Physiologically-Based Pharmacokinetic (PBPK) Modeling Providing Insights into Fentanyl Pharmacokinetics in Adults and Pediatric Patients

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Software
2.2. PBPK Model Building for Adult Populations
2.3. PBPK Modeling in Pediatrics
2.4. PBPK Model Evaluation and DDI Modeling
3. Results
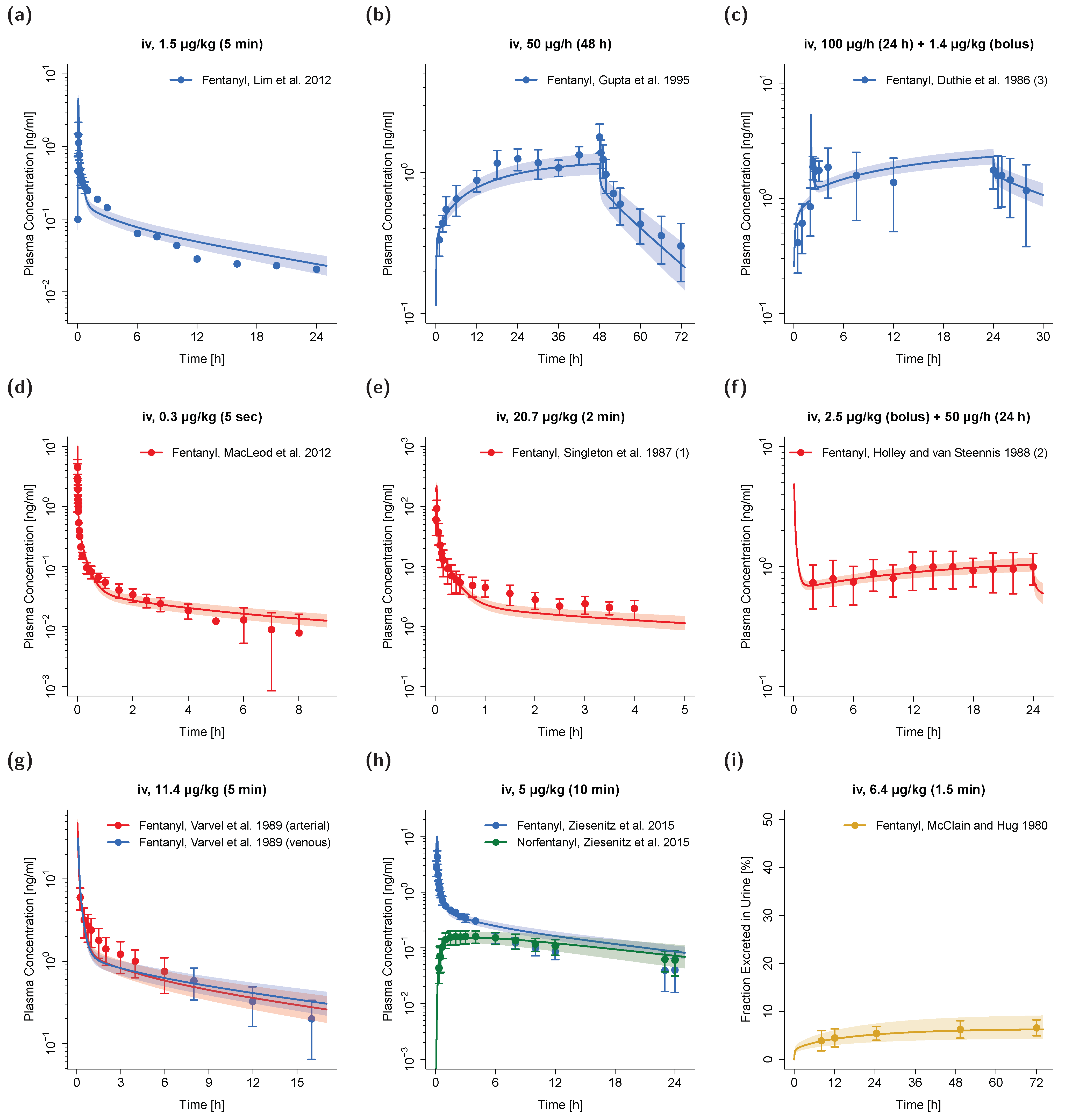
3.1. PK Data for PBPK Model Development and Pediatric Scaling
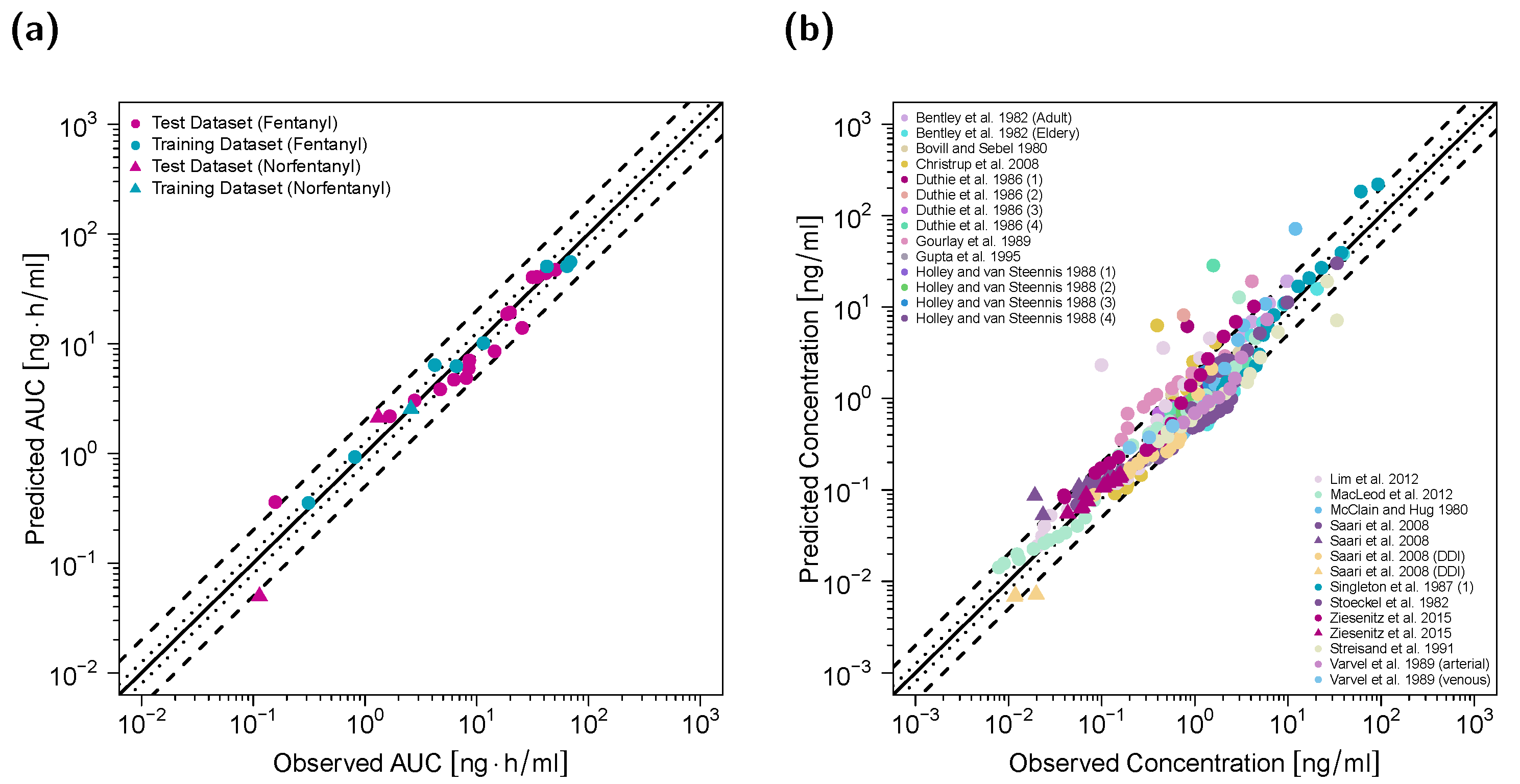
3.2. Adult PBPK Model Building and Evaluation
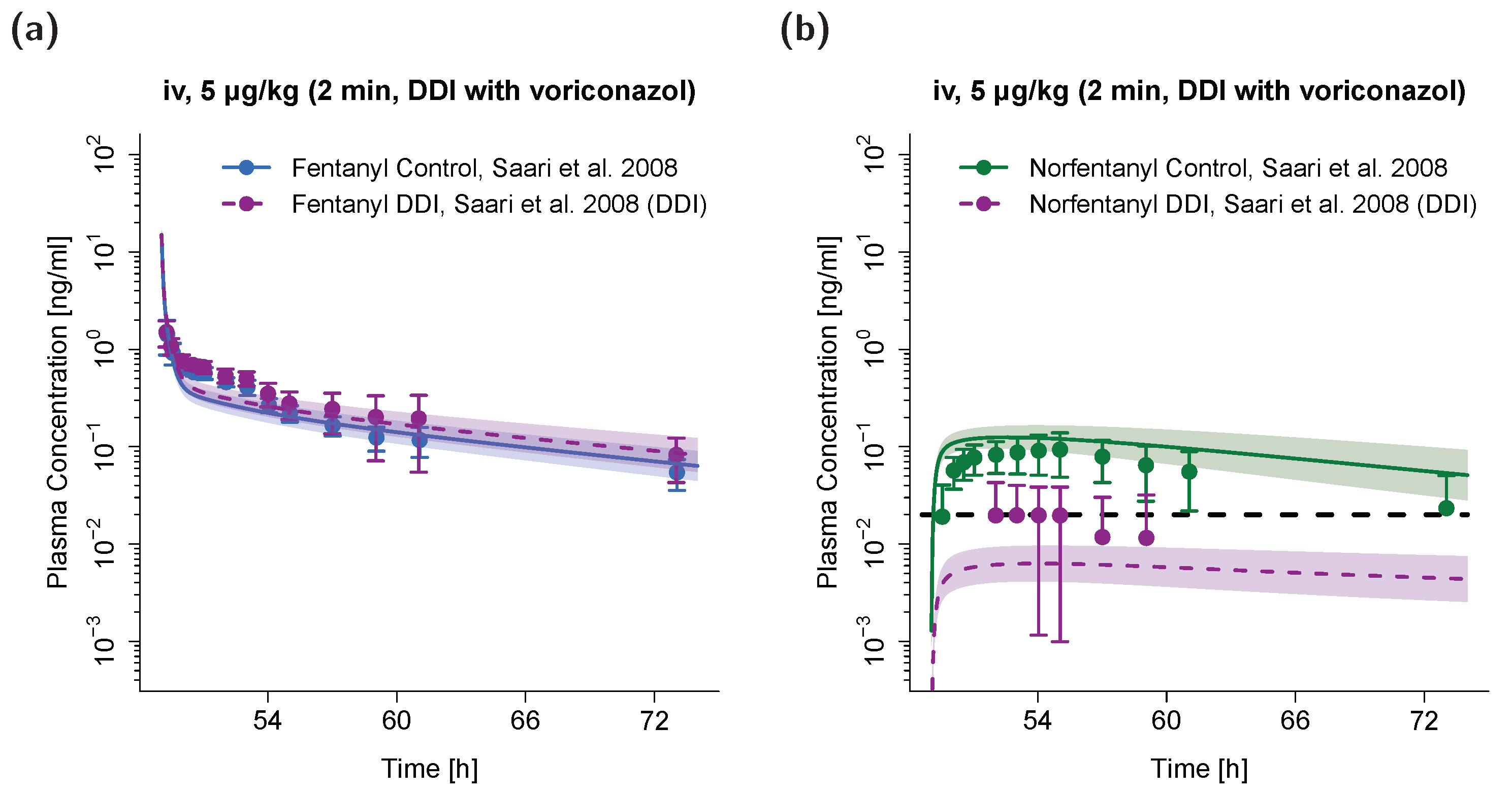
3.3. PBPK DDI Modeling
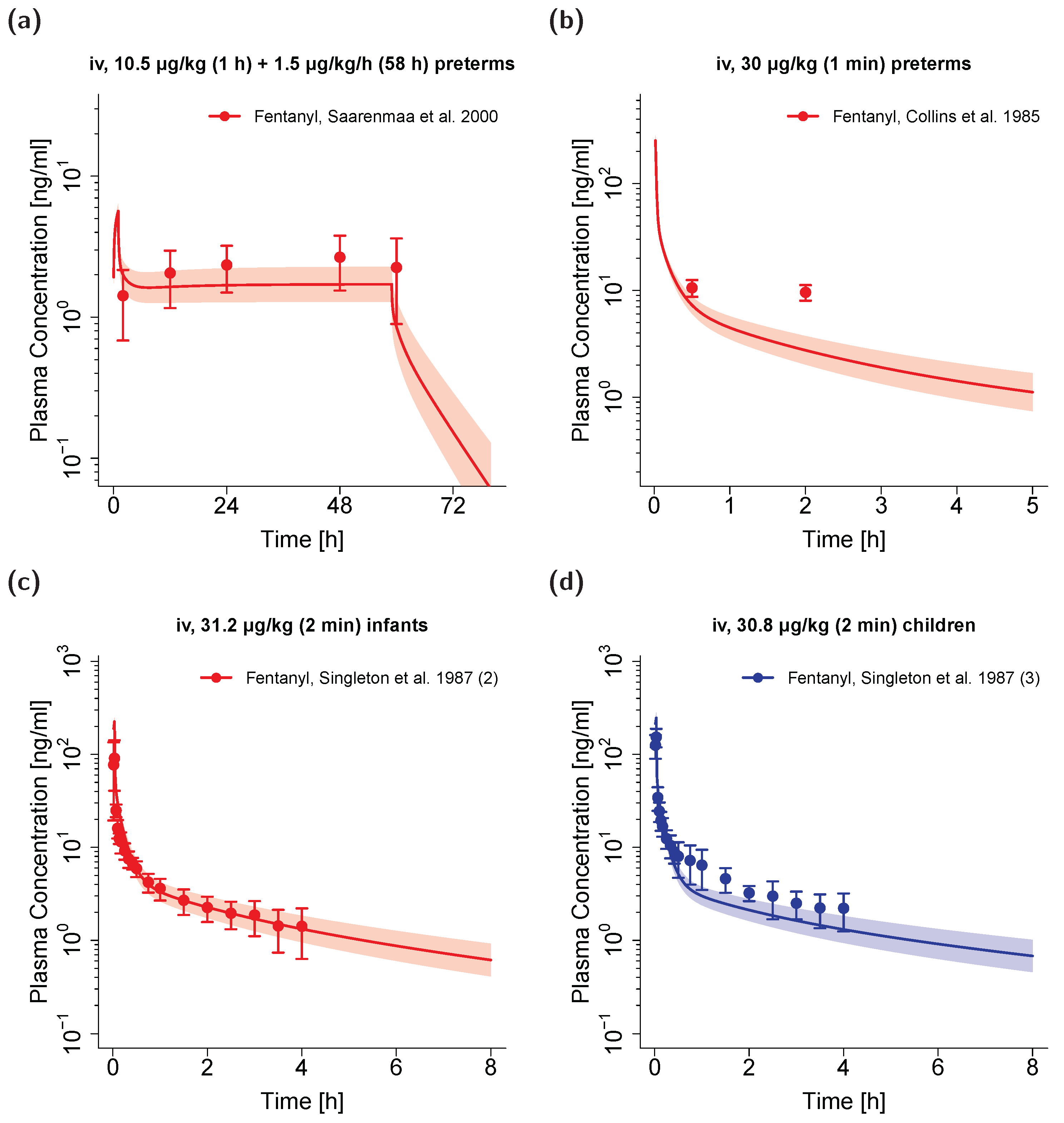
3.4. Pediatric PBPK Model Building and Evaluation
3.5. Clearance in Neonates with Increased Intraabdominal Pressure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pacifici, G.M. Clinical Pharmacology of Fentanyl in Preterm Infants. A Review. Pediatr. Neonatol. 2015, 56, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziesenitz, V.C.; Vaughns, J.D.; Koch, G.; Mikus, G.; van den Anker, J.N. Pharmacokinetics of Fentanyl and Its Derivatives in Children: A Comprehensive Review. Clin. Pharmacokinet. 2018, 57, 125–149. [Google Scholar] [CrossRef] [PubMed]
- Kuip, E.J.M.; Zandvliet, M.L.; Koolen, S.L.W.; Mathijssen, R.H.J.; van der Rijt, C.C.D. A review of factors explaining variability in fentanyl pharmacokinetics; focus on implications for cancer patients. Br. J. Clin. Pharmacol. 2017, 83, 294–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guitton, J.; Buronfosse, T.; Désage, M.; Lepape, A.; Brazier, J.L.; Beaune, P. Possible involvement of multiple cytochrome P450S in fentanyl and sufentanil metabolism as opposed to alfentanil. Biochem. Pharmacol. 1997, 53, 1613–1619. [Google Scholar] [CrossRef]
- Ziesenitz, V.C.; König, S.K.; Mahlke, N.S.; Skopp, G.; Haefeli, W.E.; Mikus, G. Pharmacokinetic interaction of intravenous fentanyl with ketoconazole. J. Clin. Pharmacol. 2015, 55, 708–717. [Google Scholar] [CrossRef]
- Saari, T.I.; Laine, K.; Neuvonen, M.; Neuvonen, P.J.; Olkkola, K.T. Effect of voriconazole and fluconazole on the pharmacokinetics of intravenous fentanyl. Eur. J. Clin. Pharmacol. 2008, 64, 25–30. [Google Scholar] [CrossRef]
