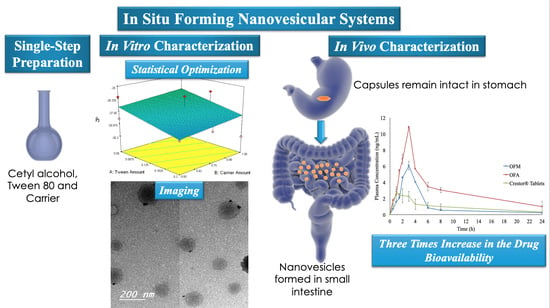
Tripling the Bioavailability of Rosuvastatin Calcium Through Development and Optimization of an In-Situ Forming Nanovesicular System

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. IFN Preparation
2.3. The Study Statistical Design
2.4. Characterization of the Prepared IFN
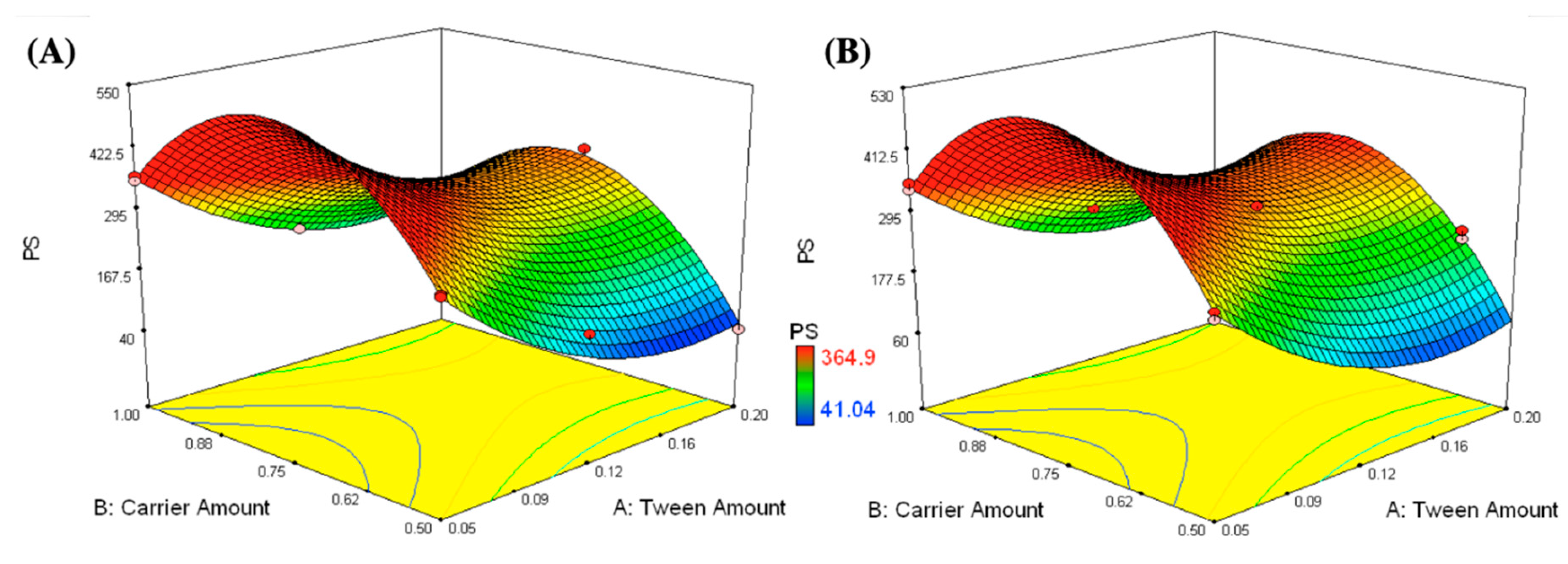
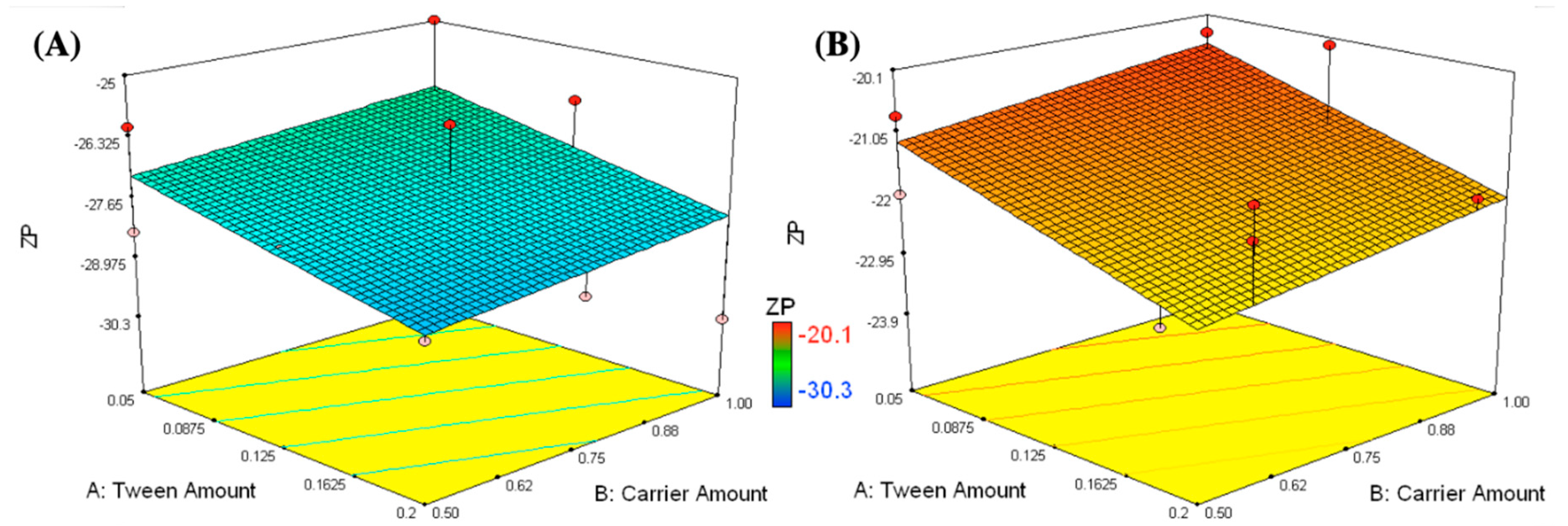
2.4.1. Measurement of PS, PDI, and ZP
2.4.2. Measurement of EE
2.4.3. Flowability of the prepared IFN
2.5. Characterization of the Optimized IFN Formulations
2.5.1. Imaging
2.5.2. In Vitro RC Release from the Optimized IFN Formulations
2.6. Preparation of Enteric-Coated Capsules Filled with the Optimized RC IFN
2.7. Characterization of the Prepared Enteric-Coated Capsules
2.7.1. Weight Variation and Content Uniformity
2.7.2. In Vitro RC Release from the Enteric-Coated Capsules
2.8. Bioavailability Study of RC
3. Results and Discussion
3.1. Characterization of the Prepared IFN
3.1.1. Measurement of PS, PDI, and ZP
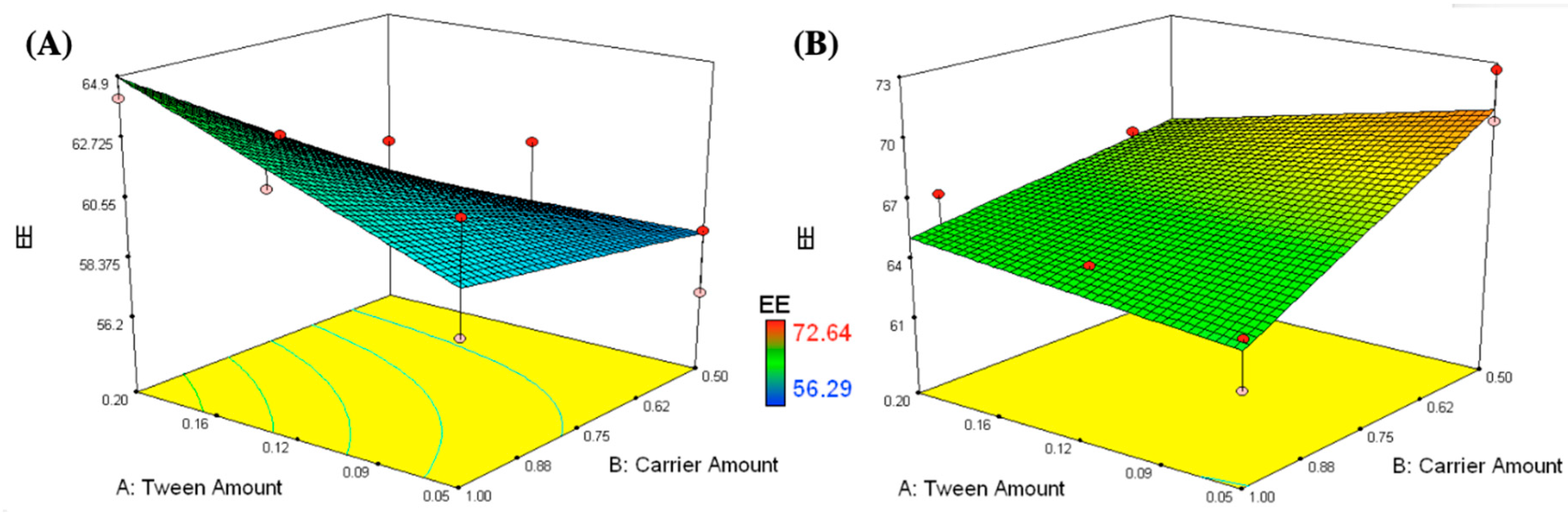
3.1.2. Measurement of EE
3.1.3. Flowability of the Prepared IFN
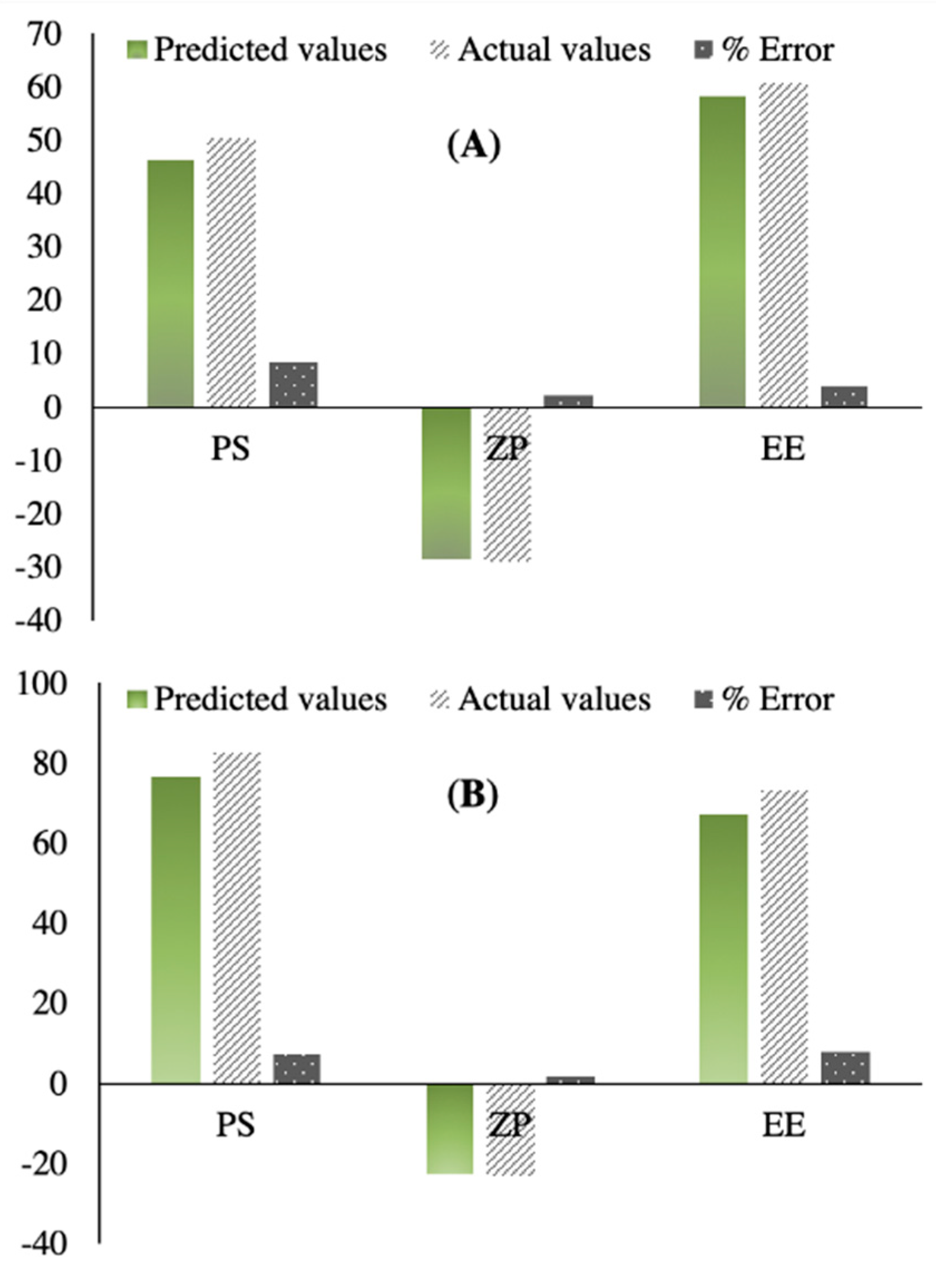
3.2. Selection of the Optimized IFN Formulations
3.3. Characterization of the Optimized IFN Formulations
3.3.1. Imaging
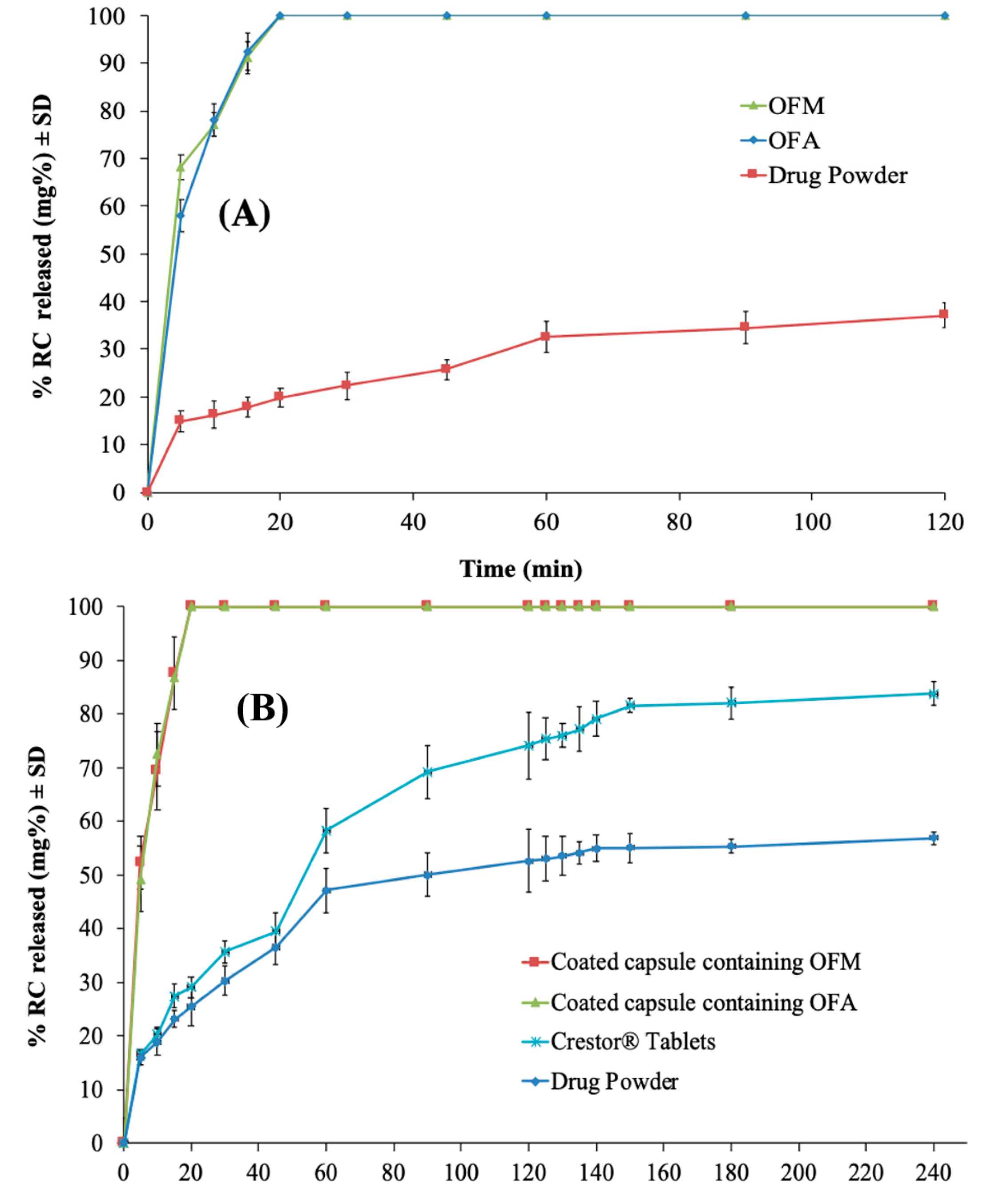
3.3.2. In Vitro RC Release from the Optimized IFN Formulations
3.4. Characterization of the Prepared Enteric-Coated Capsules
3.4.1. Weight Variation and Content Uniformity
3.4.2. In Vitro RC Release from the Enteric-Coated Capsules
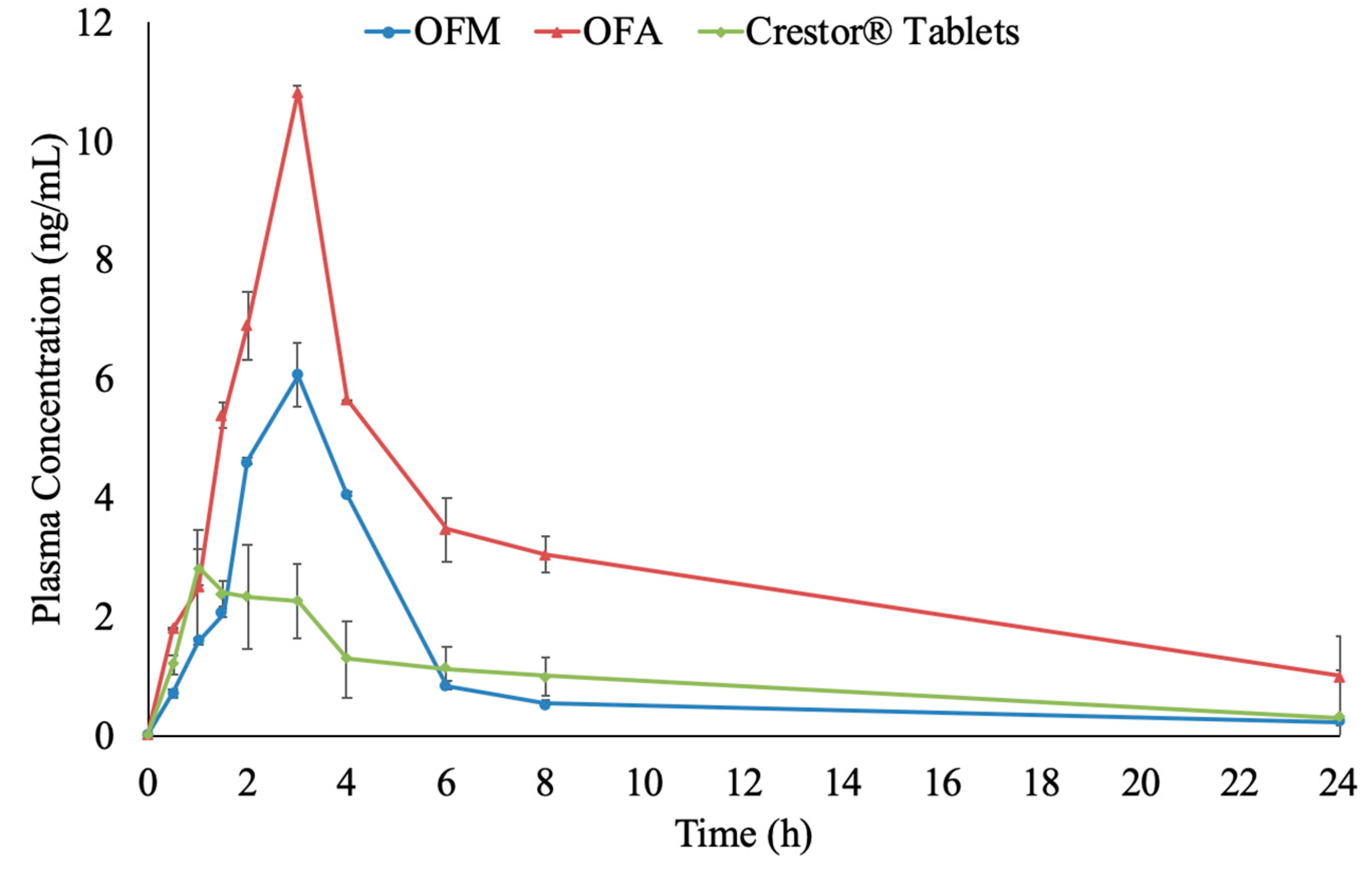
3.5. Bioavailability of RC
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lennernäs, H.; Fager, G. Pharmacodynamics and Pharmacokinetics of the HMG-CoA Reductase Inhibitors: Similarities and differences. Clin. Pharmacokinet. 1997, 32, 403–425. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, K.; Vijaya, C.; Tamil, N.; Hari, R.; Abdu, S. Self nanoemulsifying drug delivery system (SNEDDS) of Rosuvastatin calcium: Design, formulation, bioavailability and pharmacokinetic evaluation. Colloids Surf. B 2013, 112, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Zaid, A.N.; Shtayah, R.; Qadumi, A.; Ghanem, M.; Qedan, R.; Daibes, M.; Abu Awwad, S.; Jaradat, N.; Kittana, N. Stability of extemporaneously prepared rosuvastatin oral suspension. Am. J. Heal. Pharm. 2017, 74, 1579–1583. [Google Scholar] [CrossRef] [PubMed]
