1. Introduction
Recently, we reported that up to 80% of the total ATP production in melanoma and lung cancer cells [
1], and about 40% of the ATP production in pancreatic cancer cells [
2,
3], depends on cytosolic NADH and the malate-aspartate shuttle (MAS). The MAS transfers cytosolic NADH into mitochondria for ATP production through oxidative phosphorylation (OxPhos) in the mitochondrial membrane [
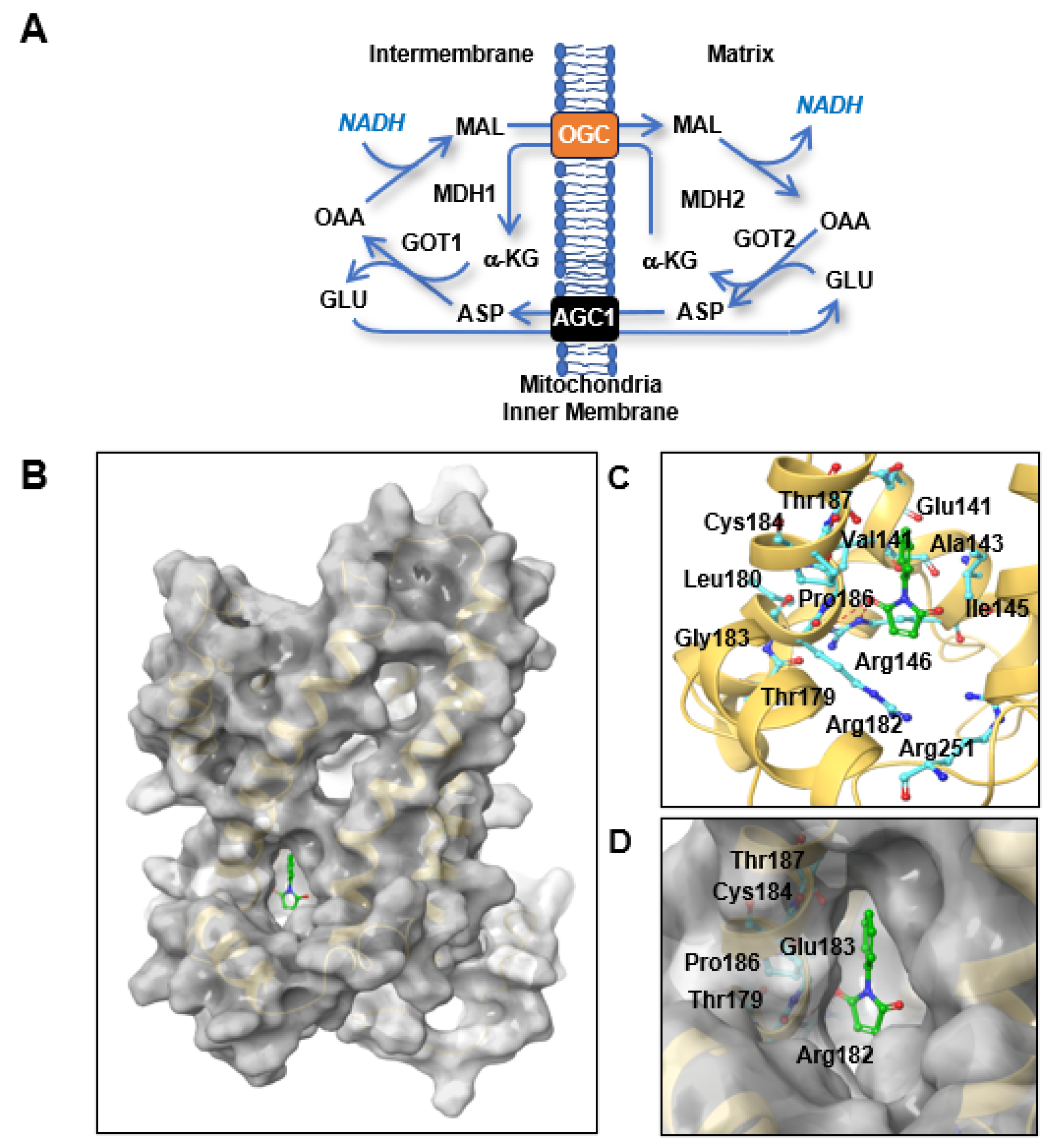
1]. The MAS consists of four metabolic enzymes, glutamic-oxaloacetic transaminase (GOT) 1 and 2 and malate dehydrogenase (MDH) 1 and 2, and two antiporters, oxoglutarate carrier (OGC, oxoglutarate/malate antiporter,
SLC25A11) and mitochondrial aspartate-glutamate carrier (AGC1) (
Figure 1A) [
1,
4]. Knock-down of OGC reduced ATP production by 80% and inhibited the growth of lung and melanoma cancer cells by over 90% [
1]. ATP depletion by more than 50% induces cell cycle arrest and cell death in a time-dependent manner in cancer cells [
5,
6,
7,
8]. Furthermore, in in vivo experiments, heterozygous OGC knock-out mice showed 50% less spontaneous tumor development in the
KRASLA2 lung cancer model [
1]. Blocking OGC may selectively inhibit cancer growth by reducing ATP production in cancer cells because cancer cells rely on the MAS for ATP generation while normal cells do not [
1,
2].
