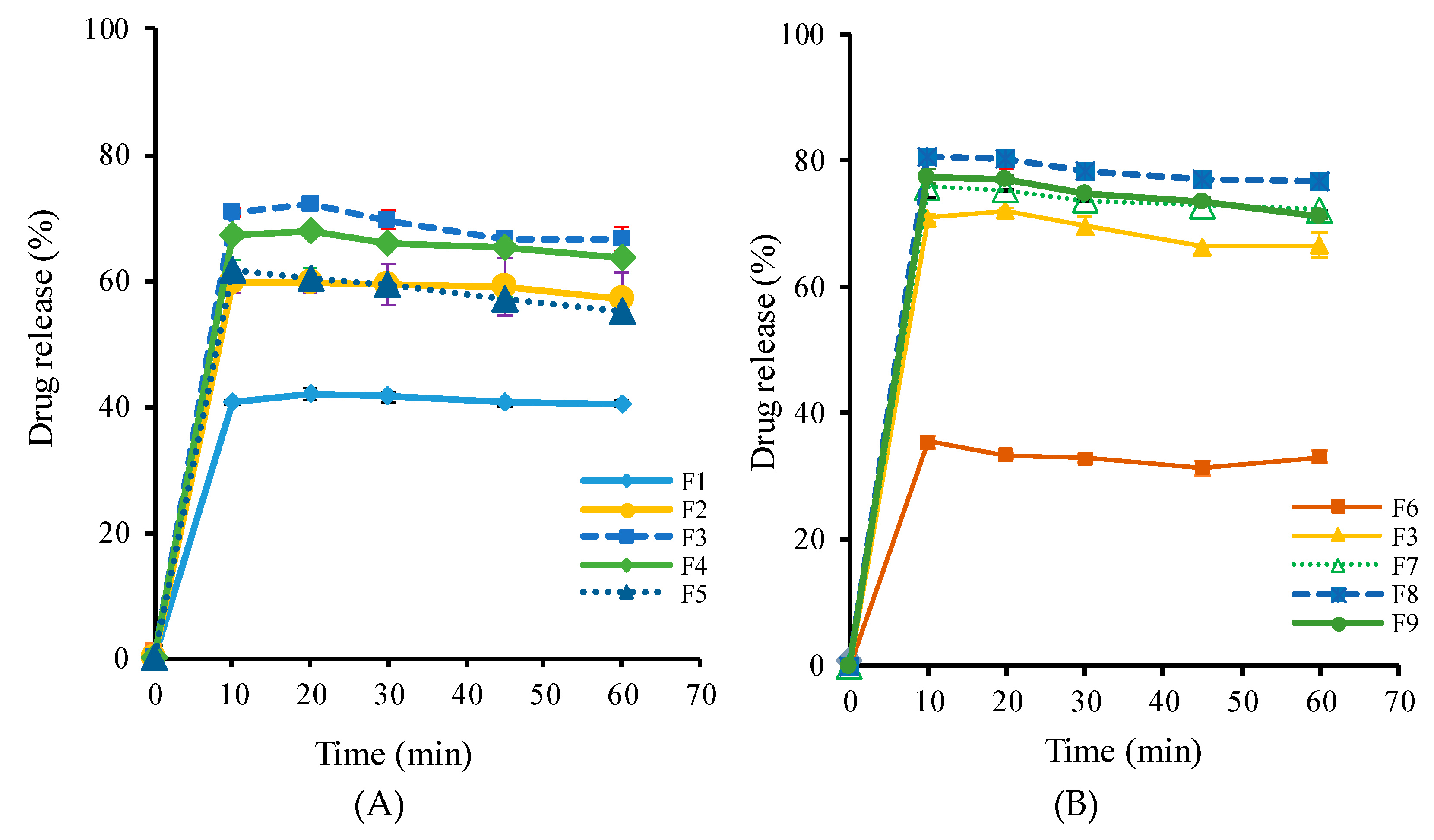
3.2. Optimization of SD Formulations
3.3. Structural and Morphological Characterization of SD Formulation
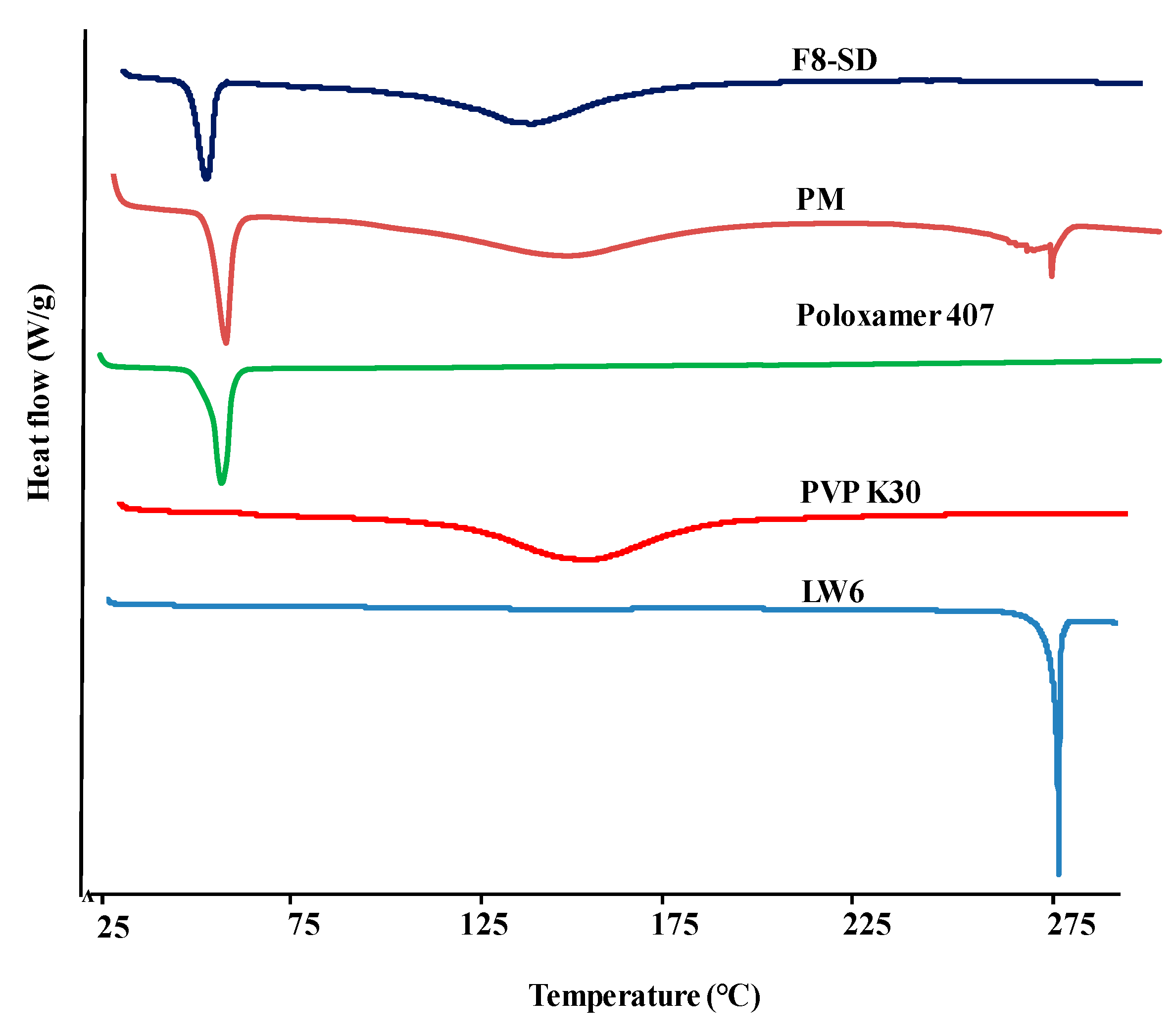
3.3.1. Differential Scanning Calorimetry (DSC)
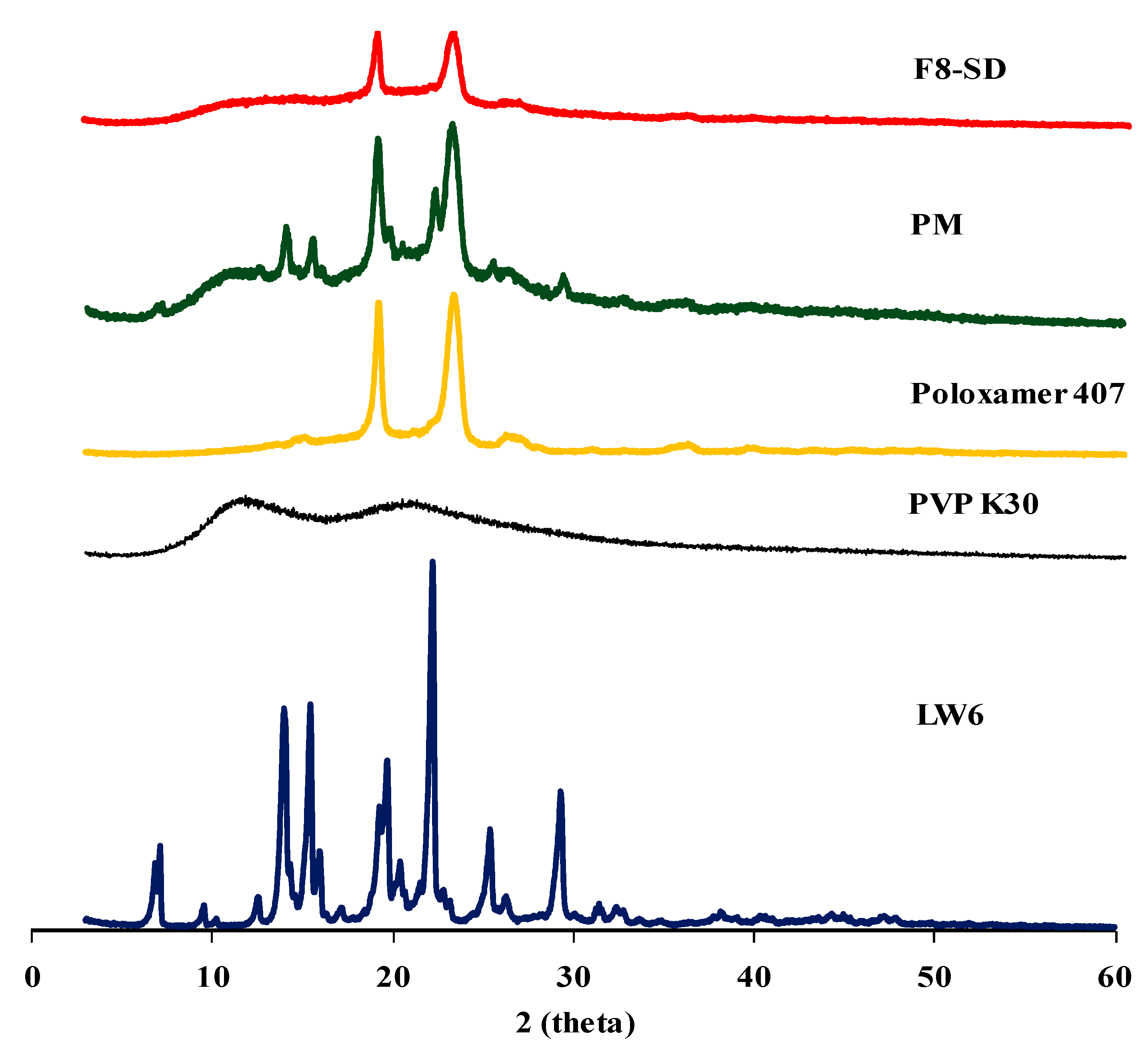
3.3.2. X-ray Powder Diffraction (XRPD) Studies
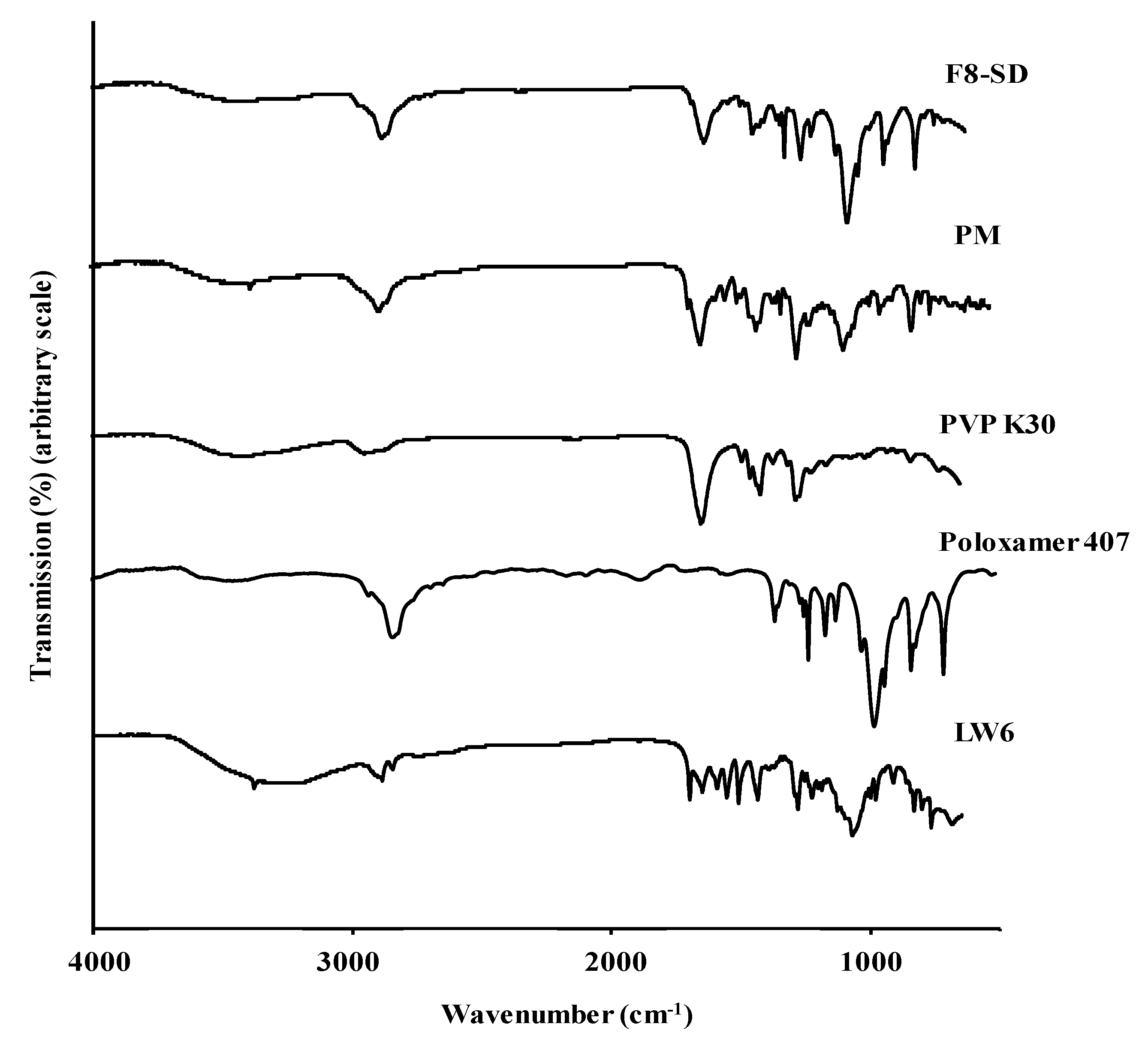
3.3.3. FT-IR
3.3.4. Scanning Electron Microscopy (SEM)
3.4. Pharmacokinetic Studies
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lee, C.A.; O’Connor, M.A.; Ritchie, T.K.; Galetin, A.; Cook, J.A.; Ragueneau-Majlessi, I.; Ellens, H.; Feng, B.; Taub, M.E.; Paine, M.F.; et al. Breast Cancer Resistance Protein (ABCG2) in Clinical Pharmacokinetics and Drug Interactions: Practical Recommendations for Clinical Victim and Perpetrator Drug-Drug Interaction Study Design. Drug Metab. Dispos. 2015, 43, 490–509. [Google Scholar] [CrossRef] [Green Version]
- Ni, Z.; Bikádi, Z.; Rosenberg, M.F.; Mao, Q. Structure and Function of the Human Breast Cancer Resistance Protein (BCRP/ABCG2). Curr. Drug Metab. 2010, 11, 603–617. [Google Scholar] [CrossRef]
- Poirier, A.; Portmann, R.; Cascais, A.-C.; Bader, U.; Walter, I.; Ullah, M.; Funk, C. The need for human breast cancer resistance protein substrate and inhibition evaluation in drug discovery and development: why, when, and how? Drug Metab. Dispos. 2014, 42, 1466–1477. [Google Scholar] [CrossRef] [PubMed]
- Song, J.G.; Lee, Y.S.; Park, J.-A.; Lim, S.-J.; Yang, S.J.; Zhao, M.; Lee, E.-H.; Lee, K.; Han, H.-K. Discovery of LW6 as a new potent inhibitor of breast cancer resistance protein. Cancer Chemother. Pharmacol. 2016, 78, 735–744. [Google Scholar] [CrossRef]