- Junghanns, J.-U.A.H.; Müller, R.H. Nanocrystal technology, drug delivery and clinical applications. Int. J. Nanomed. 2008, 3, 295. [Google Scholar]
- Junyaprasert, V.B.; Morakul, B. Nanocrystals for enhancement of oral bioavailability of poorly water-soluble drugs. Asian J. Pharm. Sci. 2015, 10, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- El-Dahmy, R.M.; Elsayed, I.; Elshafeey, A.H.; El Gawad, N.A.A.; El-Gazayerly, O.N. Optimization of long circulating mixed polymeric micelles containing vinpocetine using simple lattice mixture design, in vitro and in vivo characterization. Int. J. Pharm. 2014, 477, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Hall-Manning, T.J.; Holland, G.H.; Rennie, G.; Revell, P.; Hines, J.; Barratt, M.D.; Basketter, D.A. Skin irritation potential of mixed surfactant systems. Food Chem. Toxicol. 1998, 36, 233–238. [Google Scholar] [CrossRef]
- FDA. Defoaming agents used in coatings. In Indirect Food Additives: Paper and Paperboard Components; US Food and Drug Administration: Maryland, WA, USA, 2018. [Google Scholar]
- FDA. Polysorbate 80. In Food Additives Permitted for Direct Addition to Food for Human Consumption; US Food and Drug Administration: Maryland, WA, USA, 2018. [Google Scholar]
- Elkasabgy, N.A.; Elsayed, I.; Elshafeey, A.H. Design of lipotomes as a novel dual functioning nanocarrier for bioavailability enhancement of lacidipine: In-vitro and in-vivo characterization. Int. J. Pharm. 2014, 472, 369–379. [Google Scholar] [CrossRef]
- Capco, D.G.; Chen, Y. Nanomaterial: Impacts on Cell Biology and Medicine; Springer: Heidelberg, Germany, 2014. [Google Scholar]
- Chun, H.J.; Park, C.H.; Kwon, I.K.; Khang, G. Cutting-Edge Enabling Technologies for Regenerative Medicine; Springer: Singapore, 2018. [Google Scholar]
- Andronescu, E.; Grumezescu, A.M. Nanostructures for Oral Medicine; Elsevier Science: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Franzé, S.; Selmin, F.; Samaritani, E.; Minghetti, P.; Cilurzo, F. Lyophilization of liposomal formulations: still necessary, still challenging. Pharmaceutics 2018, 10, 139. [Google Scholar] [CrossRef]