A specific mitochondrial transport system for 2-oxoglutarate was first proposed by Dr. Chappell in 1967 [
9], and the biochemical properties of this system, including its structure and activity, were recently reviewed by Dr. Fiermonte [
10]. OGC has a similar structure to that of mitochondrial ADP/ATP carriers, although a three-dimensional (3D) study of the OGC structure by X-ray crystallography failed [
10]. Inhibitors of OGC were therefore developed based on the starting compounds of the biochemical reaction instead of the OGC structure. Most OGC inhibitors are derived from substrate analogues such as succinate, butylmalonate, and phthalonate [
11,
12], which do not bind to the translocation site but do block transport. OGC contains three cysteine residues at positions 184, 221, and 224 that can create S-S bridges with sulfhydryl reagents [
13]. Dr. Palmieri’s group found that both mercurials and maleimides integrated specifically with Cys184 [
13]. This binding was associated with inhibition of the OGC active conformation. The degree of OGC inhibition by
N-phenylmaleimide (NPM) binding to OGC was enhanced in the presence of OGC substrates [
13]. This suggests that a substrate-induced conformational change in OGC increases the reactivity of Cys184 to sulfhydryl reagents such as NPM [
13]. NPM inhibited OGC transport activity with a 50% inhibitory concentration (IC
50) of 1.25 mmol/min/g [
13], and NPM analogues inhibited the proliferation of H460 cancer cells with IC
50 values of 0.84–9 μM using in vitro assay system using reconstituted liposome with purified OGC [
14].
In this study, we investigated whether OGC inhibition with NPM inhibited cancer growth by reducing ATP production. Although NPM itself is known to have some off-target effects, this study demonstrates the potential therapeutic efficacy of OGC inhibitors.
2. Materials and Methods
2.1. Cell Culture
Tumor spheres (TS) generated from the UACC-62 and B16F10 melanoma cell lines were used in this study. UACC-62 cells were obtained from the US National Cancer Institute (NCI; Bethesda, MD, USA) (MTA 1-2702-09) and B16F10 cells (CRL-6475) were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). For TS culture, cells were cultured in TS complete media, which was composed of Dulbecco’s modified Eagle’s medium/F12 (SH30023.01, Hyclone, Logan, UT, USA), B27 supplement (17504044, Thermo Fisher Scientific, Waltham, MA, USA), 20 ng/mL basic fibroblast growth factor (F0291, Sigma-Aldrich, St. Louis, MO, USA), and 20 ng/mL epidermal growth factor (E9644, Sigma-Aldrich, St. Louis, MO, USA).
2.2. Homology Modeling and Molecular Docking
To obtain a homology model of OGC, we used the nuclear magnetic resonance (NMR)-based structure of mitochondrial uncoupling protein 2 (UCP2; Protein Data Bank (PDB) ID, 2LCK) as a template structure. The sequence alignment and homology modeling procedure were executed using Prime (Schrödinger, New York, NY, USA, 2020) [
15]. The overall quality of the modeled structure was assessed using a Ramachandran plot. The active binding sites of OGC were identified using the modeled protein structure in the SiteMap program of the Schrödinger software [
16]. To examine the binding interactions of the protein–ligand complexes, molecular docking studies were performed using the Glide software (Schrödinger, New York, NY, USA), which uses an optimized potential for liquid simulations (OPLS)-2005 force field, and refinement was carried out as per the recommendations of the Schrödinger Protein Preparation Wizard. LigPrep (Schrödinger, New York, NY, USA) was used to generate 3D structures of the ligands. The active grid was generated using the Receptor grid application in the Glide module. On a defined receptor grid, flexible docking was performed using the standard precision mode of Glide [
17]. The best docking pose for a compound was selected based on the best-scoring conformations from Glide, the binding patterns, and visual inspection.