- Birner, P.; Schindl, M.; Obermair, A.; Plank, C.; Breitenecker, G.; Oberhuber, G. Overexpression of Hypoxia-inducible Factor 1α is a Marker for an Unfavorable Prognosis in Early-Stage Invasive Cervical Cancer. Cancer Res. 2000, 60, 4693–4696. [Google Scholar]
- Sato, M.; Hirose, K.; Kashiwakura, I.; Aoki, M.; Kawaguchi, H.; Hatayama, Y.; Akimoto, H.; Narita, Y.; Takai, Y. LW6, a hypoxia-inducible factor 1 inhibitor, selectively induces apoptosis in hypoxic cells through depolarization of mitochondria in A549 human lung cancer cells. Mol. Med. Rep. 2015, 12, 3462–3468. [Google Scholar] [CrossRef] [Green Version]
- Longley, D.; Johnston, P. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Bedi, N.; Tiwary, A.K. Enhancing solubility of poorly aqueous soluble drugs: Critical appraisal of techniques. J. Pharm. Investig. 2018, 48, 509–526. [Google Scholar] [CrossRef]
- Serajuddin, A.T. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef]
- Hammond, R.B.; Pencheva, K.; Roberts, K.J.; Auffret, T. Quantifying solubility enhancement due to particle size reduction and crystal habit modification: Case study of acetyl salicylic acid. J. Pharm. Sci. 2007, 96, 1967–1973. [Google Scholar] [CrossRef]
- Herpin, M.J.; Smyth, H.D. Super-heated aqueous particle engineering (SHAPE): A novel method for the micronization of poorly water soluble drugs. J. Pharm. Investig. 2018, 48, 135–142. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef]
