Dual-Targeted Extracellular Vesicles to Facilitate Combined Therapies for Neuroendocrine Cancer Treatment



Abstract
: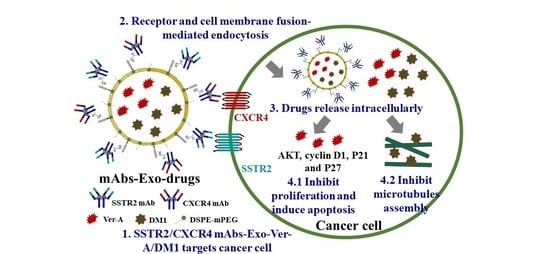
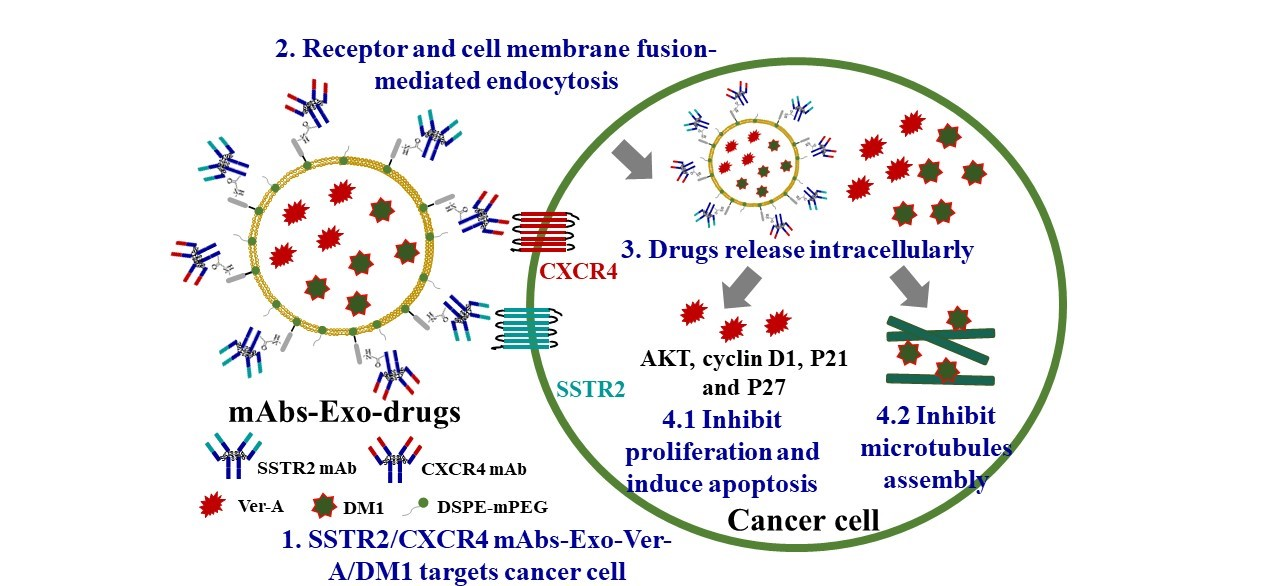{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Seed Cultures and Media
2.2. Construction of mAbs-EV-Ver-A/DM1
2.3. Packing Ver-A, DM1 and Cyanine 7 (Cy7) in mAbs-EV
2.4. Flow Cytometry
2.5. Confocal Imaging
2.6. Western Blotting
2.7. In Vivo Imaging System (IVIS) Imaging
2.8. In Vitro Anticancer Cytotoxicity
2.9. Maximum Tolerated Dose (MTD)
2.10. In Vivo Anticancer Efficacy
2.11. Hematoxylin and Eosin (H&E) Staining
2.12. Statistical Analysis
3. Results and Discussion
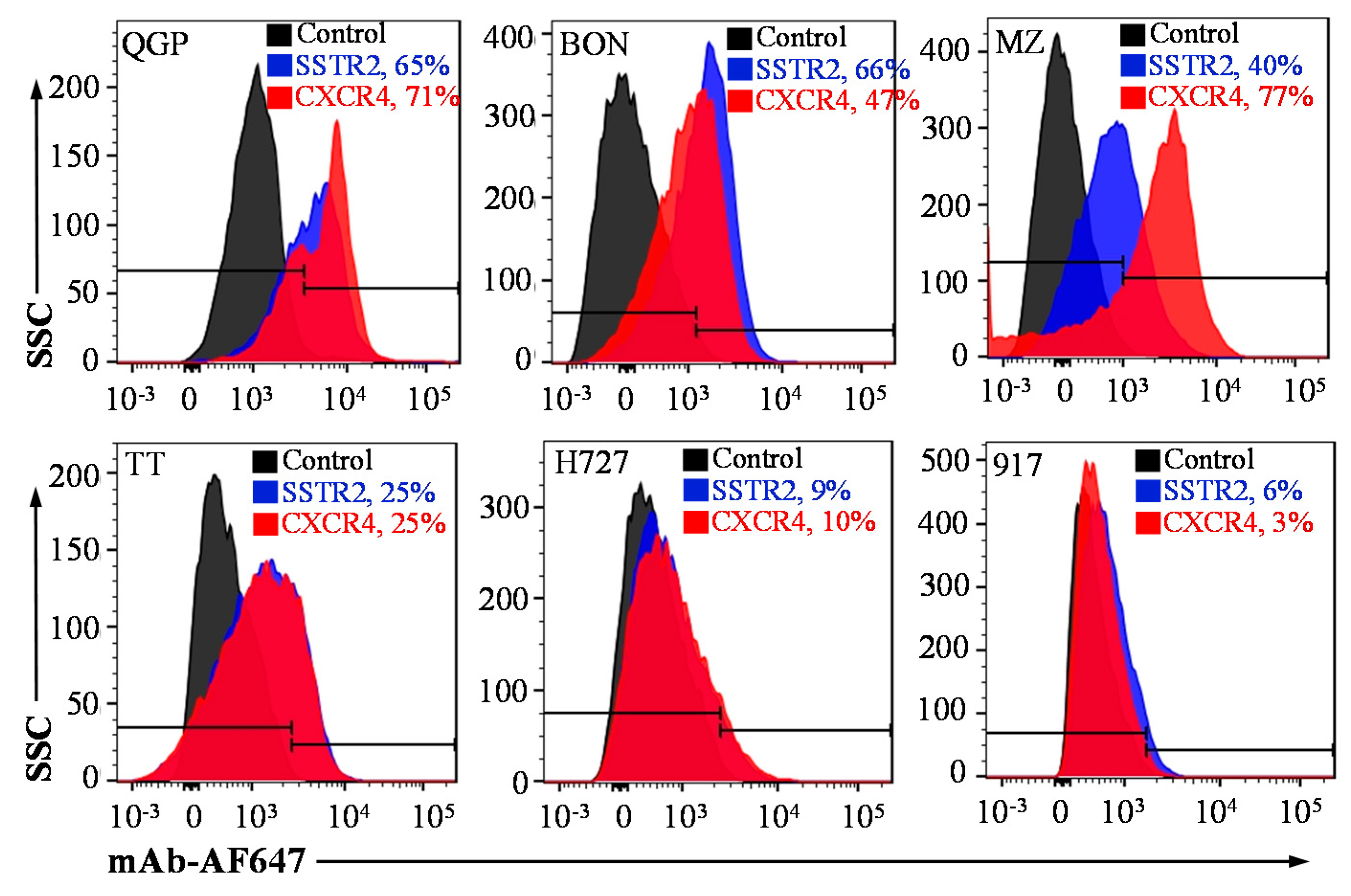
3.1. SSTR2 and CXCR4 Surface Expression and Dual Targeting
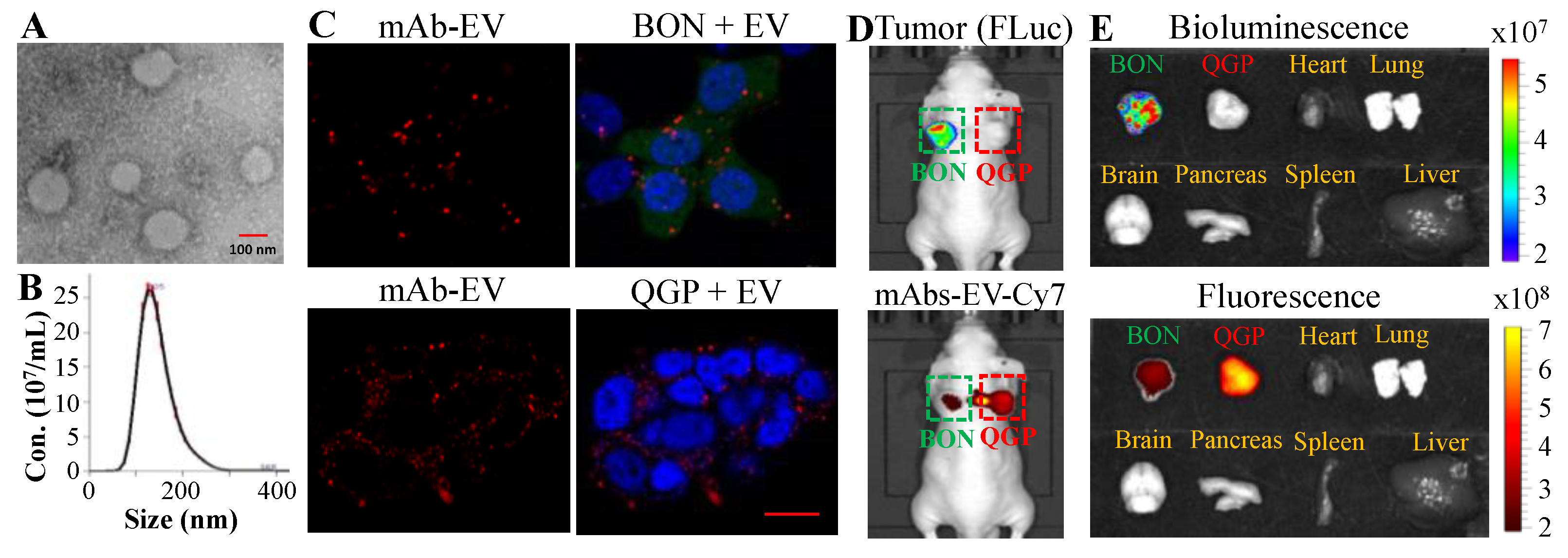
3.2. NET-Specific Targeting and Uptake of Dual-Targeted mAbs-EV
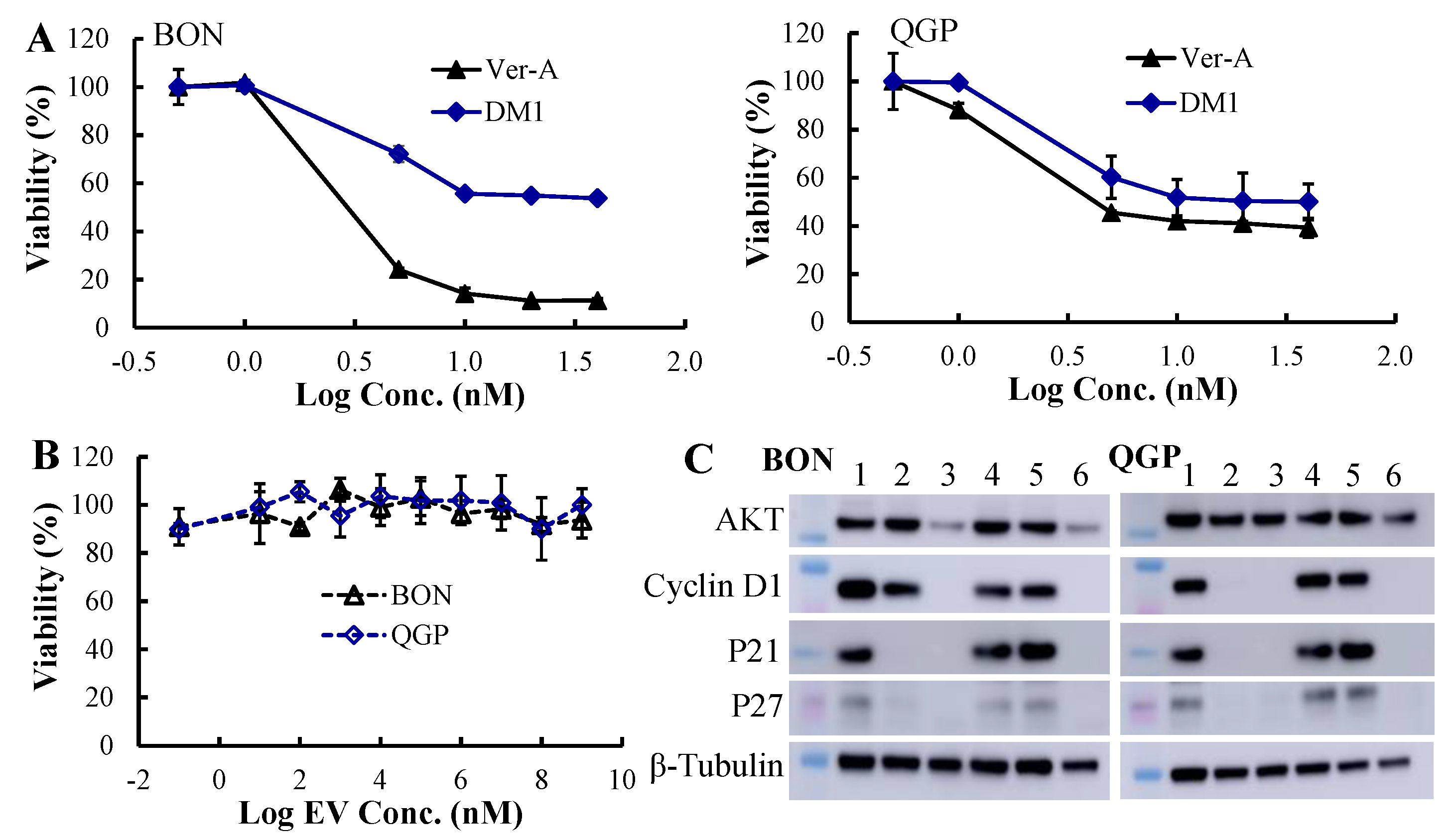
3.3. In Vitro Anticancer Cytotoxicity and Synergistic Mechanisms
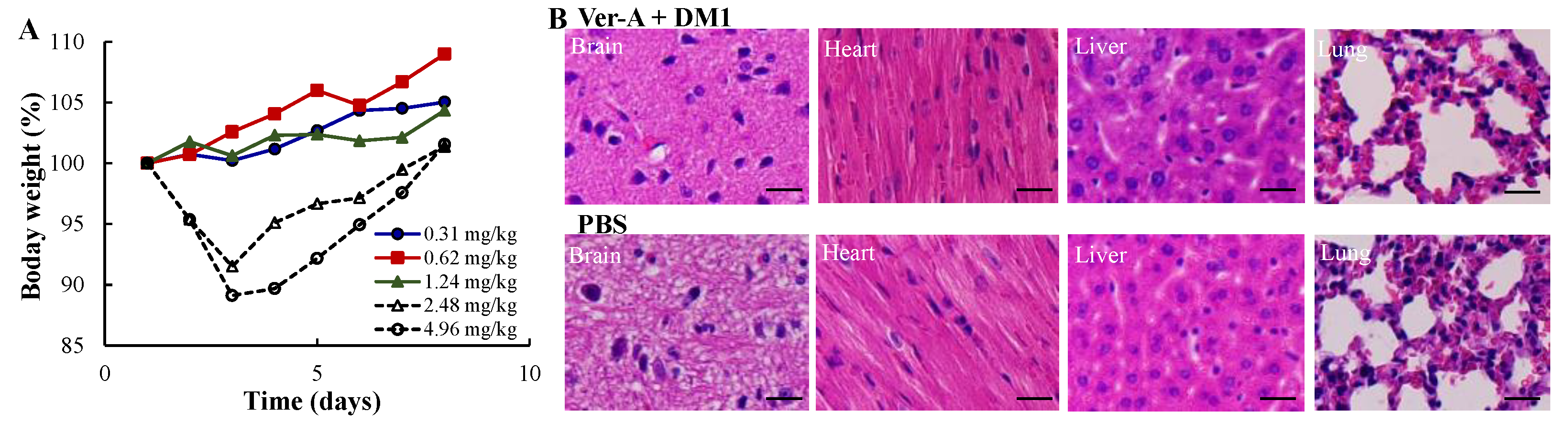
3.4. Maximum Tolerated Dosage (MTD) and Minimal Toxicity
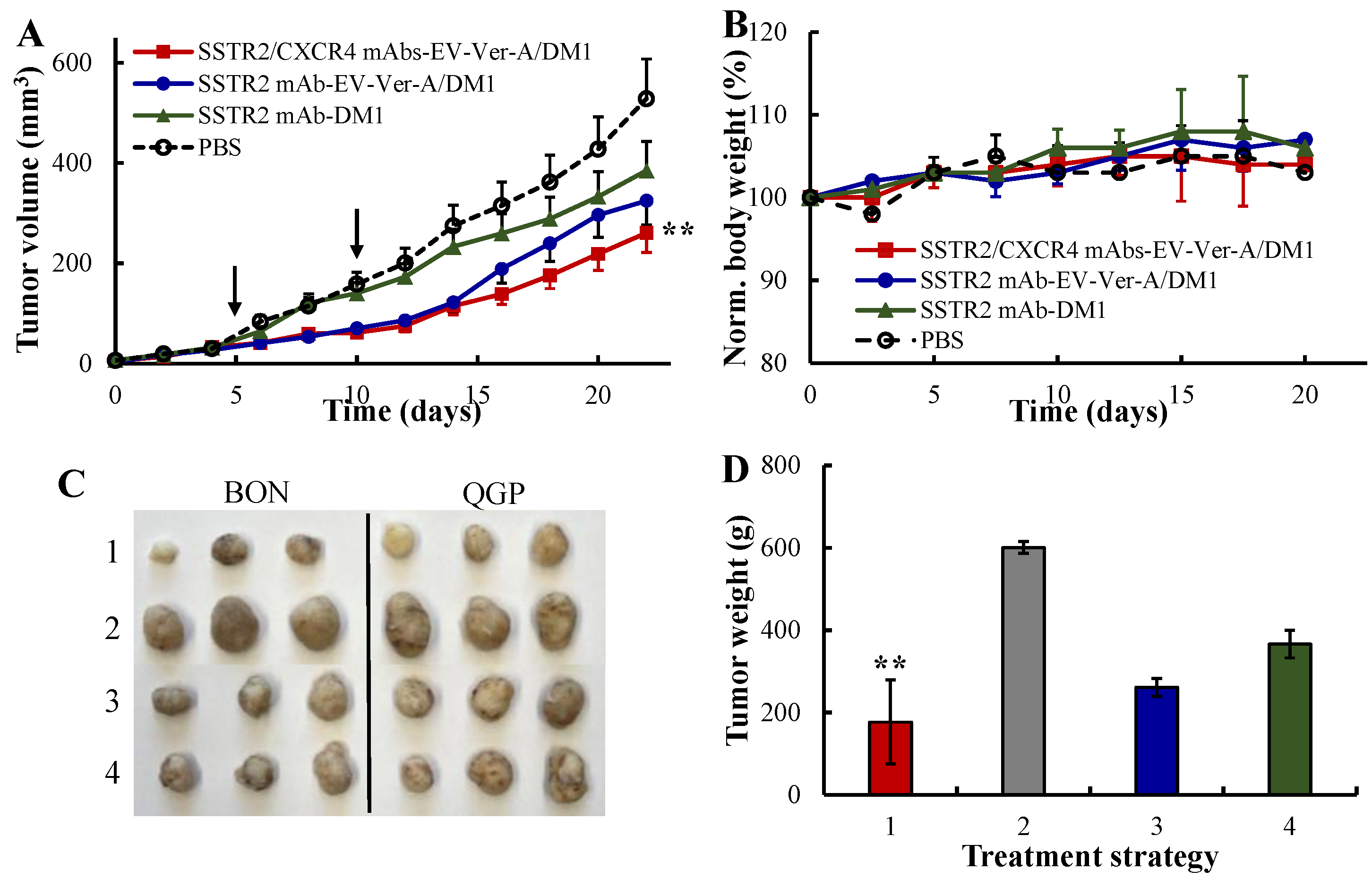
3.5. In Vivo Anti-NET Efficacy
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shiba, S.; Morizane, C.; Hiraoka, N.; Sasaki, M.; Koga, F.; Sakamoto, Y.; Kondo, S.; Ueno, H.; Ikeda, M.; Yamada, T.; et al. Pancreatic Neuroendocrine Tumors: A Single-Center 20-Year Experience with 100 Patients. Pancreatology 2016, 16, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Jaskula-Sztul, R.; Javadi, A.; Xu, W.; Eide, J.; Dammalapati, A.; Kunnimalaiyaan, M.; Chen, H.; Gong, S. Co-Delivery of Doxorubicin and SiRNA Using Octreotide-Conjugated Gold Nanorods for Targeted Neuroendocrine Cancer Therapy. Nanoscale 2012, 4, 7185–7193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Burke, J.F.; Pilla, S.; Chen, H.; Jaskula-Sztul, R.; Gong, S. Octreotide-Functionalized and Resveratrol-Loaded Unimolecular Micelles for Targeted Neuroendocrine Cancer Therapy. Nanoscale 2013, 5, 