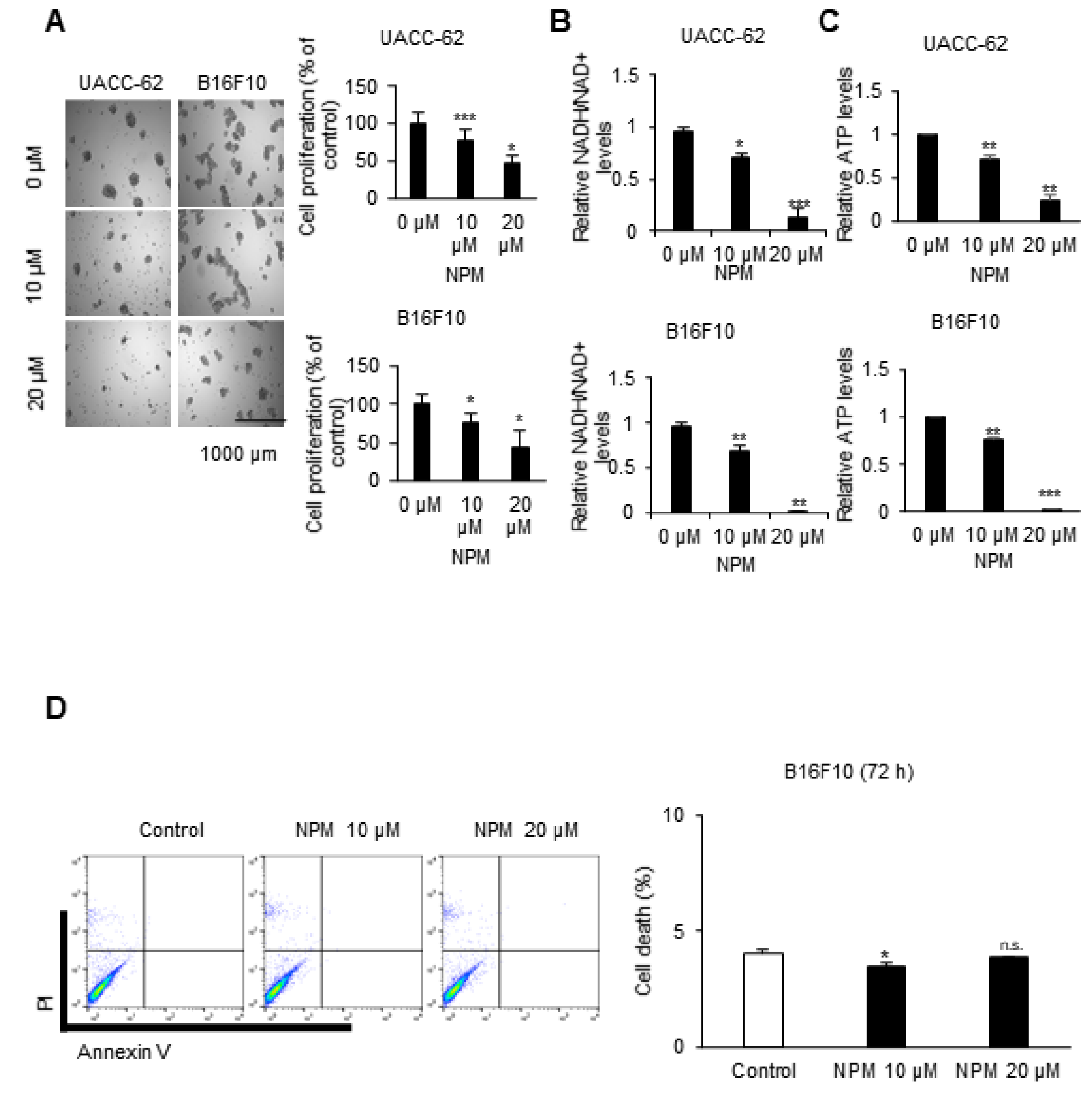
2.3. Sulforhodamine B Assay (SRB): Cell Growth Assay
Cancer cells were counted, and approximately 2 × 10
4 cells per well were seeded in 96-well cell culture plates (Corning Inc., Corning, NY, USA). After incubation at 37 °C in a humidified atmosphere with 5% CO
2 for 72 h, cells were treated with the indicated concentrations of NPM. The assay was performed following to the previously established method [
2].