- Irazuzta, J.; Pascucci, R.; Perlman, N.; Wessel, D. Effects of fentanyl administration on respiratory system compliance in infants. Crit. Care Med. 1993, 21, 1001–1004. [Google Scholar] [CrossRef]
- Dewhirst, E.; Naguib, A.; Tobias, J.D. Chest wall rigidity in two infants after low-dose fentanyl administration. Pediatr. Emerg. Care 2012, 28, 465–468. [Google Scholar] [CrossRef]
- Ward, R.M.; Sherwin, C.M.T. Ethics of Drug Studies in the Newborn. Pediatr. Drugs 2014, 17, 37–42. [Google Scholar] [CrossRef]
- Maharaj, A.R.; Edginton, A.N. Physiologically Based Pharmacokinetic Modeling and Simulation in Pediatric Drug Development. CPT Pharmacometrics Syst. Pharmacol. 2014, 3, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lippert, J.; Burghaus, R.; Edginton, A.; Frechen, S.; Karlsson, M.; Kovar, A.; Lehr, T.; Milligan, P.; Nock, V.; Ramusovic, S.; et al. Open Systems Pharmacology Community—An Open Access, Open Source, Open Science Approach to Modeling and Simulation in Pharmaceutical Sciences. CPT Pharmacometrics Syst. Pharmacol. 2019, 8, 878–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yellepeddi, V.; Rower, J.; Liu, X.; Kumar, S.; Rashid, J.; Sherwin, C.M.T. State-of-the-Art Review on Physiologically Based Pharmacokinetic Modeling in Pediatric Drug Development. Clin. Pharmacokinet. 2019, 58, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Björkman, S. Reduction and Lumping of Physiologically Based Pharmacokinetic Models: Prediction of the Disposition of Fentanyl and Pethidine in Humans by Successively Simplified Models. J. Pharmacokinet. Pharmacodyn. 2003, 30, 285–307. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, H.; Adeshina, F.; Teeguarden, J.G. Physiologically-based pharmacokinetic model for Fentanyl in support of the development of Provisional Advisory Levels. Toxicol. Appl. Pharmacol. 2013, 273, 464–476. [Google Scholar] [CrossRef]
