Octreotide Conjugates for Tumor Targeting and Imaging

 , , , , , , ,
, , , , , , ,
Abstract
:
1. Introduction
2. Materials and Methods
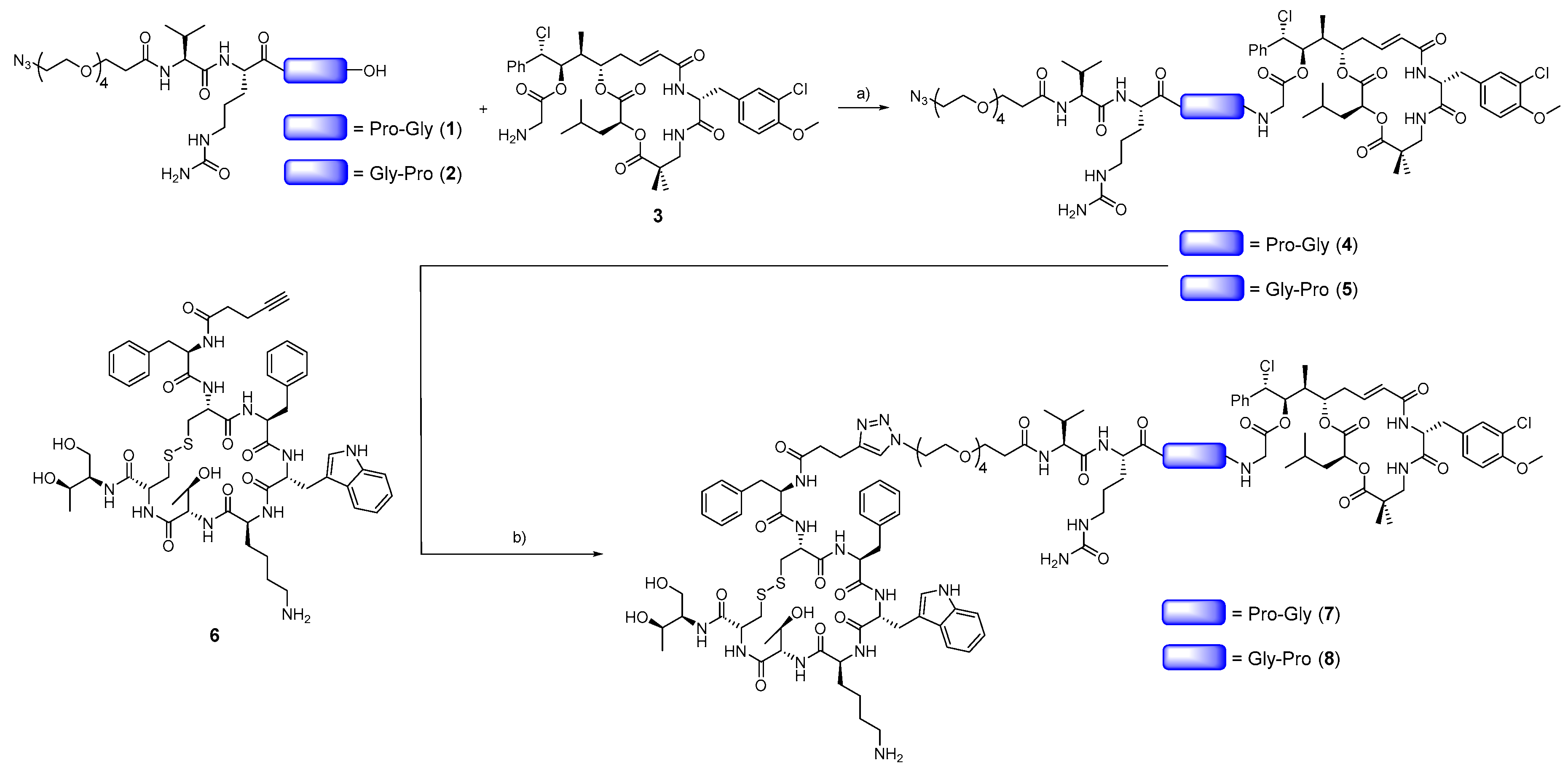
2.1. Synthesis and Structural Characterization of Compounds 1–8
2.2. Biological Evaluation
3. Results and Discussion
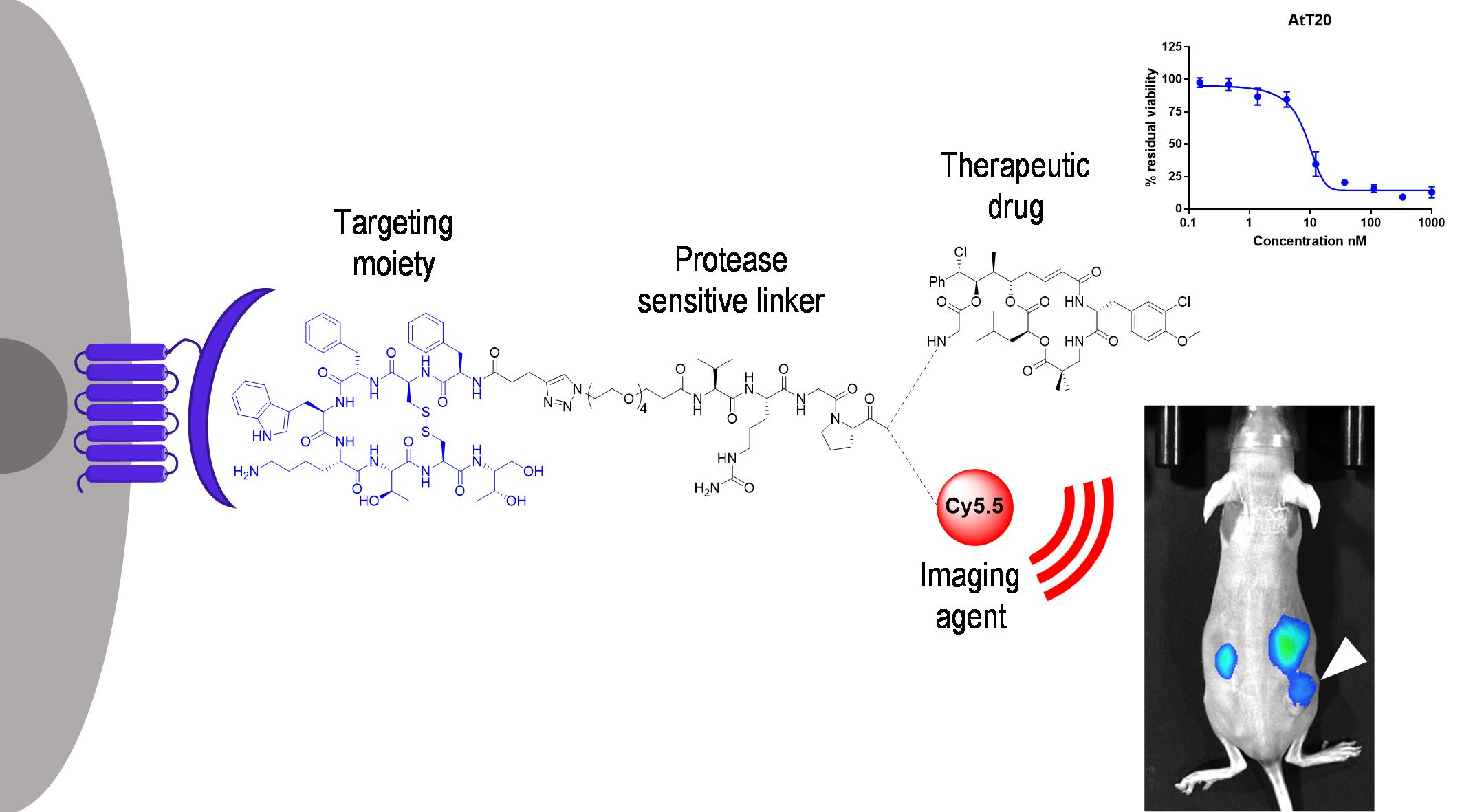
3.1. Design and Synthesis of Octreotide-cryptophycin Conjugates
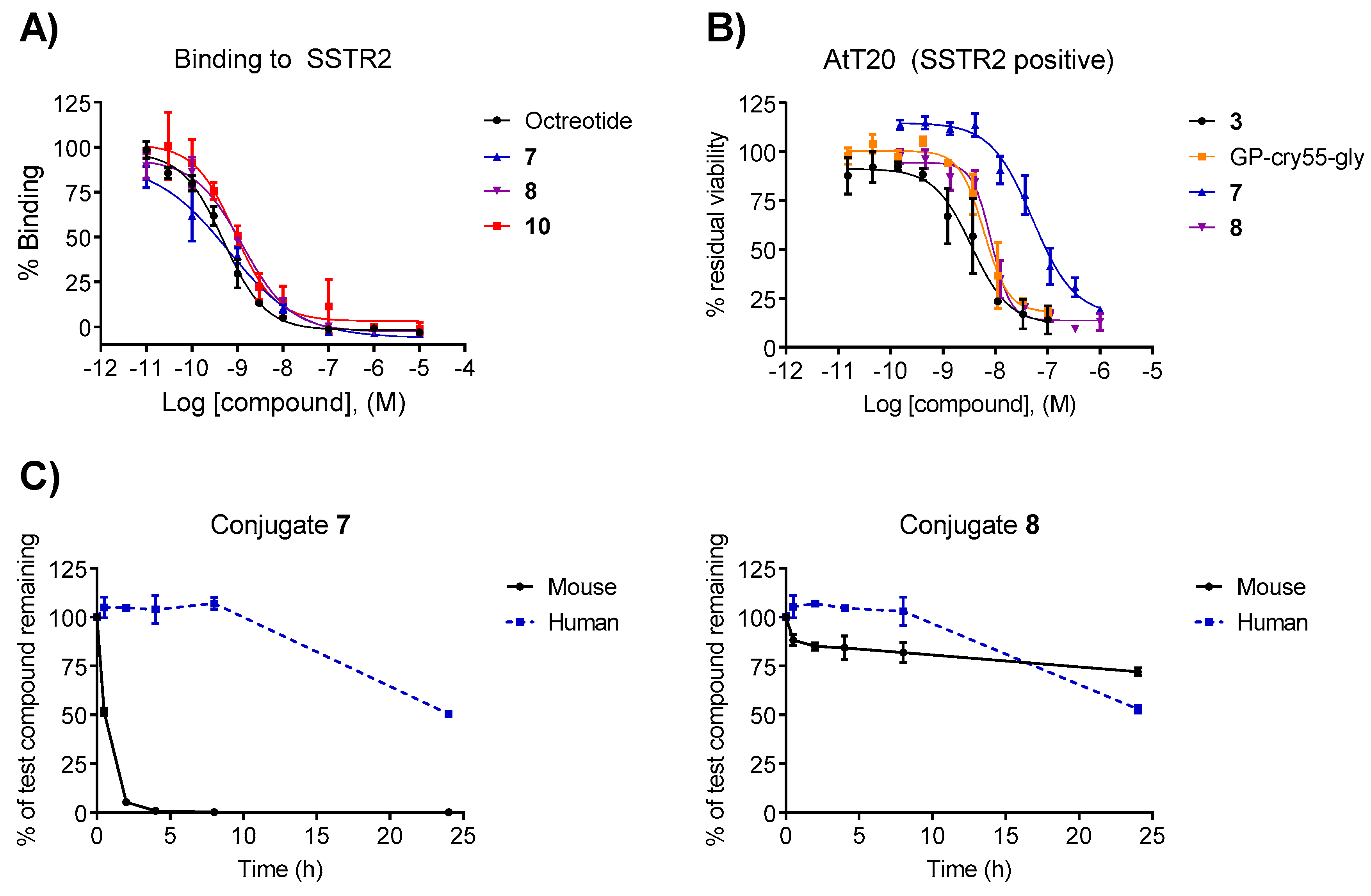
3.2. Binding Affinity
3.3. In Vitro Cytotoxicity
3.4. Plasma Stability
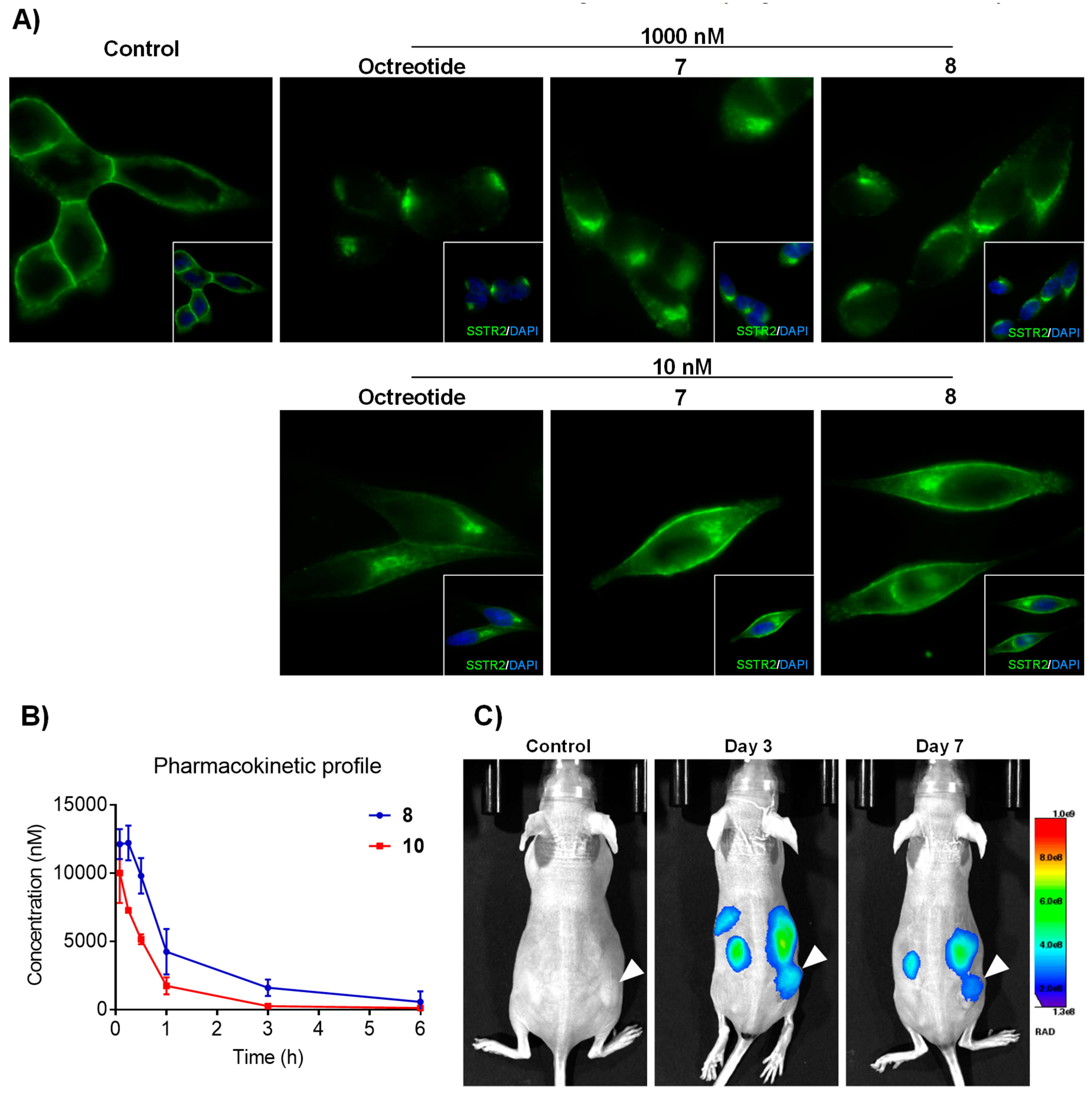
3.5. Receptor and Conjugate Internalization
3.6. In Vivo Tumor Imaging
3.7. In Vivo Antitumor Activity
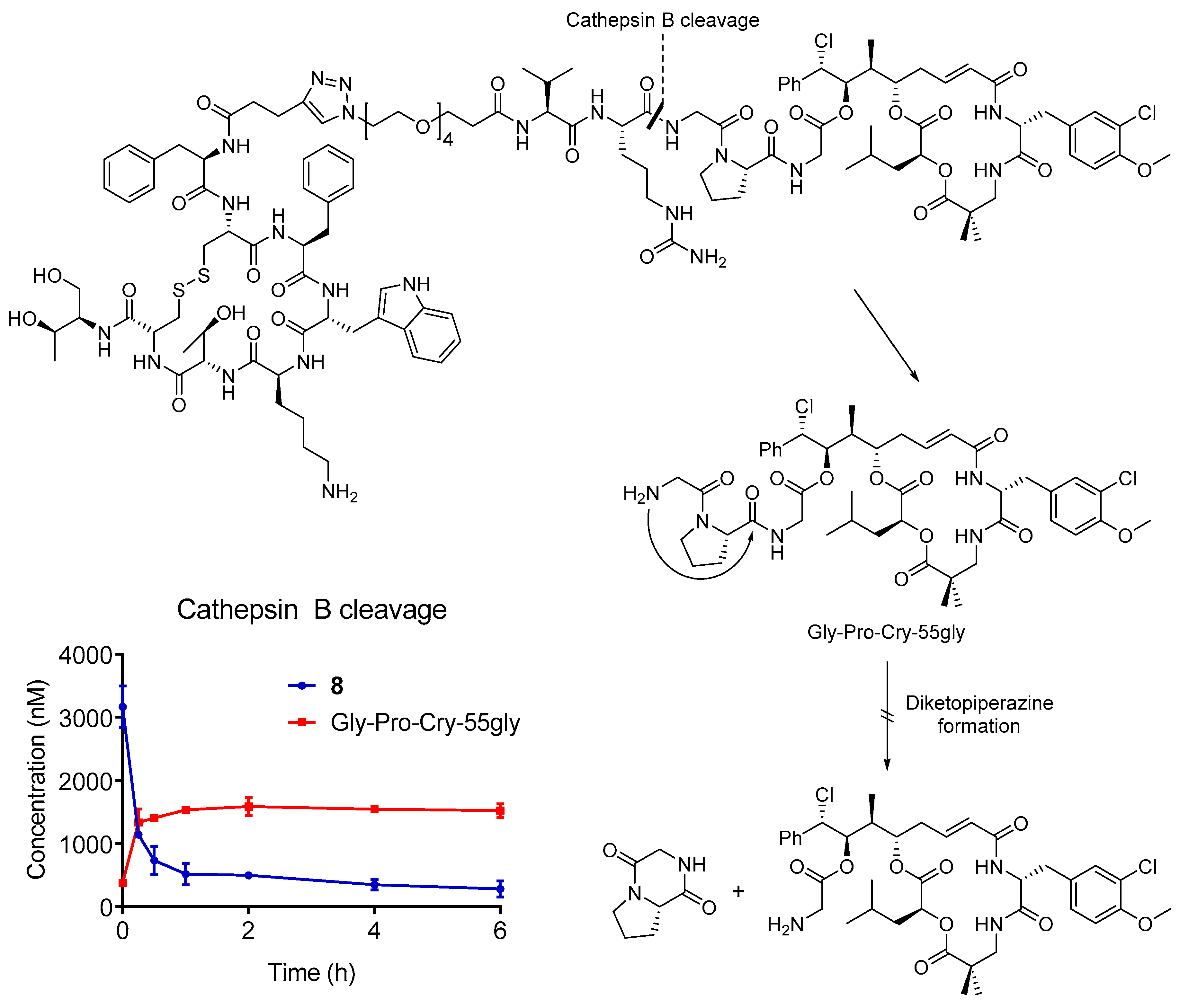
3.8. Cathepsin B Stability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Malhotra, V.; Perry, M.C. Classical chemotherapy: Mechanisms, toxicities and the therapeutic window. Cancer Biol. Ther. 2003, 2, 1–3. [Google Scholar] [CrossRef]
- Böhme, D.; Beck-Sickinger, A.G. Drug delivery and release systems for targeted tumor therapy. J. Pept. Sci. 2015, 21, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Casi, G.; Neri, D. Antibody-Drug Conjugates and Small Molecule-Drug Conjugates: Opportunities and Challenges for the Development of Selective Anticancer Cytotoxic Agents. J. Med. Chem. 2015, 58, 8751–8761. [Google Scholar] [CrossRef] [PubMed]
- Krall, N.; Scheuermann, J.; Neri, D. Small targeted cytotoxics: Current state and promises from DNA-encoded chemical libraries. Angew. Chem. Int. Ed. 2013, 52, 1384–1402. [Google Scholar] [CrossRef] [PubMed]
- Srinivasarao, M.; Low, P.S. Ligand-Targeted Drug Delivery. Chem. Rev. 2017, 117, 12133–12164. [Google Scholar] [CrossRef]
- Chatzisideri, T.; Leonidis, G.; Sarli, V. Cancer-targeted delivery systems based on peptides. Future Med. Chem. 2018, 10, 2201–2226. [Google Scholar] [CrossRef] [PubMed]