2.4. Measurement of the NADH/NAD+ Ratio and ATP Levels
To quantify NADH/NAD in cell lines, the Ultra-GloTM Recombinant Luciferase assay kit (Promega, #G9071) was used according to the manufacturer’s instructions. Briefly, the cyclic enzyme included in the kit converts NAD+ to NADH, which subsequently activates a reductase that converts pro-luciferin to luciferin. The samples were subsequently detected with Ultra-GloTM r-Luciferase. To this end, the cells were seeded into a 96-well culture plate at a density of 104 cells/well and incubated for 24 h, then treated with NPM for 72 h. Subsequently, 50 µL of NAD/NADH-GloTM Detection Reagent and an equal volume of sample were incubated at room temperature for 30 min. To measure ATP levels in cell lines, the cell titer-Glo 2.0 assay (Promega, #G9241) was used according to the manufacturer’s instructions. The cells were seeded into a 96-well culture plate at a density of 105 cells/well and incubated for 24 h and treated with NPM for 72 h. A volume of CellTiter-Glo® 2.0 Reagent equal to the volume of ATP standard present in each well was added. The mixed contents were incubated at room temperature for 10 min and luminescence was measured.
2.5. Measurement of Apoptosis
Tumor cells were incubated with or without NPM. The cells were collected, washed with cold PBS, centrifuged at 1400 rpm for 3 min, and resuspended in binding buffer from a kit (556547, BD Biosciences, San Jose, CA, USA) at a density of 1 × 106 cells/mL. Cells (1 × 105 cells in a 100 μL volume) were transferred to a 5 mL culture tube, and 5 μL each of annexin V-FITC and propidium iodide (PI) were added. The cells were gently vortexed and incubated for 15 min at room temperature in the dark. A total of 400 μL of binding buffer was added to each tube, and the samples were analyzed by flow cytometry (FACSCalibur BD Biosciences, San Jose, CA, USA).
2.6. Measurement of Mitochondrial Membrane Potential (∆ψm)