- Pilari, S.; Gaub, T.; Block, M.; Görlitz, L. Development of physiologically based organ models to evaluate the pharmacokinetics of drugs in the testes and the thyroid gland. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 532–542. [Google Scholar] [CrossRef]
- Leong, R.; Vieira, M.L.T.; Zhao, P.; Mulugeta, Y.; Lee, C.S.; Huang, S.-M.; Burckart, G.J. Regulatory experience with physiologically based pharmacokinetic modeling for pediatric drug trials. Clin. Pharmacol. Ther. 2012, 91, 926–931. [Google Scholar] [CrossRef]
- Barrett, J.S.; Della Casa Alberighi, O.; Läer, S.; Meibohm, B. Physiologically Based Pharmacokinetic (PBPK) Modeling in Children. Clin. Pharmacol. Ther. 2012, 92, 40–49. [Google Scholar] [CrossRef]
- Templeton, I.E.; Jones, N.S.; Musib, L. Pediatric Dose Selection and Utility of PBPK in Determining Dose. AAPS J. 2018, 20, 31. [Google Scholar] [CrossRef]
- Michelet, R.; Bocxlaer, J.V.; Vermeulen, A. PBPK in Preterm and Term Neonates: A Review. Curr. Pharm. Des. 2018, 23, 5943–5954. [Google Scholar] [CrossRef]
- Johnson, T.N.; Rostami-Hodjegan, A. Resurgence in the use of physiologically based pharmacokinetic models in pediatric clinical pharmacology: Parallel shift in incorporating the knowledge of biological elements and increased applicability to drug development and clinical practice. Pediatr. Anesth. 2011, 21, 291–301. [Google Scholar] [CrossRef]
- Maharaj, A.R.; Barrett, J.S.; Edginton, A.N. A workflow example of PBPK modeling to support pediatric research and development: Case study with lorazepam. AAPS J. 2013, 15, 455–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ince, I.; Solodenko, J.; Frechen, S.; Dallmann, A.; Niederalt, C.; Schlender, J.; Burghaus, R.; Lippert, J.; Willmann, S. Predictive Pediatric Modeling and Simulation Using Ontogeny Information. J. Clin. Pharmacol. 2019, 59, S95–S103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovar, L.; Schräpel, C.; Selzer, D.; Kohl, Y.; Bals, R.; Schwab, M.; Lehr, T. Physiologically-Based Pharmacokinetic (PBPK) Modeling of Buprenorphine in Adults, Children and Preterm Neonates. Pharmaceutics 2020, 12, 578. [Google Scholar] [CrossRef] [PubMed]