9924–9933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaskula-Sztul, R.; Xu, W.; Chen, G.; Harrison, A.; Dammalapati, A.; Nair, R.; Cheng, Y.; Gong, S.; Chen, H. Thailandepsin A-Loaded and Octreotide-Functionalized Unimolecular Micelles for Targeted Neuroendocrine Cancer Therapy. Biomaterials 2016, 91, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Jaskula-Sztul, R.; Harrison, A.; Dammalapati, A.; Xu, W.; Cheng, Y.; Chen, H.; Gong, S. KE108-Conjugated Unimolecular Micelles Loaded with a Novel HDAC Inhibitor Thailandepsin-A for Targeted Neuroendocrine Cancer Therapy. Biomaterials 2016, 97, 22–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.T.; Koh, B.Y.; Brody, L.A.; Getrajdman, G.I.; Susman, J.; Fong, Y.; Blumgart, L.H. Particle Embolization of Hepatic Neuroendocrine Metastases for Control of Pain and Hormonal Symptoms. J. Vasc. Interv. Radiol. 1999, 10, 397–403. [Google Scholar] [CrossRef]
- Isozaki, T.; Kiba, T.; Numata, K.; Saito, S.; Shimamura, T.; Kitamura, T.; Morita, K.; Tanaka, K.; Sekihara, H. Medullary Thyroid Carcinoma with Multiple Hepatic Metastases: Treatment with Transcatheter Arterial Embolization and Percutaneous Ethanol Injection. Intern. Med. 1999, 38, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, B.; Kloeppel, G.; Krenning, E.; Ahlman, H.; Ploeckinger, U.; Wiedenmann, B.; Arnold, R.; Auernhammer, C.J.; Koerner, M.; Rindi, G.; et al. Consensus Guidelines for the Management of Patients with Digestive Neuroendocrine Tumors–Well-Differentiated Jejunal-Ileal Tumor/Carcinoma. Neuroendocrinology 2007, 87, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Lal, A.; Chen, H. Treatment of Advanced Carcinoid Tumors. Curr. Opin. Oncol. 2006, 18, 9–15. [Google Scholar] [CrossRef]
- Lehnert, T. Liver Transplantation for Metastatic Neuroendocrine Carcinoma. Transplantation 1998, 66, 1307–1312. [Google Scholar] [CrossRef]
- Zhang, R.; Straus, F.H.; DeGroot, L.J. Effective Genetic Therapy of Established Medullary Thyroid Carcinomas with Murine Interleukin-2: Dissemination and Cytotoxicity Studies in a Rat Tumor Model. Endocrinology 1999, 140, 2152–2158. [Google Scholar] [CrossRef] [PubMed]