- Marín, D.; Alemán, A.; Sánchez-Faure, A.; Montero, P.; Gómez-Guillén, M.C. Freeze-dried phosphatidylcholine liposomes encapsulating various antioxidant extracts from natural waste as functional ingredients in surimi gels. Food Chem. 2018, 245, 525–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Grainger, D.W. Lyophilized liposome-based parenteral drug development: Reviewing complex product design strategies and current regulatory environments. Adv. Drug Deliv. Rev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Hu, Q.; Zhou, M.; Xue, J.; Luo, Y. Preparation of ultra-fine powders from polysaccharide-coated solid lipid nanoparticles and nanostructured lipid carriers by innovative nano spray drying technology. Int. J. Pharm. 2016, 511, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wei, Q.; Yang, Q.; Cao, X.; Li, Q.; Shi, F.; Tong, S.S.; Feng, C.; Yu, Q.; Yu, J.; et al. A novel formulation of [6]-gingerol: Proliposomes with enhanced oral bioavailability and antitumor effect. Int. J. Pharm. 2018, 535, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Nekkanti, V.; Venkatesan, N.; V Betageri, G. Proliposomes for oral delivery: progress and challenges. Curr. Pharm. Biotechnol. 2015, 16, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.W.; Lee, K.-H. Pro-micelle pharmaceutical compositions. U.S. Patent 6,951,655, 4 October 2005. [Google Scholar]
- Khuri, A.I.; Mukhopadhyay, S. Response surface methodology. Wiley Interdiscip. Rev. Comput. Stat. 2010, 2, 128–149. [Google Scholar] [CrossRef]
- Aburahma, M.; Abdelbary, G. Novel diphenyl dimethyl bicarboxylate provesicular powders with enhanced hepatocurative activity: Preparation, optimization, in vitro/in vivo evaluation. Int. J. Pharm. 2012, 422, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; He, L.; Nie, S. Optimized preparation of vinpocetine proliposomes by a novel method and in vivo evaluation of its pharmacokinetics in New Zealand rabbits. J. Control Release 2009, 140, 61–68. [Google Scholar] [CrossRef]
- Elhissi, A.; Hidayat, K.; David, A. Air-jet and vibrating-mesh nebulization of niosomes generated using a particulate-based proniosome technology. Int. J. Pharm. 2013, 444, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Soliman, S.M.; Abdelmalak, N.S.; El-Gazayerly, O.N.; Abdelaziz, N. Novel non-ionic surfactant proniosomes for transdermal delivery of lacidipine: Optimization using 23factorial design and in vivo evaluation in rabbits. Drug Deliv. 2016, 23, 1608–1622. [Google Scholar] [CrossRef]
- Carr, R.L. Classifying flow properties of solids. Chem. Eng. 1964, 72, 69–72. [Google Scholar]
- Hausner, H.H. friction condition in a mass of metal powders. Int. J. Powder Met. 1967, 3, 7–13. [Google Scholar]