- Hanke, N.; Kunz, C.; Thiemann, M.; Fricke, H.; Lehr, T. Translational PBPK Modeling of the Protein Therapeutic and CD95L Inhibitor Asunercept to Develop Dose Recommendations for Its First Use in Pediatric Glioblastoma Patients. Pharmaceutics 2019, 11, 152. [Google Scholar] [CrossRef] [Green Version]
- Moj, D.; Britz, H.; Burhenne, J.; Stewart, C.F.; Egerer, G.; Haefeli, W.E.; Lehr, T. A physiologically based pharmacokinetic and pharmacodynamic (PBPK/PD) model of the histone deacetylase (HDAC) inhibitor vorinostat for pediatric and adult patients and its application for dose specification. Cancer Chemother. Pharmacol. 2017, 80, 1013–1026. [Google Scholar] [CrossRef]
- Wojtyniak, J.-G.; Britz, H.; Selzer, D.; Schwab, M.; Lehr, T. Data Digitizing: Accurate and Precise Data Extraction for Quantitative Systems Pharmacology and Physiologically-Based Pharmacokinetic Modeling. CPT Pharmacomet. Syst. Pharmacol. Pharmacol. 2020. accepted for publication. [Google Scholar] [CrossRef]
- Feierman, D.E.; Lasker, J.M. Metabolism of fentanyl, a synthetic opioid analgesic, by human liver microsomes. Role of CYP3A4. Drug Metab. Dispos. 1996, 24, 932–939. [Google Scholar]
- Yu, C.; Yuan, M.; Yang, H.; Zhuang, X.; Li, H. P-glycoprotein on blood-brain barrier plays a vital role in fentanyl brain exposure and respiratory toxicity in rats. Toxicol. Sci. 2018, 164, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Lötsch, J.; Walter, C.; Parnham, M.J.; Oertel, B.G.; Geisslinger, G. Pharmacokinetics of non-intravenous formulations of fentanyl. Clin. Pharmacokinet. 2013, 52, 23–36. [Google Scholar] [CrossRef]
- Andrew Williams, J.; Ring, B.J.; Cantrell, V.E.; Jones, D.R.; Eckstein, J.; Ruterbories, K.; Hamman, M.A.; Hall, S.D.; Wrighton, S.A. Comparative metabolic capabilities of CYP3A4, CYP3A5, and CYP3A7. Drug Metab. Dispos. 2002, 30, 883–891. [Google Scholar] [CrossRef]
- Open Systems Pharmacology Suite Community Open Systems Pharmacology Suite Manual, Version 7.4. Available online: https://github.com/Open-Systems-Pharmacology/OSPSuite.Documentation/blob/master/Open Systems Pharmacology Suite.pdf (accessed on 25 March 2020).