Mitochondrial membrane potential (MMP) was assessed by measuring the mean fluorescence intensity of tetramethylrodamine ester (TMRE) loaded cells. TMRE (87917, Sigma-Aldrich, St. Louis, MO, USA) is a fluorescence probe that specifically accumulates within mitochondria in an MMP-dependent manner. Cells were plated in a 100 mm plate and treated as indicted. Twenty minutes prior to the end of each treatment, 100 nM TMRE was added to the culture medium. Cells were washed two times with ice-cold PBS. Cells were collected immediately for flow cytometric analysis (FACSCalibur, BD Biosciences, San Jose, CA, USA) of fluorescence intensity using the 585 nm (FL-2) channel.
2.7. Immunohistochemistry
Immunohistochemistry was performed on a Ventana Discovery XT automated staining instrument (Ventana Medical Systems, Tucson, AZ, USA) followed by the established method [
2]. Staining was performed with a Ki-67 antibody (ab15580; Abcam, Cambridge, UK) and KI-67 positive cells were quantified using ImageJ software (64-bit Java 1.8.0_112).
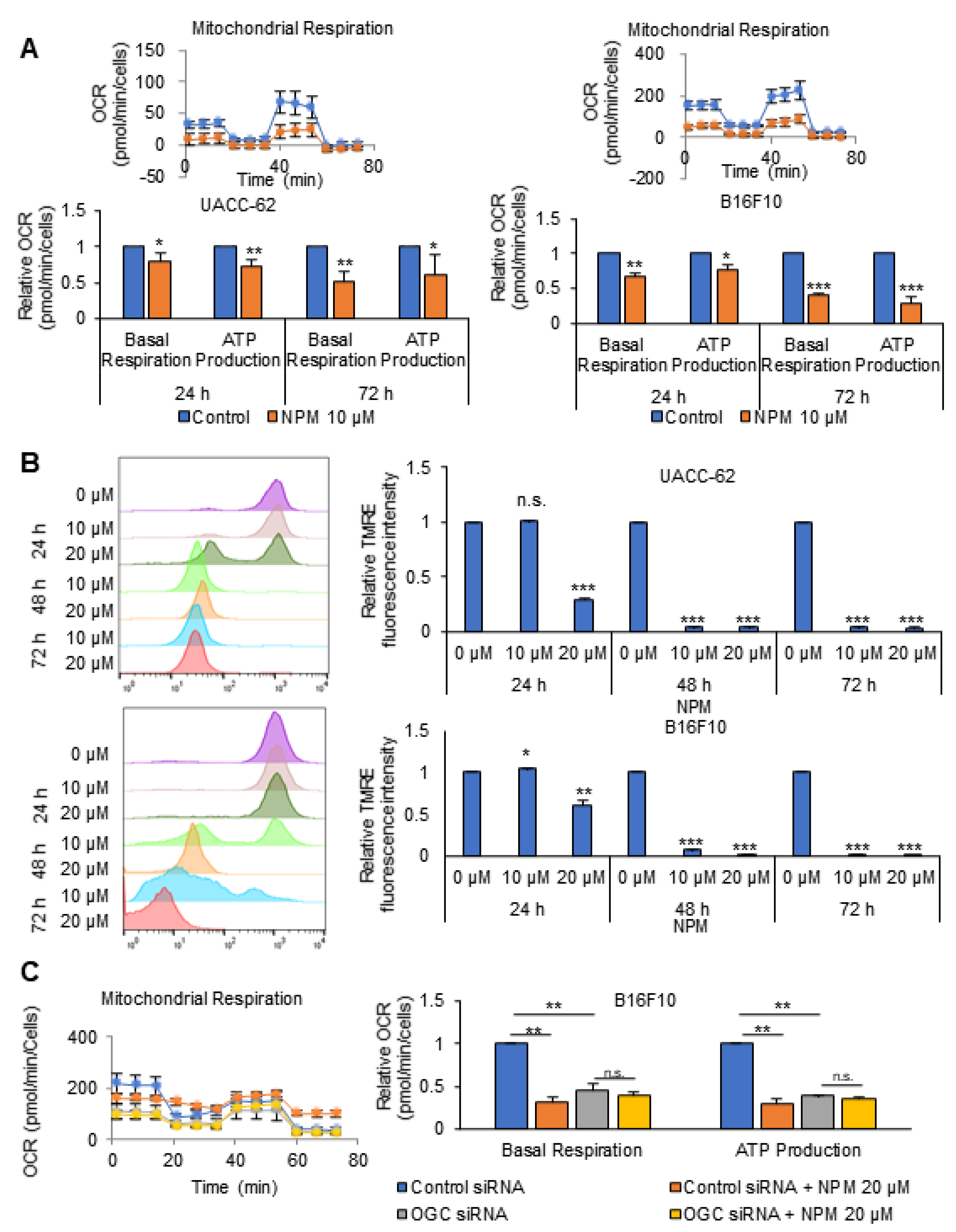
2.8. XF Cell Mito Stress Analysis
Cells were treated with the indicated drugs for 24 h. For determination of the oxygen consumption rate (OCR), cells were incubated in XF base medium supplemented with 10 mM glucose, 1 mM sodium pyruvate, and 2 mM of L-glutamine, and were equilibrated in a non-CO2 incubator for 1 h before starting the assay. The samples were incubated for 3 min and then data were acquired for 3 min using the XFe96 extracellular flux analyzer (Seahorse Bioscience, North Billerica, MA, USA). Oligomycin (0.75 µM), carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP; 1 µM), and rotenone/antimycin A (0.5 µM) (103015-100, Agilent Technologies, Santa Clara, CA, USA) were injected at the indicated time points. Finally, the OCR was normalized to the cell number, as determined by CCK-8 assay.
2.9. CCK-8 Assay
Cancer cells were counted, and approximately 5 × 103 cells per well were seeded in 96-well cell culture plates (Corning Inc., Corning, NY, USA). After incubation at 37 °C in a humidified atmosphere with 5% CO2 for 72 h, cells were treated with the indicated concentrations of NPM. At the end of XF Cell Mito Stress Analysis, 10 μL of CCK-8 reagent (ALX-850-039, Enzo Life Sciences, Farmingdale, NY, USA) was added to each well, and the optical density (OD) at 450 nm was measured using a multifunction microplate reader (Infinite M200 Pro, Tecan, Männedorf, Switzerland) after incubation for 1 h at 37 °C.