- Ahsan, M.N.; Verma, P.R.P. Enhancement of in vitro dissolution and pharmacodynamic potential of olanzapine using solid SNEDDS. J. Pharm. Investig. 2018, 48, 269–278. [Google Scholar] [CrossRef]
- Kalepu, S.; Manthina, M.; Padavala, V. Oral lipid-based drug delivery systems—An overview. Acta Pharm. Sin. B 2013, 3, 361–372. [Google Scholar] [CrossRef]
- Liu, X.; Feng, X.; Williams, R.O.; Zhang, F. Characterization of amorphous solid dispersions. J. Pharm. Investig. 2018, 48, 19–41. [Google Scholar] [CrossRef]
- Ogawa, N.; Hiramatsu, T.; Suzuki, R.; Okamoto, R.; Shibagaki, K.; Fujita, K.; Takahashi, C.; Kawashima, Y.; Yamamoto, H. Improvement in the water solubility of drugs with a solid dispersion system by spray drying and hot-melt extrusion with using the amphiphilic polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol graft copolymer and d -mannitol. Eur. J. Pharm. Sci. 2018, 111, 205–214. [Google Scholar] [CrossRef]
- Choi, J.-S.; Lee, S.-E.; Jang, W.S.; Byeon, J.C.; Park, J.-S. Solid dispersion of dutasteride using the solvent evaporation method: Approaches to improve dissolution rate and oral bioavailability in rats. Mater. Sci. Eng. C 2018, 90, 387–396. [Google Scholar] [CrossRef]
- Modi, A.; Tayade, P. Enhancement of dissolution profile by solid dispersion (kneading) technique. AAPS PharmSciTech 2017, 7, E87. [Google Scholar] [CrossRef]
- Alonzo, D.E.; Zhang, G.G.Z.; Zhou, D.; Gao, Y.; Taylor, L.S. Understanding the Behavior of Amorphous Pharmaceutical Systems during Dissolution. Pharm. Res. 2010, 27, 608–618. [Google Scholar] [CrossRef]