- PK-Sim® Ontogeny Database, Version 7.3. Available online: https://github.com/Open-Systems-Pharmacology/OSPSuite.Documentation/blob/master/PK-Sim (accessed on 25 March 2020).
- Prasad, B.; Gaedigk, A.; Vrana, M.; Gaedigk, R.; Leeder, J.; Salphati, L.; Chu, X.; Xiao, G.; Hop, C.; Evers, R.; et al. Ontogeny of Hepatic Drug Transporters as Quantified by LC-MS/MS Proteomics. Clin. Pharmacol. Ther. 2016, 100, 362–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNamara, P.J.; Alcorn, J. Protein binding predictions in infants. AAPS PharmSci 2002, 4, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.S.; Stiller, R.L.; Davis, P.J.; Fedel, G.; Chakravorti, S.; Israel, B.A.; McGowan, F.X. Fentanyl and Alfentanil Plasma Protein Binding in Preterm and Term Neonates. Anesth. Analg. 1997, 84, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Gauntlett, I.S.; Fisher, D.M.; Hertzka, R.E.; Kuhis, E.; Spellman, M.J.; Rudolph, C. Pharmacokinetics of Fentanyl in Neonatal Humans and Lambs. Anesthesiology 1988, 69, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Koehntop, D.E.; Rodman, J.H.; Brundage, D.M.; Hegland, M.G.; Buckley, J.J. Pharmacokinetics of fentanyl in neonates. Anesth. Analg. 1986, 65, 227–232. [Google Scholar] [CrossRef]
- Saarenmaa, E.; Neuvonen, P.J.; Fellman, V. Gestational age and birth weight effects on plasma clearance of fentanyl in newborn infants. J. Pediatr. 2000, 136, 767–770. [Google Scholar] [CrossRef]
- Li, X.; Frechen, S.; Moj, D.; Lehr, T.; Taubert, M.; Hsin, C.-h.; Mikus, G.; Neuvonen, P.J.; Olkkola, K.T.; Saari, T.I.; et al. A Physiologically Based Pharmacokinetic Model of Voriconazole Integrating Time-Dependent Inhibition of CYP3A4, Genetic Polymorphisms of CYP2C19 and Predictions of Drug–Drug Interactions. Clin. Pharmacokinet. 2020, 59, 781–808. [Google Scholar] [CrossRef]
- McClain, D.A.; Hug, C.C. Intravenous fentanyl kinetics. Clin. Pharmacol. Ther. 1980, 28, 106–114. [Google Scholar] [CrossRef]
- Bovill, J.G.; Sebel, P.S. Pharmacokinetics of high-dose fentanyl. Br. J. Anaesth. 1980, 52, 795–801. [Google Scholar] [CrossRef]
- Bentley, J.B.; Borel, J.D.; Nenad, R.E.; Gillespie, T.J. Age and Fentanyl Pharmacokinetics. Anesth. Analg. 1982, 61, 968–971. [Google Scholar] [CrossRef]
- Christrup, L.; Foster, D.; Popper, L.; Troen, T.; Upton, R. Pharmacokinetics, efficacy, and tolerability of fentanyl following intranasal versus intravenous administration in adults undergoing third-molar extraction: A randomized, double-blind, double-dummy, two-way, crossover study. Clin. Ther. 2008, 30, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Duthie, D.J.R.; McLaren, A.D.; Nimmo, W.S. Pharmacokinetics of fentanyl during constant rate i.v. infusion for the relief of pain after surgery. Br. J. Anaesth. 1986, 58, 950–956. [Google Scholar] [CrossRef]
- Gourlay, G.K.; Murphy, T.M.; Plummer, J.L.; Kowalski, S.R.; Cherry, D.A.; Cousins, M.J. Pharmacokinetics of fentanyl in lumbar and cervical CSF following lumbar epidural and intravenous administration. Pain 1989, 38, 253–259. [Google Scholar] [CrossRef]
- Gupta, S.K.; Southam, M.A.; Hwang, S.S. Evaluation of Diurnal Variation in Fentanyl Clearance. J. Clin. Pharmacol. 1995, 35, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Holley, F.O.; Van Steennis, C. Postoperative analgesia with fentanyl: Pharmacokinetics and pharmacodynamics of constant-rate I.V. and transdermal delivery. Br. J. Anaesth. 1988, 60, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.B.S.; Schug, S.A.; Sunderland, V.B.; Paech, M.J.; Liu, Y. A Phase I Pharmacokinetic and Bioavailability Study of a Sublingual Fentanyl Wafer in Healthy Volunteers. Anesth. Analg. 2012, 115, 1. [Google Scholar] [CrossRef]
- MacLeod, D.B.; Habib, A.S.; Ikeda, K.; Spyker, D.A.; Cassella, J.V.; Ho, K.Y.; Gan, T.J. Inhaled Fentanyl Aerosol in Healthy Volunteers. Anesth. Analg. 2012, 115, 1071–1077. [Google Scholar] [CrossRef]
- Singleton, M.A.; Rosen, J.I.; Fisher, D.M. Plasma concentrations of fentanyl in infants, children and adults. Can. J. Anaesth. 1987, 34, 152–155. [Google Scholar] [CrossRef]