- Vlahov, I.R.; Qi, L.; Kleindl, P.J.; Santhapuram, H.K.; Felten, A.; Parham, G.L.; Wang, K.; You, F.; Vaughn, J.F.; Hahn, S.J.; et al. Latent Warheads for Targeted Cancer Therapy: Design and Synthesis of pro-Pyrrolobenzodiazepines and Conjugates. Bioconj. Chem. 2017, 28, 2921–2931. [Google Scholar] [CrossRef]
- Umbricht, C.A.; Benešová, M.; Schibli, R.; Müller, C. Preclinical Development of Novel PSMA-Targeting Radioligands: Modulation of Albumin-Binding Properties to Improve Prostate Cancer Therapy. Mol. Pharm. 2018, 15, 2297–2306. [Google Scholar] [CrossRef]
- Marks, I.S.; Gardeen, S.S.; Kurdziel, S.J.; Nicolaou, S.T.; Woods, J.E.; Kularatne, S.A.; Low, P.S. Development of a Small Molecule Tubulysin B Conjugate for Treatment of Carbonic Anhydrase IX Receptor Expressing Cancers. Mol. Pharm. 2018, 15, 2289–2296. [Google Scholar] [CrossRef]
- Cazzamalli, S.; Dal Corso, A.; Widmayer, F.; Neri, D. Chemically defined antibody- and small molecule-drug conjugates for in vivo tumor targeting applications: A comparative analysis. J. Am. Chem. Soc. 2018, 140, 1617–1621. [Google Scholar] [CrossRef] [PubMed]
- Cazzamalli, S.; Dal Corso, A.; Neri, D. Acetazolamide serves as selective delivery vehicle for dipeptide-linked drugs to renal cell carcinoma. Mol. Cancer Ther. 2016, 15, 2926–2936. [Google Scholar] [CrossRef] [PubMed]
- Ginj, M.; Zhang, H.; Waser, B.; Cescato, R.; Wild, D.; Wang, X.; Erchegyi, J.; Rivier, J.; Mäcke, H.R.; Reubi, J.C. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc. Natl. Acad. Sci. USA 2006, 103, 16436–16441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reubi, J.C.; Waser, B.; Schaer, J.-C.; Laissue, J.A. Somatostatin receptor sst1–sst5 expression in normal and neoplastic human tissues using receptor autoradiography with subtype-selective ligands. Eur. J. Nucl. Med. 2001, 28, 836–846. [Google Scholar] [CrossRef]
- Froidevaux, S.; Eberle, A.N. Somatostatin analogs and radiopeptides in cancer therapy. Biopolymers 2002, 66, 161–183. [Google Scholar] [CrossRef] [PubMed]