- Boudreaux, J.P.; Putty, B.; Frey, D.J.; Woltering, E.; Anthony, L.; Daly, I.; Ramcharan, T.; Lopera, J.; Castaneda, W. Surgical Treatment of Advanced-Stage Carcinoid Tumors. Ann. Surg. 2005, 241, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.; Faraggi, M.; Giraudet, A.-L.; De Labriolle-Vaylet, C.; Aparicio, T.; Rouzet, F.; Mignon, M.; Askienazy, S.; Sobhani, I. Long-Term Efficacy of Radionuclide Therapy in Patients with Disseminated Neuroendocrine Tumors Uncontrolled by Conventional Therapy. J. Nucl. Med. 2004, 45, 1660–1668. [Google Scholar] [PubMed]
- Fiorentini, G.; Rossi, S.; Bonechi, F.; Vaira, M.; De Simone, M.; Dentico, P.; Bernardeschi, P.; Cantore, M.; Guadagni, S. Intra-Arterial Hepatic Chemoembolization in Liver Metastases from Neuroendocrine Tumors: A Phase II Study. J. Chemother. 2004, 16, 293–297. [Google Scholar] [CrossRef]
- Zuetenhorst, J.M.; Olmos, R.A.V.; Muller, M.; Hoefnagel, C.A.; Taal, B.G. Interferon and Meta-Iodobenzylguanidin Combinations in the Treatment of Metastatic Carcinoid Tumours. Endocr. Relat. Cancer 2004, 11, 553–561. [Google Scholar] [CrossRef]
- Öberg, K.; Kvols, L.; Caplin, M.; Fave, G.D.; De Herder, W.; Rindi, G.; Ruszniewski, P.; Woltering, E.A.; Wiedenmann, B. Consensus Report on the Use of Somatostatin Analogs for the Management of Neuroendocrine Tumors of the Gastroenteropancreatic System. Ann. Oncol. 2004, 15, 966–973. [Google Scholar] [CrossRef]
- Hennrich, U.; Kopka, K. Lutathera®: The First FDA- and EMA-Approved Radiopharmaceutical for Peptide Receptor Radionuclide Therapy. Pharmaceuticals 2019, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Bobrie, A.; Colombo, M.; Raposo, G.; Théry, C. Exosome Secretion: Molecular Mechanisms and Roles in Immune Responses. Traffic 2011, 12, 1659–1668. [Google Scholar] [CrossRef]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, Biogenesis and Function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the Mouse Brain by Systemic Injection of Targeted Exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- György, B.; Fitzpatrick, Z.; Crommentuijn, M.H.; Mu, D.; Maguire, C.A. Naturally Enveloped AAV Vectors for Shielding Neutralizing Antibodies and Robust Gene Delivery In Vivo. Biomaterials 2014, 35, 7598–7609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A Doxorubicin Delivery Platform Using Engineered Natural Membrane Vesicle Exosomes for Targeted Tumor Therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, K.B.; Gudbergsson, J.M.; Skov, M.N.; Pilgaard, L.; Moos, T.; Duroux, M. A Comprehensive Overview of Exosomes as Drug Delivery Vehicles—Endogenous Nanocarriers for Targeted Cancer Therapy. Biochim. Biophys. Acta 2014, 1846, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Kim, S.; Zhang, E.; Tang, Y.; Jaskula-Sztul, R.; Markert, J.M.; Chen, H.; Zhou, L.; Liu, X. Targeted Exosomes for Drug Delivery: Biomanufacturing, Surface Tagging, and Validation. Biotechnol. J. 2020, 15, e1900163. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Fani, M.; Nicolas, G.P.; Wild, D. Somatostatin Receptor Antagonists for Imaging and Therapy. J. Nucl. Med. 2017, 58, 61S–66S. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, H. Somatostatin Receptor Based Imaging and Radionuclide Therapy. BioMed Res. Int. 2015, 2015, 1–14. [Google Scholar] [CrossRef]
- Menda, Y.; O’Dorisio, T.M.; Howe, J.R.; Schultz, M.; Dillon, J.S.; Dick, D.; Watkins, G.L.; Ginader, T.; Bushnell, D.L.; Sunderland, J.J.; et al. Localization of Unknown Primary Site with 68 Ga-DOTATOC PET/CT in Patients with Metastatic Neuroendocrine Tumor. J. Nucl. Med. 2017, 58, 1054–1057. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Kan, Y.; Ge, B.H.; Yuan, L.; Li, C.; Zhao, W. Diagnostic Role of Gallium-68 DOTATOC and Gallium-68 DOTATATE PET in Patients with Neuroendocrine Tumors: A Meta-Analysis. Acta Radiol. 2014, 55, 389–398. [Google Scholar] [CrossRef]
- Pinchot, S.N.; Holen, K.; Sippel, R.S.; Chen, H. Carcinoid Tumors. Oncologist 2008, 13, 1255–1269. [Google Scholar] [CrossRef]