- Stoeckel, H.; Schüttler, J.; Magnussen, H.; Hengstmann, J.H. Plasma fentanyl concentrations and the occurrence of respiratory depression in volunteers. Br. J. Anaesth. 1982, 54, 1087–1095. [Google Scholar] [CrossRef]
- Streisand, J.B.; Varvel, J.R.; Stanski, D.R.; Le Maire, L.; Ashburn, M.A.; Hague, B.I.; Tarver, S.D.; Stanley, T.H. Absorption and Bioavailability of Oral Transmucosal Fentanyl Citrate. Anesthesiology 1991, 75, 223–229. [Google Scholar] [CrossRef]
- Varvel, J.R.; Shafer, S.L.; Hwang, S.S.; Coen, P.A.; Stanski, D.R. Absorption Characteristics of Transdermally Administered Fentanyl. Anesthesiology 1989, 70, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.; Koren, G.; Crean, P.; Klein, J.; Roy, W.L.; MacLeod, S.M. Fentanyl pharmacokinetics and hemodynamic effects in preterm infants during ligation of patent ductus arteriosus. Anesth. Analg. 1985, 64, 1078–1080. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Magee, P. Percutaneous absorption: Critical factors in transdermal transport. In Dermatotoxicology; Marzulli, F., Maibach, H., Eds.; Hemisphere Publishing Corporation: New York, NY, USA, 1991; pp. 1–35. [Google Scholar]
- Bower, S. Plasma protein binding of fentanyl. J. Pharm. Pharmacol. 1981, 33, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Bista, S.R.; Haywood, A.; Hardy, J.; Lobb, M.; Tapuni, A.; Norris, R. Protein binding of fentanyl and its metabolite nor-fentanyl in human plasma, albumin and α-1 acid glycoprotein. Xenobiotica 2015, 45, 207–212. [Google Scholar] [CrossRef]
- Jantos, R.; Schuhmacher, M.; Veldstra, J.L.; Bosker, W.M.; Klöpping-Ketelaars, I.; Touliou, K.; Sardi, G.M.; Brookhuis, K.A.; Ramaekers, J.G.; Mattern, R.; et al. Determination of blood/serum ratios of different forensically relevant analytes in authentic samples. Arch. Kriminol. 2011, 227, 188–203. [Google Scholar]
- Rodgers, T.; Rowland, M. Physiologically-based Pharmacokinetic Modeling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006, 95, 1238–1257. [Google Scholar] [CrossRef]
- Rodgers, T.; Rowland, M. Mechanistic approaches to volume of distribution predictions: Understanding the processes. Pharm. Res. 2007, 24, 918–933. [Google Scholar] [CrossRef]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically based pharmacokinetic modeling 1: Predicting the tissue distribution of moderate-to-strong bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef]
- Schmitt, W. General approach for the calculation of tissue to plasma partition coefficients. Toxicol. In Vitro 2008, 22, 457–467. [Google Scholar] [CrossRef]
- Fachinformation Fentanyl HEXAL® Injektionslösung. Available online: https://www.hexal.biz/praeparate/dokumente/fi/fentanyl_hx_inj_spc-1575534009.pdf (accessed on 19 August 2020).
- Rosaeg, O.P.; Kitts, J.B.; Koren, G.; Byford, L.J. Maternal and fetal effects of intravenous patient-controlled fentanyl analgesia during labour in a thrombocytopenic parturient. Can. J. Anaesth. 1992, 39, 277–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olkkola, K.T.; Palkama, V.J.; Neuvonen, P.J. Ritonavir’s role in reducing fentanyl clearance and prolonging its half- life. Anesthesiology 1999, 91, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Diebel, L.N.; Wilson, R.F.; Dulchavsky, S.A.; Saxe, J. Effect of increased intra-abdominal pressure on hepatic arterial, portal venous, and hepatic microcirculatory blood flow. J. Trauma 1992, 33, 279–282; discussion 282–283. [Google Scholar] [CrossRef] [PubMed]
- Thomson, M.M.S.; Hines, R.N.; Schuetz, E.G.; Meibohm, B. Expression patterns of organic anion transporting polypeptides 1B1 and 1B3 protein in human pediatric liver. Drug Metab. Dispos. 2016, 44, 999–1004. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.; Li, Q.; Gaedigk, R.; Bi, C.; De Wildt, S.N.; Leeder, J.S.; Fridley, B.L. Ontogeny related changes in the pediatric liver metabolome. Front. Pediatr. 2020, in press. [Google Scholar] [CrossRef]
- Hadley, G.; Derry, S.; Moore, R.A.; Wiffen, P.J. Transdermal fentanyl for cancer pain. Cochrane Database Syst. Rev. 2013, 28, 264–265. [Google Scholar] [CrossRef]