- Vojinović, T.; Medarević, D.; Vranić, E.; Potpara, Z.; Krstić, M.; Djuriš, J.; Ibrić, S. Development of ternary solid dispersions with hydrophilic polymer and surface adsorbent for improving dissolution rate of carbamazepine. Saudi Pharm. J. 2018, 26, 725–732. [Google Scholar] [CrossRef]
- Chaudhari, S.P.; Dugar, R.P. Application of surfactants in solid dispersion technology for improving solubility of poorly water soluble drugs. J. Drug Deliv. Sci. Technol. 2017, 41, 68–77. [Google Scholar] [CrossRef]
- Ye, L.; Shi, J.; Wan, S.; Yang, X.; Wang, Y.; Zhang, J.; Zheng, D.; Liu, Z. Development and validation of a liquid chromatography-tandem mass spectrometry method for topotecan determination in beagle dog plasma and its application in a bioequivalence study. Biomed. Chromatogr. 2013, 27, 1532–1539. [Google Scholar] [CrossRef]
- ChemSpider Search and Share Chemistry. Available online: http://www.chemspider.com (accessed on 10 May 2018).
- Sethia, S.; Squillante, E. Solid dispersion of carbamazepine in PVP K30 by conventional solvent evaporation and supercritical methods. Int. J. Pharm. 2004, 272, 1–10. [Google Scholar] [CrossRef]
- Ghebremeskel, A.N.; Vemavarapu, C.; Lodaya, M. Use of Surfactants as Plasticizers in Preparing Solid Dispersions of Poorly Soluble API: Stability Testing of Selected Solid Dispersions. Pharm. Res. 2006, 23, 1928–1936. [Google Scholar] [CrossRef]