- Zatelli, M.C.; Tagliati, F.; Taylor, J.E.; Rossi, R.; Culler, M.D.; Uberti, E.C. Somatostatin Receptor Subtypes 2 and 5 Differentially Affect Proliferation In Vitro of the Human Medullary Thyroid Carcinoma Cell Line TT. J. Clin. Endocrinol. Metab. 2001, 86, 2161–2169. [Google Scholar] [CrossRef]
- Sun, L.; Coy, D.H. Somatostatin Receptor-Targeted Anti-Cancer Therapy. Curr. Drug Deliv. 2011, 8, 2–10. [Google Scholar] [CrossRef]
- Leijon, H.; Remes, S.; Hagström, J.; Louhimo, J.; Mäenpää, H.; Schalin-Jäntti, C.; Miettinen, M.; Haglund, C.; Arola, J.; Phl, S.R. Variable Somatostatin Receptor Subtype Expression in 151 Primary Pheochromocytomas and Paragangliomas. Hum. Pathol. 2019, 86, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Fotouhi, O.; Zedenius, J.; Höög, A.; Juhlin, C.C. Regional Differences in Somatostatin Receptor 2 (SSTR2) Immunoreactivity Is Coupled to Level of Bowel Invasion in Small Intestinal Neuroendocrine Tumors. Neuro-Endocrinol. Lett. 2018, 39, 305–309. [Google Scholar] [PubMed]
- Kimura, N.; Pilichowska, M.; Date, F.; Kimura, I.; Schindler, M. Immunohistochemical Expression of Somatostatin Type 2A Receptor in Neuroendocrine Tumors. Clin. Cancer Res. 1999, 5, 3483–3487. [Google Scholar] [PubMed]
- Si, Y.; Kim, S.; Ou, J.; Lu, Y.; Ernst, P.; Chen, K.; Whitt, J.; Carter, A.M.; Markert, J.M.; Bibb, J.A.; et al. Anti-SSTR2 Antibody-Drug Conjugate for Neuroendocrine Tumor Therapy. Cancer Gene Ther. 2020, 1–14. [Google Scholar] [CrossRef]
- Werner, R.A.; Weich, A.; Higuchi, T.; Schmid, J.S.; Schirbel, A.; Lassmann, M.; Wild, V.; Rudelius, M.; Kudlich, T.; Herrmann, K.; et al. Imaging of Chemokine Receptor 4 Expression in Neuroendocrine Tumors —A Triple Tracer Comparative Approach. Theranostics 2017, 7, 1489–1498. [Google Scholar] [CrossRef]
- Kaemmerer, D.; Träger, T.; Hoffmeister, M.; Sipos, B.; Hommann, M.; Sänger, J.; Schulz, S.; Lupp, A. Inverse Expression of Somatostatin and CXCR4 Chemokine Receptors in Gastroenteropancreatic Neuroendocrine Neoplasms of Different Malignancy. Oncotarget 2015, 6, 27566–27579. [Google Scholar] [CrossRef]