- DURAGESIC® (Fentanyl Transdermal System) Full Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2005/19813s039lbl.pdf (accessed on 14 August 2020).
- Kovar, L.; Selzer, D.; Britz, H.; Benowitz, N.; St. Helen, G.; Kohl, Y.; Bals, R.; Lehr, T. Comprehensive Parent–Metabolite PBPK/PD Modeling Insights into Nicotine Replacement Therapy Strategies. Clin. Pharmacokinet. 2020. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
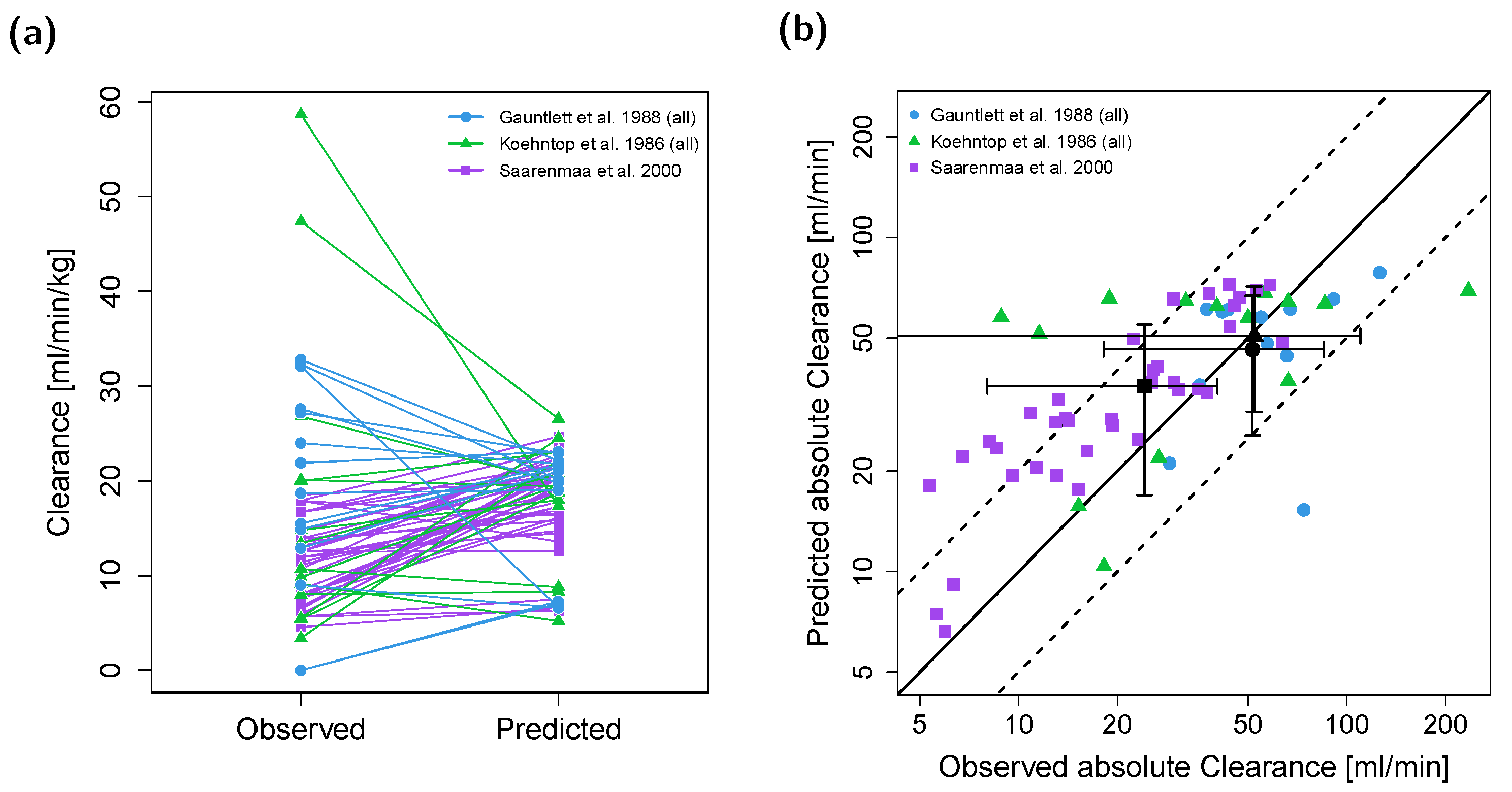{kind=link}
{kind=link}
| Clinical Study ID | Dose a [µg/kg] | Dose b [µg/h] | Administration | n | Female [%] | Age [Years] | Weight [kg] | Blood Sample | Surgery | Dataset | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bentley et al., 1982 (Adult) | 10.0 | iv (bolus) | 5 | 100 | 36 ± 4 | 64 ± 3 | arterial | yes | e | [42] | |
| Bentley et al., 1982 (Eldery) | 10.0 | iv (bolus) | 4 | 100 | 67 ± 2 | 68 ± 7 | arterial | yes | e | [42] | |
| Bovill and Sebel 1980 | 60.0 | iv (2 min) | 5 | 40 | 57 (45–65) | 71 (53–87) | venous | yes | e | [41] | |
| Christrup et al., 2008 | 1.5 | iv (-) | 7 | 43 | 24 (22–28) | 68 (52–82) | venous | yes | i | [43] | |
| Duthie et al., 1986 (1) | 1.4 | 100.0 | iv (24 h + bolus) | 10 | - | 61 ± 8 | 69 ± 12 | venous | yes | e | [44] |
| Duthie et al., 1986 (2) | 1.5 | 100.0 | iv (24 h + bolus) | 13 | - | 49 ± 14 | 65 ± 14 | venous | yes | e | [44] |
| Duthie et al., 1986 (3) | 1.4 | 100.0 | iv (24 h + bolus) | 10 | - | 58 ± 11 | 69 ± 11 | venous | yes | e | [44] |
| Duthie et al., 1986 (4) | 7.2 | 100.0 | iv (26 h + bolus) | 12 | - | 55 ± 12 | 69 ± 9 | venous | yes | i | [44] |
| Gourlay et al., 1989 | 1.0 | iv (1 min) | 6 | - | - | 70 (40–85) | venous c | no | e | [45] | |
| Gupta et al., 1995 | 50.0 | iv (48 h) | 6 | 0 | - | - | venous d | no | i | [46] | |
| Holley and van Steennis 1988 (1) | 1.3 | 25.0 | iv (loading dose + 24 h) | 10 | 0 | 54 ± 12 | 76 ± 12 | arterial | yes | i | [47] |
| Holley and van Steennis 1988 (2) | 2.5 | 50.0 | iv (loading dose + 24 h) | 10 | 0 | 44 ± 15 | 81 ± 16 | arterial | yes | e | [47] |
| Holley and van Steennis 1988 (3) | 5.0 | 100.0 | iv (loading dose + 24 h) | 10 | 0 | 56 ± 12 | 80 ± 17 | arterial | yes | e | [47] |
| Holley and van Steennis 1988 (4) | 6.5 | 125.0 | iv (loading dose + 24 h) | 9 | 0 | 54 ± 12 | 77 ± 5 | arterial | yes | i | [47] |
| Lim et al., 2012 | 1.5 | iv (5 min) | 22 | 58 | 23 (19–32) | 67 (51–101) | venous | no | e | [48] | |
| MacLeod et al., 2012 | 0.3 | iv (5 sec) | 10 | 51 | 25 (18–55) | 73 ± 13 | arterial | no | i | [49] | |
| McClain and Hug 1980 | 6.4 | iv (1.5 min) | 5 | 0 | - (22–29) | 75 (65–85) | arterial | no | i | [40] | |
| Saari et al., 2008 e | 5.0 | iv (2 min) | 12 | 42 | - | - | venous | no | e | [6] | |
| Saari et al., 2008 (DDI) e,f | 5.0 | iv (2 min) | 12 | 42 | - | - | venous | no | e | [6] | |
| Singleton et al., 1987 (1) | 20.7 | iv (2 min) | 7 | - | 33 (18–41) | - | arterial | yes | e | [50] | |
| Stoeckel et al., 1982 | 7.6 | iv (bolus) | 3 | 33 | 22 (20–26) | 66 (59–77) | venous | no | e | [51] | |
| Streisand et al., 1991 | 15.0 | iv (8 min) | 10 | 0 | 27 (23–31) | 76 (68–85) | arterial | no | e | [52] | |
| Varvel et al., 1989 | 11.4 | iv (5 min) | 8 | 63 | 45 (33–57) | 68 (52–100) | arterial/venous | yes | e | [53] | |
| Ziesenitz et al., 2015 e | 5.0 | iv (10 min) | 16 | 25 | 33 (22–49) | 73 (61–85) | venous | no | i | [5] |
| Clinical Study ID | Dose a [µg/kg] | Dose b [µg/kg/h] | Administration | n | Female [%] | Chronological Age | Gestational Age | Weight [kg] | Blood Sample | Surgery | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Collins et al., 1985 | 30.0 | iv (1 min) | 9 | 22 | - | 32 (23–38) | 1.1 (0.7–1.6) | arterial | yes | [54] | |
| Gauntlett et al., 1988 (1) | 52.5 | iv (2 min) | 1 | - | 1 day | 38 | 2.8 | arterial | yes | [36] | |
| Gauntlett et al., 1988 (2) | 56.5 | iv (2 min) | 1 | - | 3 days | 40 | 2.5 | arterial | yes | [36] | |
| Gauntlett et al., 1988 (all) c | 54.1 ± 2.3 | iv (2 min) | 14 | - | 18.0 (1–71) days | 38 (32–40) | 2.7 (1.9–3.9) | arterial | yes | [36] | |