- Ben Osman, Y.; Liavitskaya, T.; Vyazovkin, S. Polyvinylpyrrolidone affects thermal stability of drugs in solid dispersions. Int. J. Pharm. 2018, 551, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Dumortier, G.; Grossiord, J.L.; Agnely, F.; Chaumeil, J.C. A Review of Poloxamer 407 Pharmaceutical and Pharmacological Characteristics. Pharm. Res. 2006, 23, 2709–2728. [Google Scholar] [CrossRef]
- Singh-Joy, S.D.; McLain, V.C. Safety assessment of poloxamers 101, 105, 108, 122, 123, 124, 181, 182, 183, 184, 185, 188, 212, 215, 217, 231, 234, 235, 237, 238, 282, 284, 288, 331, 333, 334, 335, 338, 401, 402, 403, and 407, poloxamer 105 benzoate, and poloxamer 182 dibenzoate as used in cosmetics. Int. J. Toxicol. 2008, 27, 93–128. [Google Scholar]
- Prasad, D.; Chauhan, H.; Atef, E. Amorphous Stabilization and Dissolution Enhancement of Amorphous Ternary Solid Dispersions: Combination of Polymers Showing Drug–Polymer Interaction for Synergistic Effects. J. Pharm. Sci. 2014, 103, 3511–3523. [Google Scholar] [CrossRef] [PubMed]
- Newa, M.; Bhandari, K.H.; Oh, D.H.; Kim, Y.R.; Sung, J.H.; Kim, J.O.; Woo, J.S.; Choi, H.G.; Yong, C.S. Enhanced dissolution of ibuprofen using solid dispersion with poloxamer 407. Arch. Pharm. Res. 2008, 31, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Fakhari, A.; Corcoran, M.; Schwarz, A. Thermogelling properties of purified poloxamer 407. Heliyon 2017, 3, e00390. [Google Scholar] [CrossRef]
- Kaewnopparat, N.; Ingkatawornwong, S.; Tantishaiyakul, V. Properties of solid dispersions of piroxicam in polyvinylpyrrolidone. Int. J. Pharm. 1999, 181, 143–151. [Google Scholar]