2.10. Invasion Assay
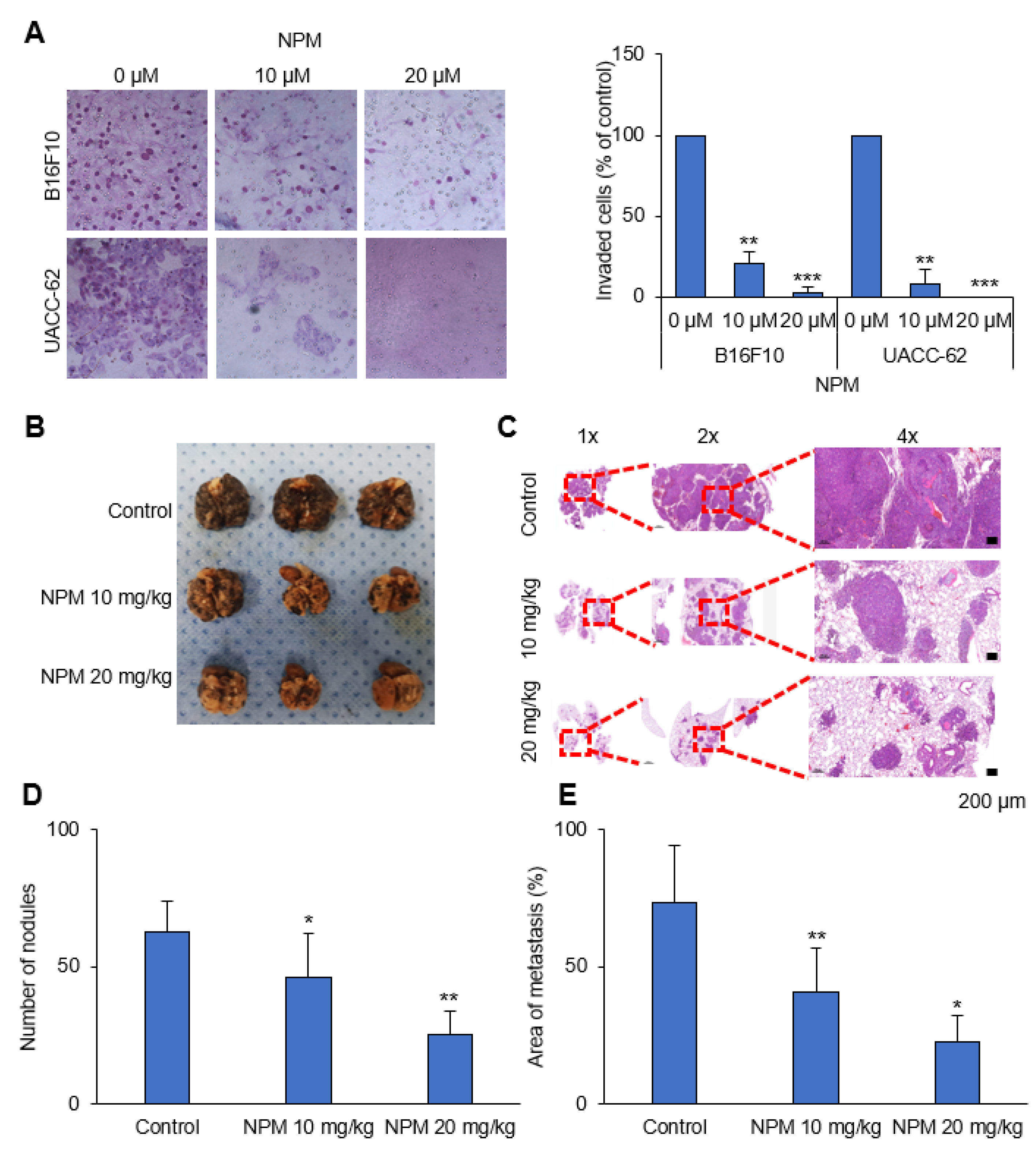
The upper compartments of 8 mm Transwells (6.5 mm diameter; Coastar Corp., Cambridge, MA, USA) were precoated with Matrigel (1 mg/mL). Cells (105 cells) were suspended in DMEM and placed in the upper compartments of the Transwells, and the lower compartments were filled with DMEM supplemented with 3% FBS. After 24 h, the filters were washed with PBS and fixed with methanol. Migrated cells on the filter membrane were stained using a Diff-Quik staining kit (38721, Sysmex, Kobe, Japan). Each assay was conducted at least three times, and three random fields under 20× magnification were analyzed for each filter membrane.