| Koehntop et al., 1986 (1) | 25.0 | iv (1–3 min) | 1 | - | 2 days | - | 2.0 | arterial | yes | [37] | |
| Koehntop et al., 1986 (2) | 50.0 | iv (1–3 min) | 1 | - | 2 days | - | 3.5 | arterial | yes | [37] | |
| Koehntop et al., 1986 (all) c | 10.0–50.0 | iv (1–3 min) | 14 | - | 3.0 (0.5–14) days | - | 2.9 (1.9–3.8) | arterial | yes | [37] | |
| Saarenmaa et al., 2000 c | 10.5 | 1.5 | iv (1 h + 58 h) | 38 | 26 | 0.4 (0–2) days | 32 (26–42) | 1.8 (0.9–3.6) d | arterial | yes | [38] |
| Singleton et al., 1987 (2) | 31.2 | iv (2 min) | 7 | - | 6.5 (3–10) months | - | - | arterial | yes | [50] | |
| Singleton et al., 1987 (3) | 30.8 | iv (2 min) | 7 | - | 2.7 (1–9) years | - | - | venous e | yes | [50] |
| Parameter | Fentanyl | Norfentanyl | Description | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Value | Unit | Source | Literature | Reference | Value | Unit | Source | Literature | Reference | ||
| MW | 336.5 | g/mol | lit. | 336.5 | [55] a | 232.3 | g/mol | lit. | 232.3 | [55] b | Molecular weight |
| pKa (base) | 8.99 | lit. | 8.99 | [55] a | 10.03 | lit. | 10.03 | [55] b | Acid dissociation constant | ||
| logP | 3.49 | lit. | 3.49 | [56] | 2.00 | lit. | 2.00 | [55] b | Lipophilicity | ||
| fu (adults) | 20.8 | % | lit. | 20.8 | [57] | 81.9 | % | lit. | 81.9 | [58] | Unbound fraction |
| fu (pediatrics) | 29.0–33.0 | % | calc. | [34] | Unbound fraction | ||||||
| CYP3A4 Km → norfen | 117 | µmol/L | lit. | 117 | [27] | Michaelis–Menten constant | |||||
| CYP3A4 kcat → norfen | 20.6 | 1/min | optim. | - | - | Catalytic rate constant | |||||
| CYP3A7 Km → norfen | 596 | µmol/L | calc.c | - | - | Michaelis–Menten constant | |||||
| CYP3A7 kcat → norfen | 5.22 | 1/min | calc.c | - | - | Catalytic rate constant | |||||
| Unspecific hepatic clearance → undef | 1.46 | 1/min | lit. | - | - | Elimination from plasma (first-order process in the liver) | |||||
| P-gp Km | 5.72 | µmol/L | optim. | - | - | Michaelis–Menten constant | |||||
| P-gp kcat | 1.71 | 1/min | optim. | - | - | Transport rate constant | |||||
| B/P ratio | 0.87 | lit. | 0.87 | [59] | 1.26 | calc. | - | - | Blood-to-plasma ratio | ||
| GFR fraction | 1.00 | asm. | - | - | 4.30 | optim. | - | - | Filtered drug in the urine | ||
| Partition coefficients | Diverse d | calc. | R&R | [60,61,62] | Diverse d | calc. | Schmitt | [63] | Cell to plasma partitioning | ||
| Cellular permeability | Diverse d | cm/min | calc. | Ch.-dep. Schmitt | [31] | 1.80 × 10−2 | cm/min | calc. | PK-Sim | [31] | Perm. into the cellular space |
| CYP3A4 KI of voriconazole | 9.33 | µmol/L | lit. | 9.33 | [39] | The inhibitor concentration when reaching half of kinact | |||||
| CYP3A4 kinact of voriconazole | 0.015 | 1/min | lit. | 0.015 | [39] | The maximum inactivation rate constant | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovar, L.; Weber, A.; Zemlin, M.; Kohl, Y.; Bals, R.; Meibohm, B.; Selzer, D.; Lehr, T. Physiologically-Based Pharmacokinetic (PBPK) Modeling Providing Insights into Fentanyl Pharmacokinetics in Adults and Pediatric Patients. Pharmaceutics 2020, 12, 908. https://doi.org/10.3390/pharmaceutics12100908
Kovar L, Weber A, Zemlin M, Kohl Y, Bals R, Meibohm B, Selzer D, Lehr T. Physiologically-Based Pharmacokinetic (PBPK) Modeling Providing Insights into Fentanyl Pharmacokinetics in Adults and Pediatric Patients. Pharmaceutics. 2020; 12(10):908. https://doi.org/10.3390/pharmaceutics12100908
Chicago/Turabian StyleKovar, Lukas, Andreas Weber, Michael Zemlin, Yvonne Kohl, Robert Bals, Bernd Meibohm, Dominik Selzer, and Thorsten Lehr. 2020. "Physiologically-Based Pharmacokinetic (PBPK) Modeling Providing Insights into Fentanyl Pharmacokinetics in Adults and Pediatric Patients" Pharmaceutics 12, no. 10: 908. https://doi.org/10.3390/pharmaceutics12100908
APA StyleKovar, L., Weber, A., Zemlin, M., Kohl, Y., Bals, R., Meibohm, B., Selzer, D., & Lehr, T. (2020). Physiologically-Based Pharmacokinetic (PBPK) Modeling Providing Insights into Fentanyl Pharmacokinetics in Adults and Pediatric Patients. Pharmaceutics, 12(10), 908. https://doi.org/10.3390/pharmaceutics12100908