- Di Cianni, A.; Carotenuto, A.; Brancaccio, D.; Novellino, E.; Reubi, J.C.; Beetschen, K.; Papini, A.M.; Ginanneschi, M. Novel octreotide dicarba-analogues with high affinity and different selectivity for somatostatin receptors. J. Med. Chem. 2010, 53, 6188–6197. [Google Scholar] [CrossRef] [PubMed]
- Barragán, F.; Carrion-Salip, D.; Gómez-Pinto, I.; González-Cantó, A.; Sadler, P.J.; De Llorens, R.; Moreno, V.; González, C.; Massaguer, A.; Marchán, V. Somatostatin subtype-2 receptor-targeted metal-based anticancer complexes. Bioconj. Chem. 2012, 23, 1838–1855. [Google Scholar] [CrossRef] [PubMed]
- Pratesi, A.; Ginanneschi, M.; Lumini, M.; Papini, A.M.; Novellino, E.; Brancaccio, D.; Carotenuto, A. DOTA-Derivatives of Octreotide Dicarba-Analogs with High Affinity for Somatostatin sst2,5 Receptors. Front. Chem. 2017, 5. [Google Scholar] [CrossRef]
- Yin, T.; Wu, Q.; Wang, L.; Yin, L.; Zhou, J.; Huo, M. Well-Defined Redox-Sensitive Polyethene Glycol-Paclitaxel Prodrug Conjugate for Tumor-Specific Delivery of Paclitaxel Using Octreotide for Tumor Targeting. Mol. Pharm. 2015, 12, 3020–3031. [Google Scholar] [CrossRef] [PubMed]
- Lelle, M.; Kaloyanova, S.; Freidel, C.; Theodoropoulou, M.; Musheev, M.; Niehrs, C.; Stalla, G.; Peneva, K. Octreotide-Mediated Tumor-Targeted Drug Delivery via a Cleavable Doxorubicin-Peptide Conjugate. Mol. Pharm. 2015, 12, 4290–4300. [Google Scholar] [CrossRef]
- Zhang, H.-Y.; Xu, W.-Q.; Zheng, Y.-Y.; Omari-Siaw, E.; Zhu, Y.; Cao, X.; Tong, S.-S.; Yu, J.-N.; Xu, X.-M. Octreotide-periplocymarin conjugate prodrug for improving targetability and anti-tumor efficiency: Synthesis, in vitro and in vivo evaluation. Oncotarget 2016, 7, 86326–86338. [Google Scholar] [CrossRef]
- Zhang, H.-Y.; Xu, W.-Q.; Wang, Y.-W.; Omari-Siaw, E.; Wang, Y.; Zheng, Y.-Y.; Cao, X.; Tong, S.-S.; Yu, J.-N.; Xu, X.-M. Tumor targeted delivery of octreotide-periplogenin conjugate: Synthesis, in vitro and in vivo evaluation. Int. J. Pharm. 2016, 502, 98–106. [Google Scholar] [CrossRef]