- Aggarwal, G.; Chandel, P.; Harikumar, S.; Bansal, S. Design and development of cefdinir niosomes for oral delivery. J. Pharm. Bioallied. Sci. 2013, 5, 318. [Google Scholar] [CrossRef] [PubMed]
- Kapure, V.J.; Pande, V.V.; Deshmukh, P.K. Dissolution enhancement of rosuvastatin calcium by liquisolid compact technique. J. Pharm. 2013. [Google Scholar] [CrossRef] [PubMed]
- Vraníková, B.; Gajdziok, J.; Doležel, P. The effect of superdisintegrants on the properties and dissolution profiles of liquisolid tablets containing rosuvastatin. Pharm. Dev. Technol. 2015, 22, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Sinha, V.R.; Kumria, R. Coating polymers for colon specific drug delivery: A comparative in vitro evaluation. Acta Pharm. 2003, 53, 41–47. [Google Scholar] [PubMed]
- British Pharmacopoeia Commission. The British Pharmacopoeia; The Stationary Office: London, UK, 2008. [Google Scholar]
- Dhoranwala, K.A.; Shah, P.; Shah, S. Formulation Optimization of Rosuvastatin Calcium- Loaded Solid Lipid Nanoparticles by 3 2 Full-Factorial Design. NanoWorld J. 2015, 1, 110–119. [Google Scholar] [CrossRef]
- Kulkamj, N.S.; Dhole, S.N.; Ranpise, N.S. Development and evaluation of self emulsifying drug delivery system for Rosuvastatin Calcium. Int. J. Res. Dev. Pharm. Life Sci. 2013, 2, 531–537. [Google Scholar]
- Kumar, A.A.; Kumari, M.S.; Surekha, K.; Prasad, C.; Suresh, S. Formulation and evaluation of sustained release Valsartan matrix tablets by using natural polymers. Int. J. Pharm. Chem. Biol. Sci. 2012, 2, 146–150. [Google Scholar]
- Abo Enin, H.A. Self-nanoemulsifying drug-delivery system for improved oral bioavailability of rosuvastatin using natural oil antihyperlipdemic. Drug Dev. Ind. Pharm. 2015, 41, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Ponnuraj, R.K.J.; Gopalakrishnan, S.; Arumugam, A. Formulation and Development of Capsules Containing Rosuvastatin calcium nanoparticles and epigallocatechin gallate nanoparticles. Indo Am. J. Pharm. Res. 2015, 5, 2217–2231. [Google Scholar]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed]