- Mai, R.; Kaemmerer, D.; Träger, T.; Neubauer, E.; Sänger, J.; Baum, R.P.; Schulz, S.; Lupp, A. Different Somatostatin and CXCR4 Chemokine Receptor Expression in Gastroenteropancreatic Neuroendocrine Neoplasms Depending on Their Origin. Sci. Rep. 2019, 9, 4339. [Google Scholar] [CrossRef]
- Kajtazi, Y.; Kaemmerer, D.; Sänger, J.; Schulz, S.; Lupp, A. Somatostatin and Chemokine CXCR4 Receptor Expression in Pancreatic Adenocarcinoma Relative to Pancreatic Neuroendocrine Tumours. J. Cancer Res. Clin. Oncol. 2019, 145, 2481–2493. [Google Scholar] [CrossRef]
- Werner, R.A.; Kircher, S.; Higuchi, T.; Kircher, M.; Schirbel, A.; Wester, H.-J.; Buck, A.K.; Pomper, M.G.; Rowe, S.P.; Lapa, C. CXCR4-Directed Imaging in Solid Tumors. Front. Oncol. 2019, 9, 770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, F.M.; Vesely, C.; Childs, A.; Marafioti, T.; Khan, M.S.; Mandair, D.; Cives, M.; Ensell, L.; Lowe, H.; Akarca, A.U.; et al. Circulating Tumour Cells and Their Association With Bone Metastases in Patients with Neuroendocrine Tumours. Br. J. Cancer 2019, 120, 294–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhou, L. Novel Anti-SSTR2 Monoclonal Antibody-Based Therapy to Treat Neuroendocrine Cancer. Provision Patent TH Docket No. 222119-8030, 10 November 2018. [Google Scholar]
- Ghobrial, I.M.; Liu, C.-J.; Redd, R.A.; Perez, R.P.; Baz, R.; Zavidij, O.; Sklavenitis-Pistofidis, R.; Richardson, P.G.; Anderson, K.C.; Laubach, J.P.; et al. A Phase Ib/II Trial of the First-in-Class Anti-CXCR4 Antibody Ulocuplumab in Combination with Lenalidomide or Bortezomib Plus Dexamethasone in Relapsed Multiple Myeloma. Clin. Cancer Res. 2020, 26, 344–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashyap, M.K.; Amaya-Chanaga, C.I.; Kumar, D.; Simmons, B.; Huser, N.; Gu, Y.; Hallin, M.; Lindquist, K.; Yafawi, R.; Choi, M.Y.; et al. Targeting the CXCR4 Pathway Using a Novel Anti-CXCR4 IgG1 Antibody (PF-06747143) in Chronic Lymphocytic Leukemia. J. Hematol. Oncol. 2017, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.A.; Ellison, E.C. Therapeutic Alternatives in Metastatic Neuroendocrine Tumors. Surg. Oncol. Clin. N. Am. 1998, 7, 863–879. [Google Scholar] [CrossRef]