- Maxwell, J.E.; Howe, J.R. Imaging in neuroendocrine tumors: An update for the clinician. Int. J. Endocr. Oncol. 2015, 2, 159–168. [Google Scholar] [CrossRef]
- Pauwels, E.; Cleeren, F.; Bormans, G.; Deroose, C.M. Somatostatin receptor PET ligands—The next generation for clinical practice. Am. J. Nucl. Med. Mol. Imaging 2018, 8, 311–331. [Google Scholar]
- Weiss, C.; Figueras, E.; Borbely, A.N.; Sewald, N. Cryptophycins: Cytotoxic cyclodepsipeptides with potential for tumor targeting. J. Pept. Sci. 2017, 23, 514–531. [Google Scholar] [CrossRef]
- Schwartz, R.E.; Hirsch, C.F.; Sesin, D.F.; Flor, J.E.; Chartrain, M.; Fromtling, R.E.; Harris, G.H.; Salvatore, M.J.; Liesch, J.M.; Yudin, K. Pharmaceuticals from cultured algae. J. Ind. Microbiol. 1990, 5, 113–124. [Google Scholar] [CrossRef]
- Smith, C.D.; Zhang, X.; Mooberry, S.L.; Patterson, G.M.L.; Moore, R.E. Cryptophycin: A New Antimicrotubule Agent Active against Drug-resistant Cells. Cancer Res. 1994, 54, 3779–3784. [Google Scholar] [PubMed]
- Edelman, M.J.; Gandara, D.R.; Hausner, P.; Israel, V.; Thornton, D.; DeSanto, J.; Doyle, L.A. Phase 2 study of cryptophycin 52 (LY355703) in patients previously treated with platinum based chemotherapy for advanced non-small cell lung cancer. Lung Cancer 2003, 39, 197–199. [Google Scholar] [CrossRef]
- D’Agostino, G.; Del Campo, J.; Mellado, B.; Izquierdo, M.A.; Minarik, T.; Cirri, L.; Marini, L.; Perez-Gracia, J.L.; Scambia, G. A multicenter phase II study of the cryptophycin analog LY355703 in patients with platinum-resistant ovarian cancer. Int. J. Gynecol. Cancer 2006, 16, 71–76. [Google Scholar] [CrossRef]
- Eißler, S.; Bogner, T.; Nahrwold, M.; Sewald, N. Efficient synthesis of cryptophycin-52 and novel para-Alkoxymethyl unit A analogues. Chem. A Eur. J. 2009, 15, 11273–11287. [Google Scholar] [CrossRef]
- Wen, L.L.; Jian, C.Z.; Fa, Q.J.; Fu, L. Synthesis and cytotoxicity studies of new cryptophycin analogues. Arch. Pharm. 2009, 342, 577–583. [Google Scholar]