- El-Kommos, M.E.; Mohamed, N.A.; Ali, H.R.H.; Hakiem, A.F.A. Micellar electrokinetic chromatographic determination of rosuvastatin in rabbit plasma and evaluation of its pharmacokinetics and interaction with niacin. Biomed. Chromatogr. 2014, 28, 1828–1838. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.P.; Midha, K.K.; Dighe, S.; McGilveray, I.J.; Skelly, J.P.; Yacobi, A.; Layloff, T.; Viswanathan, C.T.; Cook, C.E.; McDowall, R.D. Analytical methods validation: Bioavailability, bioequivalence and pharmacokinetic studies. Conference report. Eur. J. Drug Metab. Pharmacokinet. 1991, 16, 249. [Google Scholar]
- Shargel, L.; Wu-Pong, S.; Yu, A. Applied Biopharmaceutics & Pharmacokinetics, 6th ed.; McGraw Hill Professional: New York, NY, USA, 2012. [Google Scholar]
- Negi, P.; Ahmad, F.; Ahmad, D. Development of a novel formulation for transdermal delivery of an antidepressant drug. Int. J. Pharm. Sci. Res. 2011, 2, 1766–1771. [Google Scholar]
- Rahman, S.; Abdelmalak, N.; Badawi, A. Formulation of tretinoin-loaded topical proniosomes for treatment of acne: in-vitro characterization, skin irritation test and comparative clinical study. Drug Deliv. 2014, 22, 731–739. [Google Scholar] [CrossRef]
- Huang, S.-L.; MacDonald, R.C. Acoustically active liposomes for drug encapsulation and ultrasound-triggered release. Biochim. Biophys. Acta Biomembr. 2004, 1665, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Varshosaz, J.; Eskandari, S.; Tabbakhian, M. Freeze-drying of nanostructure lipid carriers by different carbohydrate polymers used as cryoprotectants. Carbohydr. Polym. 2012, 88, 1157–1163. [Google Scholar] [CrossRef]
- Cho, H.J.; Park, J.W.; Yoon, I.S.; Kim, D.D. Surface-modified solid lipid nanoparticles for oral delivery of docetaxel: enhanced intestinal absorption and lymphatic uptake. Int. J. Nanomed. 2014, 9, 495–504. [Google Scholar] [Green Version]
- Wissing, S.A.; Kayser, O.; Muller, R.H. Solid lipid nanoparticles for parenteral drug delivery. Adv. Drug Deliv. Rev. 2004, 56, 1257–1272. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Hsu, C.; Zhu, L.; Tseng, S.; Hsu, J.P. Influence of metal oxide nanoparticles concentration on their zeta potential. J. Colloid Interface Sci. 2013, 407, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Gawali, S.L.; Barick, B.K.; Barick, K.C.; Hassan, P.A. Effect of sugar alcohol on colloidal stabilization of magnetic nanoparticles for hyperthermia and drug delivery applications. J. Alloy. Compd. 2017, 725, 800–806. [Google Scholar] [CrossRef]
- de Lima, L.S.; Araujo, M.D.; Quináia, S.P.; Migliorine, D.W.; Garcia, J.R. Adsorption modeling of Cr, Cd and Cu on activated carbon of different origins by using fractional factorial design. Chem. Eng. J. 2011, 166, 881–889. [Google Scholar] [CrossRef]