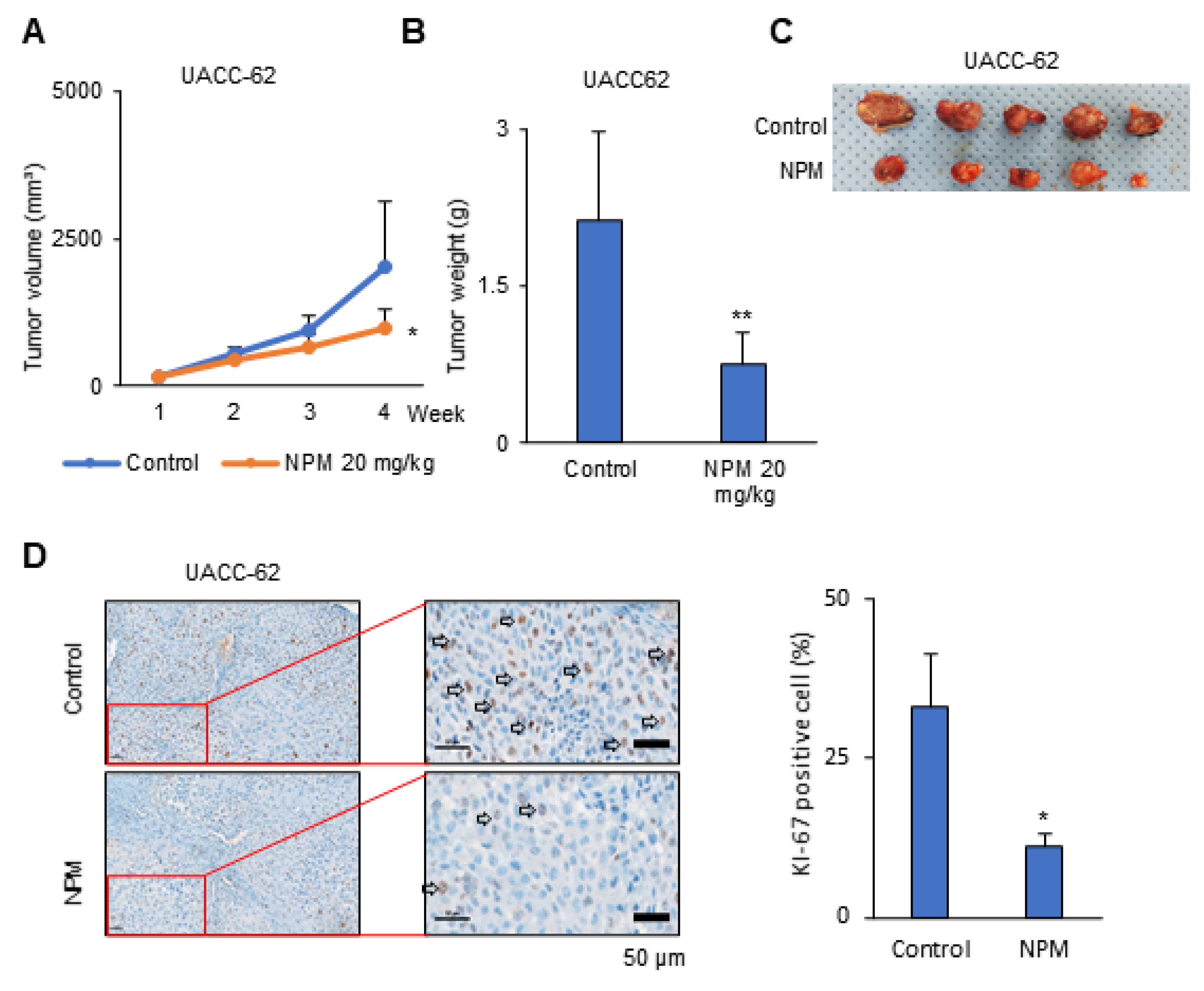
2.11. Preclinical Xenograft Model
Balb/c-nu mice (Orient, Seoul, Korea) were between 6 and 8 weeks of age before tumor induction. This study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the National Cancer Center Research Institute, which is an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International-accredited facility that abides by the Institute of Laboratory Animal Resources guidelines (protocol NCC-19-195). Mice were inoculated with UACC-62 cells (1 × 107) in 100 μL PBS subcutaneously using a 1 mL syringe. After 1 week, the mice were divided into two groups, a control group treated with vehicle and an NPM-treated group (n = 5 mice/group). Vehicle (5% dimethyl sulfoxide (DMSO) and 10% Kolliphor in PBS; 100 μL) and NPM (20 mg/kg) were administered orally once per day, 5 days/week, for 3 weeks. The primary tumor size was measured every week using calipers. The tumor volume was calculated using the formula V = (A × B2)/2, where V is the volume (mm3), A is the long diameter, and B is the short diameter.
2.12. Syngeneic Lung Tumor Metastasis Model
This study was reviewed and approved by the IACUC of the National Cancer Center Research Institute (protocol NCC-20-557). To determine the effect of NPM on tumor metastasis in vivo, the formation of lung metastases was assessed in C57BL/6 mice injected intravenously with B16F10 cells (1 × 105) via the tail vein. Three weeks after injection of B16F10 cells, the mice were sacrificed by CO2 asphyxiation, images of the lungs were captured, and the lung metastases were counted. Tissue specimens from each group were fixed in formalin and embedded in paraffin for histologic examination. Metastatic lesions were quantified using ImageJ software (64-bit Java 1.8.0_112).