- Nahrwold, M.; Bogner, T.; Eissler, S.; Verma, S.; Sewald, N. “Clicktophycin-52”: A bioactive cryptophycin-52 triazole analogue. Org. Lett. 2010, 12, 1064–1067. [Google Scholar] [CrossRef] [PubMed]
- Weiß, C.; Bogner, T.; Sammet, B.; Sewald, N. Total synthesis and biological evaluation of fluorinated cryptophycins. Beilstein J. Org. Chem. 2012, 8, 2060–2066. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Kumar, M.; Sharma, S.; Guru, S.K.; Bhushan, S.; Shah, B.A. Design and synthesis of a new class of cryptophycins based tubulin inhibitors. Eur. J. Med. Chem. 2015, 93, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Al-Awar, R.S.; Corbett, T.H.; Ray, J.E.; Polin, L.; Kennedy, J.H.; Wagner, M.M.; Williams, D.C. Biological evaluation of cryptophycin 52 fragment A analogues: Effect of the multidrug resistance ATP binding cassette transporters on antitumor activity. Mol. Cancer Ther. 2004, 3, 1061–1067. [Google Scholar]
- Kotoku, N.; Kato, T.; Narumi, F.; Ohtani, E.; Kamada, S.; Aoki, S.; Okada, N.; Nakagawa, S.; Kobayashi, M. Synthesis of 15,20-triamide analogue with polar substituent on the phenyl ring of arenastatin A, an extremely potent cytotoxic spongean depsipeptide. Bioorg. Med. Chem. 2006, 14, 7446–7457. [Google Scholar] [CrossRef]
- Sammet, B.; Bogner, T.; Nahrwold, M.; Weiss, C.; Sewald, N. Approaches for the synthesis of functionalized cryptophycins. J. Org. Chem. 2010, 75, 6953–6960. [Google Scholar] [CrossRef]
- Nahrwold, M.; Weiß, C.; Bogner, T.; Mertink, F.; Conradi, J.; Sammet, B.; Palmisano, R.; Gracia, S.R.; Preuße, T.; Sewald, N. Conjugates of Modified Cryptophycins and RGD-Peptides Enter Target Cells by Endocytosis. J. Med. Chem. 2013, 56, 1853–1864. [Google Scholar] [CrossRef]
- Weiss, C.; Sammet, B.; Sewald, N. Recent approaches for the synthesis of modified cryptophycins. Nat. Prod. Rep. 2013, 30, 924–940. [Google Scholar] [CrossRef]
- Figueras, E.; Borbély, A.; Ismail, M.; Frese, M.; Sewald, N. Novel unit B cryptophycin analogues as payloads for targeted therapy. Beilstein J. Org. Chem. 2018, 14, 1281–1286. [Google Scholar] [CrossRef]