- Annadurai, G.; Ling, L.Y.; Lee, J.F. Statistical optimization of medium components and growth conditions by response surface methodology to enhance phenol degradation by Pseudomonas putida. J. Hazard. Mater. 2008, 151, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Etman, M.A.; Naggar, V.F. Thermodynamics of paracetamol solubility in sugar-water cosolvent systems. Int. J. Pharm. 1990. [Google Scholar] [CrossRef]
- Wells, J.; Aulton, M. Pharmaceutics the Science of Dosage Form Design, 2nd ed.; Churchill Livingstone: Edinburgh, UK, 2002. [Google Scholar]
- Kamel, A.O.; Mahmoud, A.A. Enhancement of human oral bioavailability and in vitro antitumor activity of rosuvastatin via spray dried self-nanoemulsifying drug delivery system. J. Biomed. Nanotechnol. 2013, 9, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Banker, G.S.; Siepmann, J.; Rhodes, C. Drugs and the Pharmaceutical Sciences. In Modern Pharmaceutics; CRC Press: Boca Raton, FL, USA, 2002. [Google Scholar]
- Bremmell, K.E.; Tan, A.; Martin, A.; Prestidge, C.A. Tableting Lipid-Based Formulations for Oral Drug Delivery: A Case Study with Silica Nanoparticle–Lipid–Mannitol Hybrid Microparticles. J. Pharm. Sci. 2013, 102, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Pai, R.S.; Devi, V.K. Optimization of pellets containing solid dispersion prepared by extrusion/spheronization using central composite design and desirability function. J. Young Pharm. 2012, 4, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.D.; Wen, H.; Taylor, L.S. Non-sink dissolution conditions for predicting product quality and in vivo performance of supersaturating drug delivery systems. J. Pharm. Sci. 2016, 105, 2477–2488. [Google Scholar] [CrossRef] [PubMed]
- Trasi, N.S.; Purohit, H.S.; Wen, H.; Sun, D.D.; Taylor, L.S. Non-sink dissolution behavior and solubility limit of commercial tacrolimus amorphous formulations. J. Pharm. Sci. 2017, 106, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.B.; Barnes, T.J.; Prestidge, C.A. Silica nanoparticle stabilization of liquid crystalline lipid dispersions: impact on enzymatic digestion and drug solubilization. Curr. Drug Deliv. 2015, 12, 47–55. [Google Scholar] [CrossRef] [PubMed]
- FDA. Dissolution Methods. Available online: https://www.accessdata.fda.gov/scripts/cder/dissolution/dsp_SearchResults.cfm (accessed on 14 January 2019).