- Simonazzi, A.; Davies, C.; Cid, A.G.; Gonzo, E.; Parada, L.; Bermúdez, J.M. Preparation and Characterization of Poloxamer 407 Solid Dispersions as an Alternative Strategy to Improve Benznidazole Bioperformance. J. Pharm. Sci. 2018, 107, 2829–2836. [Google Scholar] [CrossRef] [PubMed]
- A Nagourney, R.; Sommers, B.L.; Harper, S.M.; Radecki, S.; Evans, S.S. Ex vivo analysis of topotecan: advancing the application of laboratory-based clinical therapeutics. Br. J. Cancer 2003, 89, 1789–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagata, T.; Morishita, M.; Kusuhara, H.; Takayama, K.; Benameur, H.; Sugiyama, Y. Characterization of the inhibition of breast cancer resistance protein-mediated efflux of mitoxantrone by pharmaceutical excipients. Int. J. Pharm. 2009, 370, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, T.; Kusuhara, H.; Morishita, M.; Takayama, K.; Benameur, H.; Sugiyama, Y. Effect of excipients on breast cancer resistance protein substrate uptake activity. J. Control. Release 2007, 124, 1–5. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carriers | Solubility (µg/mL) |
|---|---|
| PEG 6000 | 0.057 ± 0.078 |
| PVP K12 | 1.659 ± 0.729 |
| HPMC E5 | 9.470 ± 2.874 |
| Poloxamer 407 | 5.057 ± 1.105 |
| PVP K30 | 19.20 ± 3.112 |
| Group | Formulation | Weight Ratio (w/w/w) | ||
|---|---|---|---|---|
| LW6 | PVP K30 | Poloxamer 407 | ||
| F1 | 1 | 5 | 1 | |
| F2 | 1 | 5 | 3 | |
| 1 | F3 | 1 | 5 | 5 |
| F4 | 1 | 5 | 6 | |
| F5 | 1 | 5 | 7 | |
| F6 | 1 | 1 | 5 | |
| F3 | 1 | 5 | 5 | |
| 2 | F7 | 1 | 7 | 5 |
| F8 | 1 | 8 | 5 | |
| F9 | 1 | 9 | 5 | |
| Parameters | Topotecan | Topotecan + LW6 | Topotecan + F8-SD |
|---|---|---|---|