- Liang, J.; Moore, R.E.; Moher, E.D.; Munroe, J.E.; Al-Awar, R.S.; Hay, D.A.; Varie, D.L.; Zhang, T.Y.; Aikins, J.A.; Martinelli, M.J.; et al. Cryptophycins-309, 249 and other cryptophycin analogs: Preclinical efficacy studies with mouse and human tumors. Investig. New Drugs 2005, 23, 213–224. [Google Scholar] [CrossRef]
- Bouchard, H.; Brun, M.-P.; Commerçon, A.; Zhang, J. Novel Conjugates, Preparation Thereof, and Therapeutic Use Thereof. WO 2011/001052, 6 January 2011. [Google Scholar]
- Verma, V.A.; Pillow, T.H.; Depalatis, L.; Li, G.; Phillips, G.L.; Polson, A.G.; Raab, H.E.; Spencer, S.; Zheng, B. The cryptophycins as potent payloads for antibody drug conjugates. Bioorg. Med. Chem. Lett. 2015, 25, 864–868. [Google Scholar] [CrossRef]
- Chen, H.; Lin, Z.; Arnst, K.E.; Miller, D.D.; Li, W. Tubulin inhibitor-based antibody-drug conjugates for Cancer Therapy. Molecules 2017, 22, 1281. [Google Scholar] [CrossRef] [PubMed]
- Bigot, A.; Bouchard, H.; Brun, M.-P.; Clerc, F.; Zhang, J. Novel Cryptophycin Compounds and Conjugates, Their Preparation and Their Therapeutic Use. WO 2017/076998 A1, 11 May 2017. [Google Scholar]
- Su, D.; Kozak, K.R.; Sadowsky, J.; Yu, S.F.; Fourie-O’Donohue, A.; Nelson, C.; Vandlen, R.; Ohri, R.; Liu, L.; Ng, C.; et al. Modulating Antibody-Drug Conjugate Payload Metabolism by Conjugation Site and Linker Modification. Bioconj. Chem. 2018, 29, 1155–1167. [Google Scholar] [CrossRef]
- Leamon, C.P.; Vlahov, I.R.; You, F.; Kleindl, P.J.; Santhapuram, H.K.R. Conjugates Containing Hydrophilic Spacer Linkers. WO 2009/002993 A1, 31 December 2008. [Google Scholar]
- Cazzamalli, S.; Figueras, E.; Pethő, L.; Borbély, A.; Steinkühler, C.; Neri, D.; Sewald, N. In Vivo Antitumor Activity of a Novel Acetazolamide–Cryptophycin Conjugate for the Treatment of Renal Cell Carcinomas. ACS Omega 2018, 3, 14726–14731. [Google Scholar] [CrossRef] [PubMed]