- Sarfraz, R.M.; Ahmad, M.; Mahmood, A.; Minhas, M.U.; Yaqoob, A. Development and evaluation of rosuvastatin calcium based microparticles for solubility enhancement: an in vitro study. Adv. Polym. Technol. 2017, 36, 433–441. [Google Scholar] [CrossRef]
- Butler, J.; Devane, J.; Stark, P. Pharmaceutical formulations for carrier-mediated transport statins and uses thereof. U.S. Patent 8,987,322, 24 March 2015. [Google Scholar]
- Bando, H.; McGinity, J.W. Physicochemical properties of enteric films prepared from aqueous dispersions and organic solutions. Int. J. Pharm. 2006, 313, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Garg, B.; Beg, S.; Kaur, R.; Kumar, R.; Katare, O.P.; Singh, B. Long-chain triglycerides-based self-nanoemulsifying oily formulations (SNEOFs) of darunavir with improved lymphatic targeting potential. J. Drug Target. 2018, 26, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Garg, B.; Katare, O.P.; Beg, S.; Lohan, S.; Singh, B. Systematic development of solid self-nanoemulsifying oily formulations (S-SNEOFs) for enhancing the oral bioavailability and intestinal lymphatic uptake of lopinavir. Colloids Surf. B 2016, 141, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.-S.; Cho, C.-W. Surface modification of solid lipid nanoparticles for oral delivery of curcumin: Improvement of bioavailability through enhanced cellular uptake, and lymphatic uptake. Eur. J. Pharm. Biopharm. 2017, 117, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Makwana, V.; Jain, R.; Patel, K.; Nivsarkar, M.; Joshi, A. Solid lipid nanoparticles (SLN) of Efavirenz as lymph targeting drug delivery system: Elucidation of mechanism of uptake using chylomicron flow blocking approach. Int. J. Pharm. 2015, 495, 439–446. [Google Scholar] [CrossRef]
- Siram, K.; Marslin, G.; Raghavan, C.V.; Balakumar, K.; Rahman, H.; Franklin, G. A brief perspective on the diverging theories of lymphatic targeting with colloids. Int. J. Nanomedicine. 2016, 11, 2867. [Google Scholar]
- Porter, C.J.; Charman, W.N. Intestinal lymphatic drug transport: An update. Adv. Drug Deliv. Rev. 2001, 50, 61–80. [Google Scholar] [PubMed]
- Schiller, L.R. Evaluation of chronic diarrhea and irritable bowel syndrome with diarrhea in adults in the era of precision medicine. Am. J. Gastroenterol 2018, 113, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Quaresma, A.B.; Briancini, G.; Chiesa, T.; Monteiro, S.; Mergener, R.A. Intestinal preparations for colonoscopy. Comparative study: Mannitol, picosulphate and macrogol. JCOL 2018, 38, 105–110. [Google Scholar] [CrossRef]
- Adkin, D.A.; Davis, S.S.; Sparrow, R.A.; Huckle, P.D.; Phillips, A.J.; Wilding, I.R. The effect of different concentrations of mannitol in solution on small intestinal transit: Implications for drug absorption. Pharm. Res. 1995, 12, 393–396. [Google Scholar] [CrossRef]
- Dupont, C.; Vernisse, B. Anti-Diarrheal Effects of Diosmectite in the Treatment of Acute Diarrhea in Children. Paediatr Drugs 2009, 11, 89–99. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulations | X1: Tween 80 Amount (g) | X2: Carrier Amount (g) | X3: Carrier Type | Y1: PS (nm) | Y2: ZP (mV) | PDI | Y3: EE (%w/w) | CI (%) | HR |
|---|---|---|---|---|---|---|---|---|---|
| F1 | 0.05 | 0.5 | Mannitol | 304.7 ± 11.6 | −28.4 ± 1.1 | 0.325 ± 0.05 | 56.3 ± 3.5 | 12.5 ± 0.2 | 1.1 ± 0.02 |
| 299.4 ± 10.4 | −26.1 ± 1.0 | 0.299 ± 0.03 | 58.7 ± 4.2 | 12.4 ± 0.1 | 1.2 ± 0.01 | ||||
| F2 | 0.05 | 1 | 364.9 ± 9.1 | −25.0 ± 0.5 | 0.275 ± 0.03 | 58.5 ± 4.1 | 18.7 ± 0.3 | 1.2 ± 0.02 | |