- Amagata, T.; Rath, C.; Rigot, J.F.; Tarlov, N.; Tenney, K.; Valeriote, F.A.; Crews, P. Structures and Cytotoxic Properties of Trichoverroids and Their Macrolide Analogues Produced by Saltwater Culture ofMyrotheciumverrucaria. J. Med. Chem. 2003, 46, 4342–4350. [Google Scholar] [CrossRef]
- Woldemichael, G.M.; Turbyville, T.J.; Vasselli, J.R.; Linehan, W.M.; McMahon, J.B. Lack of a Functional VHL Gene Product Sensitizes Renal Cell Carcinoma Cells to the Apoptotic Effects of the Protein Synthesis Inhibitor Verrucarin A. Neoplasia 2012, 14, 771-IN28. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Yu, Y.; Chow, D.-C.; Palzkill, T.; Madoux, F.; Hodder, P.; Chase, P.; Griffin, P.R.; O’Malley, B.W.; Lonard, D.M. Identification of Verrucarin A as a Potent and Selective Steroid Receptor Coactivator-3 Small Molecule Inhibitor. PLoS ONE 2014, 9, e95243. [Google Scholar] [CrossRef] [Green Version]
- Palanivel, K.; Kanimozhi, V.; Kadalmani, B. Verrucarin A Alters Cell-Cycle Regulatory Proteins and Induces Apoptosis Through Reactive Oxygen Species-Dependent p38MAPK Activation in the Human Breast Cancer Cell Line MCF-7. Tumor Biol. 2014, 35, 10159–10167. [Google Scholar] [CrossRef]
- Xu, N.; Ou, J.; Si, Y.; Goh, K.Y.; Flanigan, D.D.; Han, X.; Yang, Y.; Yang, S.; Zhou, L.; Liu, X. Proteomics Insight Into the Production of Monoclonal Antibody. Biochem. Eng. J. 2019, 145, 177–185. [Google Scholar] [CrossRef]
- Ou, J.; Si, Y.; Goh, K.; Yasui, N.; Guo, Y.; Song, J.; Wang, L.; Jaskula-Sztul, R.; Fan, J.; Zhou, L.; et al. Bioprocess Development of Antibody-Drug Conjugate Production for Cancer Treatment. PLoS ONE 2018, 13, e0206246. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Xu, Y.; Guan, J.; Chen, K.; Kim, S.; Yang, E.S.; Zhou, L.; Liu, X. Anti-EGFR Antibody-Drug Conjugate for Triple-Negative Breast Cancer Therapy. Eng. Life Sci. 2020. [Google Scholar] [CrossRef]