- Borbély, A.; Figueras, E.; Martins, A.; Esposito, S.; Auciello, G.; Monteagudo, E.; Di Marco, A.; Summa, V.; Cordella, P.; Perego, R.; et al. Synthesis and Biological Evaluation of RGD-Cryptophycin Conjugates for Targeted Drug Delivery. Pharmaceutics 2019, 11, 151. [Google Scholar] [CrossRef] [PubMed]
- Morpurgo, M.; Monfardini, C.; Hofland, L.J.; Sergi, M.; Orsolini, P.; Dumont, J.M.; Veronese, F.M. Selective alkylation and acylation of α and ε amino groups with PEG in a somatostatin analogue: Tailored chemistry for optimized bioconjugates. Bioconj. Chem. 2002, 13, 1238–1243. [Google Scholar] [CrossRef]
- Esposito, S.; Mele, R.; Ingenito, R.; Bianchi, E.; Bonelli, F.; Monteagudo, E.; Orsatti, L. An efficient liquid chromatography-high resolution mass spectrometry approach for the optimization of the metabolic stability of therapeutic peptides. Anal. Bioanal. Chem. 2017, 409, 2685–2696. [Google Scholar] [CrossRef]
- Manabe, S.; Machida, H.; Aihara, Y.; Yasunaga, M.; Ito, Y.; Matsumura, Y. Development of a diketopiperazine-forming dipeptidyl Gly-Pro spacer for preparation of an antibody-drug conjugate. MedChemComm 2013, 4, 792–796. [Google Scholar] [CrossRef]
- Dimarchi, R.D.; Brook, G.S. Selective Chemical Removal of a Protein Amino-Terminal Residue. U.S. Patent 4782139, 1 November 1988. [Google Scholar]
- Battersby, J.E.; Hancock, W.S.; Canova-Davis, E.; Oeswein, J.; O’connor, B. Diketopiperazine formation and N-terminal degradation in recombinant human growth hormone. Int. J. Pept. Protein Res. 1994, 44, 215–222. [Google Scholar] [CrossRef]
- Liu, Q.; Cescato, R.; Dewi, D.A.; Rivier, J.; Reubi, J.-C.; Schonbrunn, A. Receptor signaling and endocytosis are differentially regulated by somatostatin analogs. Mol. Pharmacol. 2005, 68, 90–101. [Google Scholar] [CrossRef]
- Cescato, R.; Schulz, S.; Waser, B.; Eltschinger, V.; Rivier, J.E.; Wester, H.-J.; Culler, M.; Ginj, M.; Liu, Q.; Schonbrunn, A.; et al. Internalization of sst2, sst3, and sst5 Receptors: Effects of Somatostatin Agonists and Antagonists. J. Nucl. Med. 2006, 47, 502–511. [Google Scholar] [PubMed]