| 354.8 ± 6.5 | −27.0 ± 0.0 | 0.127 ± 0.05 | 62.9 ± 5.5 | 16.7 ± 1.0 | 1.2 ± 0.04 | ||||
| F3 | 0.12 | 0.81 | 333.6 ± 13.9 | −26.2 ± 1.0 | 0.241 ± 0.02 | 60.9 ± 1.8 | 20.0 ± 0.4 | 1.2 ± 0.03 | |
| F4 | 0.12 | 0.52 | 124.6 ± 1.0 | −27.9 ± 0.8 | 0.236 ± 0.03 | 60.8 ± 5.7 | 10.0 ± 0.3 | 1.1 ± 0.02 | |
| F5 | 0.12 | 1 | 171.3 ± 14.5 | −26.2 ± 1.1 | 0.127 ± 0.05 | 63.4 ± 3.3 | 17.6 ± 0.6 | 1.2 ± 0.01 | |
| F6 | 0.2 | 0.5 | 41.0 ± 0.9 | −28.7 ± 0.6 | 0.221 ± 0.04 | 59.1 ± 2.7 | 20.0 ± 0.4 | 1.2 ± 0.04 | |
| F7 | 0.2 | 0.75 | 348.2 ± 14.5 | −28.8 ± 1.1 | 0.316 ± 0.05 | 64.4 ± 3.3 | 17.6 ± 0.6 | 1.2 ± 0.01 | |
| F8 | 0.2 | 1 | 180.5 ± 0.9 | −30.3 ± 0.6 | 0.174 ± 0.04 | 72.1 ± 2.7 | 20.0 ± 0.4 | 1.2 ± 0.04 | |
| F9 | 0.05 | 0.5 | Aerosil | 278.3 ± 10.6 | −20.8 ± 0.1 | 0.271 ± 0.01 | 70.0 ± 3.5 | 18.9 ± 0.1 | 1.2 ± 0.01 |
| 264.7 ± 12.4 | −22.1 ± 0.2 | 0.249 ± 0.01 | 72.6 ± 1.6 | 17.6 ± 0.1 | 1.1 ± 0.02 | ||||
| F10 | 0.05 | 1 | 337.9 ± 2.7 | −21.8 ± 0.7 | 0.197 ± 0.04 | 61.7 ± 1.2 | 10.2 ± 0.3 | 1.2 ± 0.02 | |
| 351.2 ± 3.5 | −20.4 ± 0.8 | 0.267 ± 0.03 | 64.2 ± 0.8 | 11.6 ± 0.3 | 1.2 ± 0.01 | ||||
| F11 | 0.12 | 0.96 | 240.0 ± 6.9 | −20.1 ± 0.5 | 0.313 ± 0.02 | 65.2 ± 2.1 | 13.8 ± 0.5 | 1.2 ± 0.04 | |
| F12 | 0.13 | 0.68 | 329.8 ± 9.5 | −23.9 ± 0.1 | 0.304 ± 0.05 | 66.8 ± 6.3 | 5.4 ± 0.3 | 1.0 ± 0.03 | |
| F13 | 0.2 | 0.58 | 235.5 ± 11.7 | −21.1 ± 1.0 | 0.245 ± 0.01 | 65.2 ± 3.9 | 3.3 ± 0.7 | 1.0 ± 0.05 | |
| 219.4 ± 7.0 | −21.6 ± 1.1 | 0.249 ± 0.01 | 67.2 ± 7.0 | 3.5 ± 0.7 | 1.1 ± 0.04 | ||||
| F14 | 0.2 | 0.95 | 266.0 ± 5.7 | −21.9 ± 0.4 | 0.233 ± 0.06 | 66.8 ± 1.1 | 10.2 ± 0.1 | 1.1 ± 0.01 |
| Pharmacokinetics Parameters | Treatment (Mean ± SD) a | ||
|---|---|---|---|
| OFM | OFA | The Market Product | |
| Cmax (ng/mL) | 6.08 ± 0.52 | 10.84 ± 1.02 | 2.83 ± 0.94 |
| AUC0-24 (ng.h/mL) | 26.22 ± 1.87 | 71.71 ± 3.98 | 23.02 ± 1.36 |
| AUC0-∞ (ng.h/mL) | 29.92 ± 1.24 | 86.16 ± 5.21 | 27.38 ± 2.09 |
| Tmax (h) | 3.00 | 3.00 | 1.00 |
| Ke (l/h) | 0.06 ± 0.00 | 0.07 ± 0.00 | 0.07 ± 0.00 |
| t½e (h) | 10.92 ± 1.13 | 10.01 ± 1.89 | 9.63 ± 0.82 |
| MRT | 9.80 | 12.66 | 12.17 |
| % Relative Bioavailability | 109.28% | 314.68% | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsayed, I.; El-Dahmy, R.M.; Elshafeey, A.H.; Abd El Gawad, N.A.; El Gazayerly, O.N. Tripling the Bioavailability of Rosuvastatin Calcium Through Development and Optimization of an In-Situ Forming Nanovesicular System. Pharmaceutics 2019, 11, 275. https://doi.org/10.3390/pharmaceutics11060275
Elsayed I, El-Dahmy RM, Elshafeey AH, Abd El Gawad NA, El Gazayerly ON. Tripling the Bioavailability of Rosuvastatin Calcium Through Development and Optimization of an In-Situ Forming Nanovesicular System. Pharmaceutics. 2019; 11(6):275. https://doi.org/10.3390/pharmaceutics11060275
Chicago/Turabian StyleElsayed, Ibrahim, Rania Moataz El-Dahmy, Ahmed Hassen Elshafeey, Nabaweya Abdelaziz Abd El Gawad, and Omaima Naim El Gazayerly. 2019. "Tripling the Bioavailability of Rosuvastatin Calcium Through Development and Optimization of an In-Situ Forming Nanovesicular System" Pharmaceutics 11, no. 6: 275. https://doi.org/10.3390/pharmaceutics11060275
APA StyleElsayed, I., El-Dahmy, R. M., Elshafeey, A. H., Abd El Gawad, N. A., & El Gazayerly, O. N. (2019). Tripling the Bioavailability of Rosuvastatin Calcium Through Development and Optimization of an In-Situ Forming Nanovesicular System. Pharmaceutics, 11(6), 275. https://doi.org/10.3390/pharmaceutics11060275