| Cmax (ng/mL) | 31.3 ± 7.1 | 91.5 ± 17.7 * | 326 ± 25.2 * |
| Tmax (h) | 0.4 ± 0.1 | 0.4 ± 0.1 | 1.0 * |
| AUC (ng*h/mL) | 204 ± 11.1 | 598 ± 188 * | 2140 ± 46.6 * |
| T1/2 (h) | 2.0 ± 0.4 | 9.3 ± 4.8 * | 8.8 ± 1.5 * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bajracharya, R.; Lee, S.H.; Song, J.G.; Kim, M.; Lee, K.; Han, H.-K. Development of a Ternary Solid Dispersion Formulation of LW6 to Improve the In Vivo Activity as a BCRP Inhibitor: Preparation and In Vitro/In Vivo Characterization. Pharmaceutics 2019, 11, 206. https://doi.org/10.3390/pharmaceutics11050206
Bajracharya R, Lee SH, Song JG, Kim M, Lee K, Han H-K. Development of a Ternary Solid Dispersion Formulation of LW6 to Improve the In Vivo Activity as a BCRP Inhibitor: Preparation and In Vitro/In Vivo Characterization. Pharmaceutics. 2019; 11(5):206. https://doi.org/10.3390/pharmaceutics11050206
Chicago/Turabian StyleBajracharya, Rajiv, Sang Hoon Lee, Jae Geun Song, Minkyoung Kim, Kyeong Lee, and Hyo-Kyung Han. 2019. "Development of a Ternary Solid Dispersion Formulation of LW6 to Improve the In Vivo Activity as a BCRP Inhibitor: Preparation and In Vitro/In Vivo Characterization" Pharmaceutics 11, no. 5: 206. https://doi.org/10.3390/pharmaceutics11050206
APA StyleBajracharya, R., Lee, S. H., Song, J. G., Kim, M., Lee, K., & Han, H.-K. (2019). Development of a Ternary Solid Dispersion Formulation of LW6 to Improve the In Vivo Activity as a BCRP Inhibitor: Preparation and In Vitro/In Vivo Characterization. Pharmaceutics, 11(5), 206. https://doi.org/10.3390/pharmaceutics11050206