- Fani, M.; Nicolas, G.P.; Wild, D. Somatostatin Receptor Antagonists for Imaging and Therapy. J. Nucl. Med. 2017, 58, 61S–66S. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | Binding Affinity (IC50 in nM) | Cytotoxicity in AtT20 (IC50 in nM) | Plasma Stability (t1/2 in h) | |
|---|---|---|---|---|---|
| Murine | Human | ||||
| 1 | Cry-55 gly (3) | n.d. | 3.53 | > 24 [48] | > 24 [48] |
| 2 | Oct-PG-Cry (7) | 0.6 | 51.23 | 0.5 | 23 |
| 3 | Oct-GP-Cry (8) | 1.3 | 8.37 | > 24 | 24 |
| 4 | GP-Cry-55 gly | n.d. | 6.05 | n.d. | n.d. |
| 5 | Oct-GP-Cy5.5 (10) | 0.9 | n.d. | n.d. | n.d. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figueras, E.; Martins, A.; Borbély, A.; Le Joncour, V.; Cordella, P.; Perego, R.; Modena, D.; Pagani, P.; Esposito, S.; Auciello, G.; et al. Octreotide Conjugates for Tumor Targeting and Imaging. Pharmaceutics 2019, 11, 220. https://doi.org/10.3390/pharmaceutics11050220
Figueras E, Martins A, Borbély A, Le Joncour V, Cordella P, Perego R, Modena D, Pagani P, Esposito S, Auciello G, et al. Octreotide Conjugates for Tumor Targeting and Imaging. Pharmaceutics. 2019; 11(5):220. https://doi.org/10.3390/pharmaceutics11050220
Chicago/Turabian StyleFigueras, Eduard, Ana Martins, Adina Borbély, Vadim Le Joncour, Paola Cordella, Raffaella Perego, Daniela Modena, Paolo Pagani, Simone Esposito, Giulio Auciello, and et al. 2019. "Octreotide Conjugates for Tumor Targeting and Imaging" Pharmaceutics 11, no. 5: 220. https://doi.org/10.3390/pharmaceutics11050220
APA StyleFigueras, E., Martins, A., Borbély, A., Le Joncour, V., Cordella, P., Perego, R., Modena, D., Pagani, P., Esposito, S., Auciello, G., Frese, M., Gallinari, P., Laakkonen, P., Steinkühler, C., & Sewald, N. (2019). Octreotide Conjugates for Tumor Targeting and Imaging. Pharmaceutics, 11(5), 220. https://doi.org/10.3390/pharmaceutics11050220