2.13. hERG K+ Channel Binding Assay
E-4031 (M5060, Sigma-Aldrich, St. Louis, MO, USA) compound was used as a positive control. Membrane containing the hERG channel was mixed with the tracer for 4 h. The fluorescence intensity in the presence of NPT (excitation at 530 nm, emission at 590 nm) was measured by Synergy Neo (Biotek, Winooski, VT, USA) using the hERG Fluorescence Polarization Assay kit (PV5365, Thermo Fisher Scientific) and compared with the fluorescence intensity of the DMSO solvent control. The hERG assay was performed by a licensed contract research organization, Daegu-Gyeongbuk Medical Innovation Foundation (Daegu, Korea).
2.14. Statistical Analysis
Statistical analysis was performed using the Student’s t-test as appropriate. Tumor growth and tumor weight was analyzed statistically by two-way analysis of variance (ANOVA) tests using GraphPad PRISM 5 (GraphPad Software, San Diego, CA, USA).
4. Discussion
In in vitro experiments with cultured melanoma tumor spheres, NPM suppressed mitochondrial activity, oxygen consumption, and ATP production in a dose-dependent manner by inhibiting OGC, resulting in decreased melanoma cell proliferation. NPM also reduced melanoma cell invasion by over 70% in a human melanoma metastasis model. Therefore, blocking OGC activity with NPM may be a useful approach for inhibiting cancer growth.
However, there are reports of the inhibitory effects of NPM on pyruvate transport, telomerase activity, and Bak protein activity. It is known that pyruvate transport may be inhibited by thiol-blocking reagents such as iodoacetate and NPM [
20]. However, in cancer cells, pyruvate transport into mitochondria is limited because cancer cells have higher expression of lactate dehydrogenase (LDHA), which catalyzes pyruvate to lactate, compared with normal cells [
21]. Furthermore, cancer cells do not rely on the tricarboxylic acid (TCA) cycle to produce NADH, while normal cells depend completely on the TCA cycle for NADH production [
2]. Therefore, the decrease in ATP production observed in response to NPM likely does not occur through inhibition of pyruvate transport. Indeed, OCR and ATP production were measured in B16F10 cells treated with the indicated concentration of pyruvate inhibitor UK-5099 for 24 h. The analysis showed no decrease of these parameters. (
Supplementary Figure S2). NPM is also known to inhibit telomerase activity, which plays a key role in maintaining telomerase length [
22]. In in vitro assays, NPM inhibited telomerase activity. However, the IC
50 of NPM for inhibition of telomerase activity was approximately 2 μM, at which concentration there was no telomerase inhibition in cancer cells [
22]. This implies that the cytotoxicity of NPM is unrelated to inhibition of telomerase activity. Finally, NPM has been reported to induce apoptosis through Bak oligomerization in Jurkat cells at a concentration of 0.5 μM [
23]. However, we did not observe apoptosis by FACS analysis of melanoma cell lines treated with 20 μM NPM for 72 h.
Maleimide is formed with a -C(O)NHC(O)- functional group from the reaction of maleic acid and imide. NPM is a derivative of maleimide in which the NH group of maleimide is replaced with the aryl group of phenyl. NPM can cause skin and eye irritation and showed acute toxicity, with a 50% lethal dose (LD
50) of 78 mg/kg in mice when administered orally (
https://www.cdc.gov/niosh-rtecs/ON5ACA30.html). In this study, we did not observe any death or weight loss in mice treated with 20 mg/kg NPM. Although NPM is considered a nonspecific cytotoxic agent, NPM also did not show any cardiotoxicity by hERG assay (
Supplementary Figure S3). The hERG channel inhibition assay is a highly sensitive assay that will identify compounds exhibiting cardiotoxicity related to hERG inhibition in vivo. The IC
50 value of NPM in that assay was 78.75 μM, which indicates that it shows no hERG channel inhibition.
OGC with NPM significantly reduced ATP production in melanoma cells by decreasing NADH production. Treatment of mice with NPM reduced tumor growth and tumor invasion by 60% and 80%, respectively, in a human melanoma xenograft model. These data suggest that OGC inhibition combined with cytotoxic anticancer therapy may have synergistic effects on tumor growth.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