- Sherman, S.K.; Carr, J.C.; Wang, D.; O’Dorisio, M.S.; O’Dorisio, T.M.; Howe, J.R. Gastric Inhibitory Polypeptide Receptor (GIPR) Is a Promising Target for Imaging and Therapy in Neuroendocrine Tumors. Surgery 2013, 154, 1206–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeb, D.; Gao, X.; Liu, Y.; Zhang, Y.; Shaw, J.; Valeriote, F.A.; Gautam, S.C. The Inhibition of Cell Proliferation and Induction of Apoptosis in Pancreatic Ductal Adenocarcinoma Cells by Verrucarin A, a Macrocyclic Trichothecene, Is Associated with the Inhibition of Akt/NF-κB/mTOR Prosurvival Signaling. Int. J. Oncol. 2016, 49, 1139–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Gao, X.; Deeb, D.; Zhang, Y.; Shaw, J.A.; Valeriote, F.; Gautam, S.C. Mycotoxin Verrucarin A Inhibits Proliferation and Induces Apoptosis in Prostate Cancer Cells by Inhibiting Prosurvival Akt/NF-kB/mTOR signaling. J. Exp. Ther. Oncol. 2016, 11, 251–260. [Google Scholar] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Si, Y.; Guan, J.; Xu, Y.; Chen, K.; Kim, S.; Zhou, L.; Jaskula-Sztul, R.; Liu, X.M. Dual-Targeted Extracellular Vesicles to Facilitate Combined Therapies for Neuroendocrine Cancer Treatment. Pharmaceutics 2020, 12, 1079. https://doi.org/10.3390/pharmaceutics12111079
Si Y, Guan J, Xu Y, Chen K, Kim S, Zhou L, Jaskula-Sztul R, Liu XM. Dual-Targeted Extracellular Vesicles to Facilitate Combined Therapies for Neuroendocrine Cancer Treatment. Pharmaceutics. 2020; 12(11):1079. https://doi.org/10.3390/pharmaceutics12111079
Chicago/Turabian StyleSi, Yingnan, JiaShiung Guan, Yuanxin Xu, Kai Chen, Seulhee Kim, Lufang Zhou, Renata Jaskula-Sztul, and X. Margaret Liu. 2020. "Dual-Targeted Extracellular Vesicles to Facilitate Combined Therapies for Neuroendocrine Cancer Treatment" Pharmaceutics 12, no. 11: 1079. https://doi.org/10.3390/pharmaceutics12111079
APA StyleSi, Y., Guan, J., Xu, Y., Chen, K., Kim, S., Zhou, L., Jaskula-Sztul, R., & Liu, X. M. (2020). Dual-Targeted Extracellular Vesicles to Facilitate Combined Therapies for Neuroendocrine Cancer Treatment. Pharmaceutics, 12(11), 1079. https://doi.org/10.3390/pharmaceutics12111079