Topical Delivery of Carvedilol Loaded Nano-Transfersomes for Skin Cancer Chemoprevention

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Compounds
2.2. Preparation of Carvedilol Loaded Transfersomes
2.3. Determination of Particle Size and Zeta Potential
2.4. Determination of Drug Encapsulation Efficiency (EE)
2.5. HPLC Analysis
2.6. In Vitro Drug Release Study
2.7. Ex Vivo Skin Permeation Study
2.8. Cell Lines and Cytotoxicity Assay
2.9. Intracellular Uptake of Transfersomes
2.10. UV Light Source
2.11. 3Dimensional (3D) Human Reconstituted Skin Model
2.12. Slot Blot Analysis for Cyclobutane Pyrimidine dimer (CPD) and Pyrimidine (6-4) Pyrimidone Photoproducts (6-4PP)
2.13. Quantitative RT-PCR Analysis
2.14. Histological Analysis and Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Assay
2.15. Statistical Analysis
3. Results
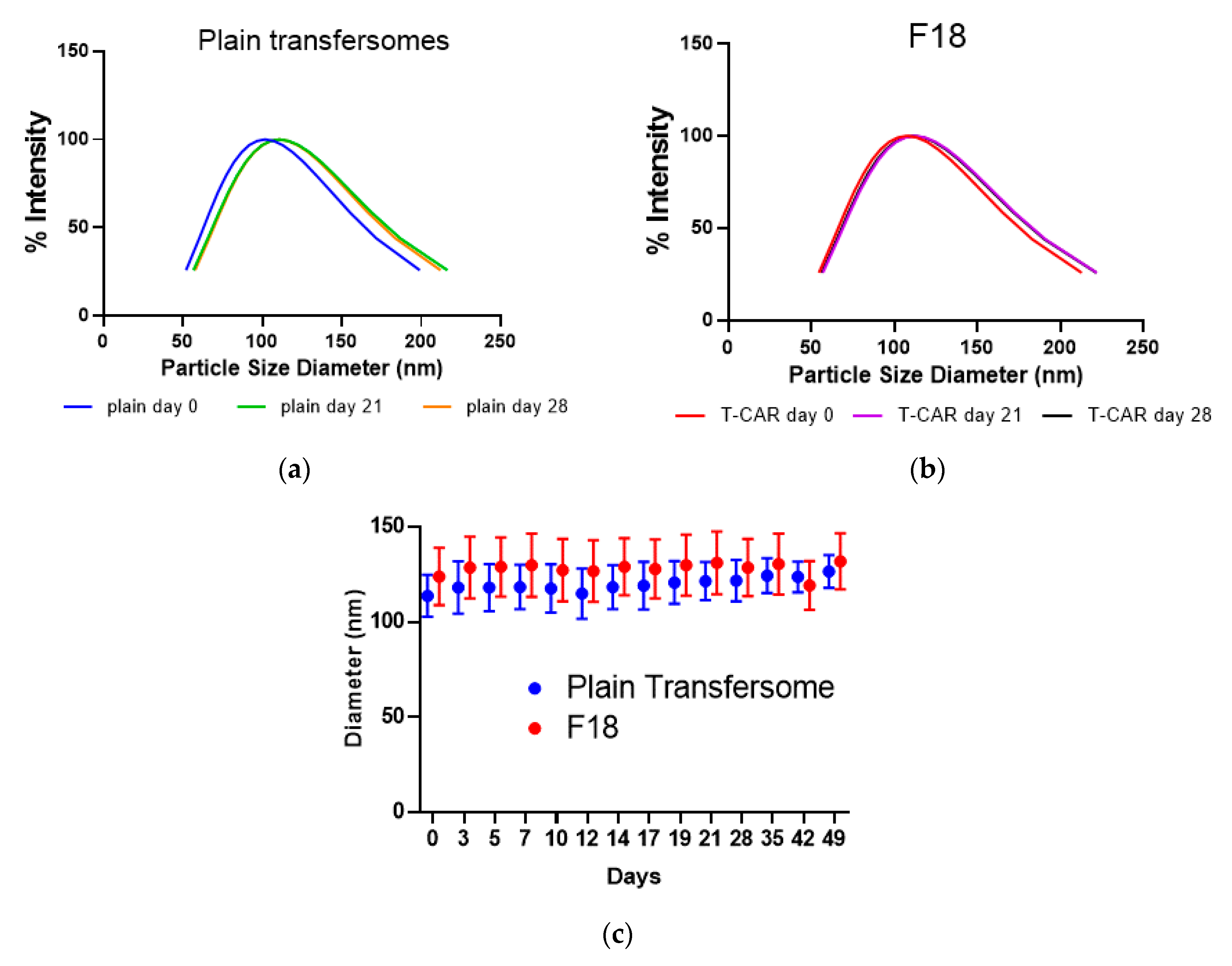
3.1. Formulation Preparation, Characterization, and Optimization
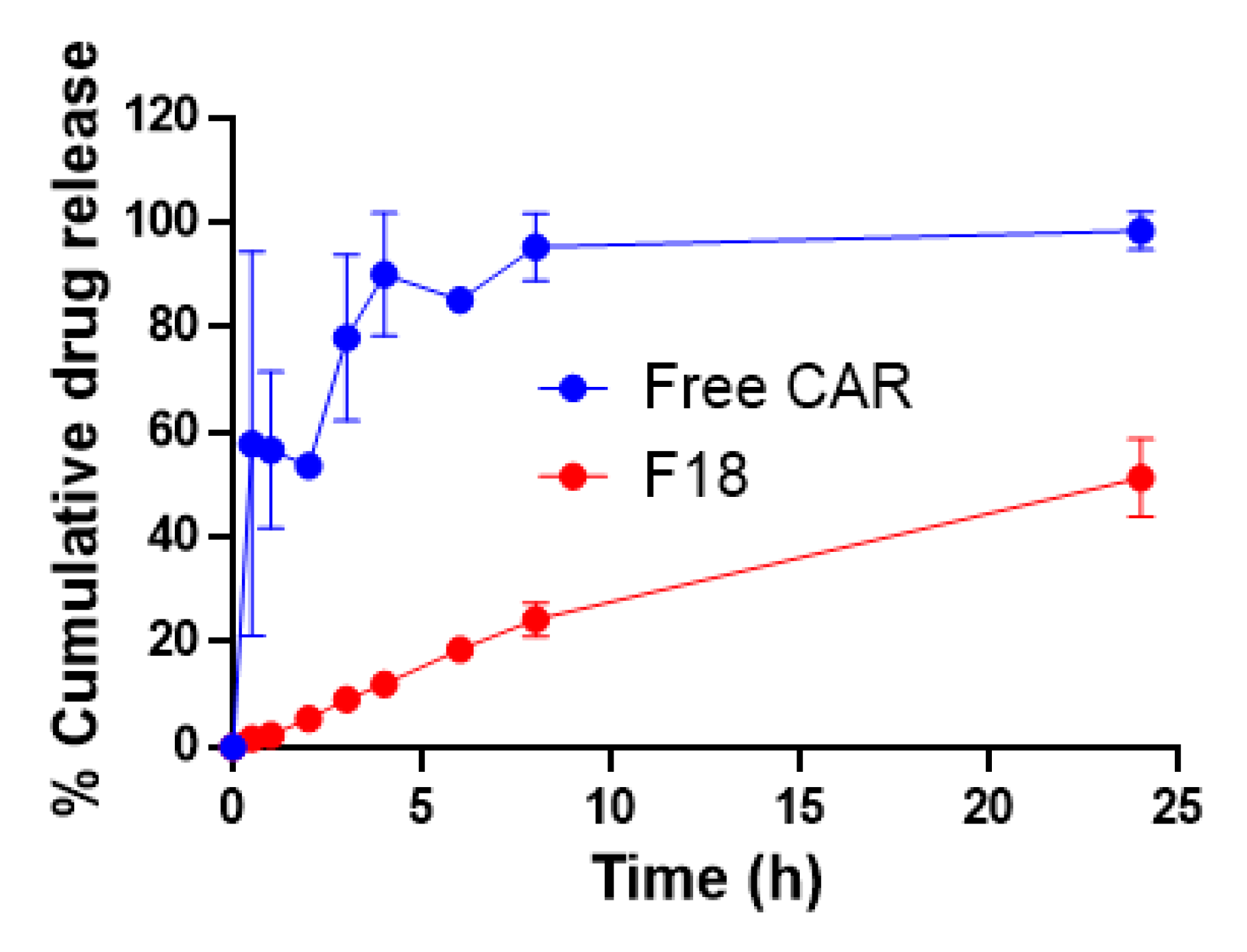
3.2. In Vitro Drug Release Kinetics
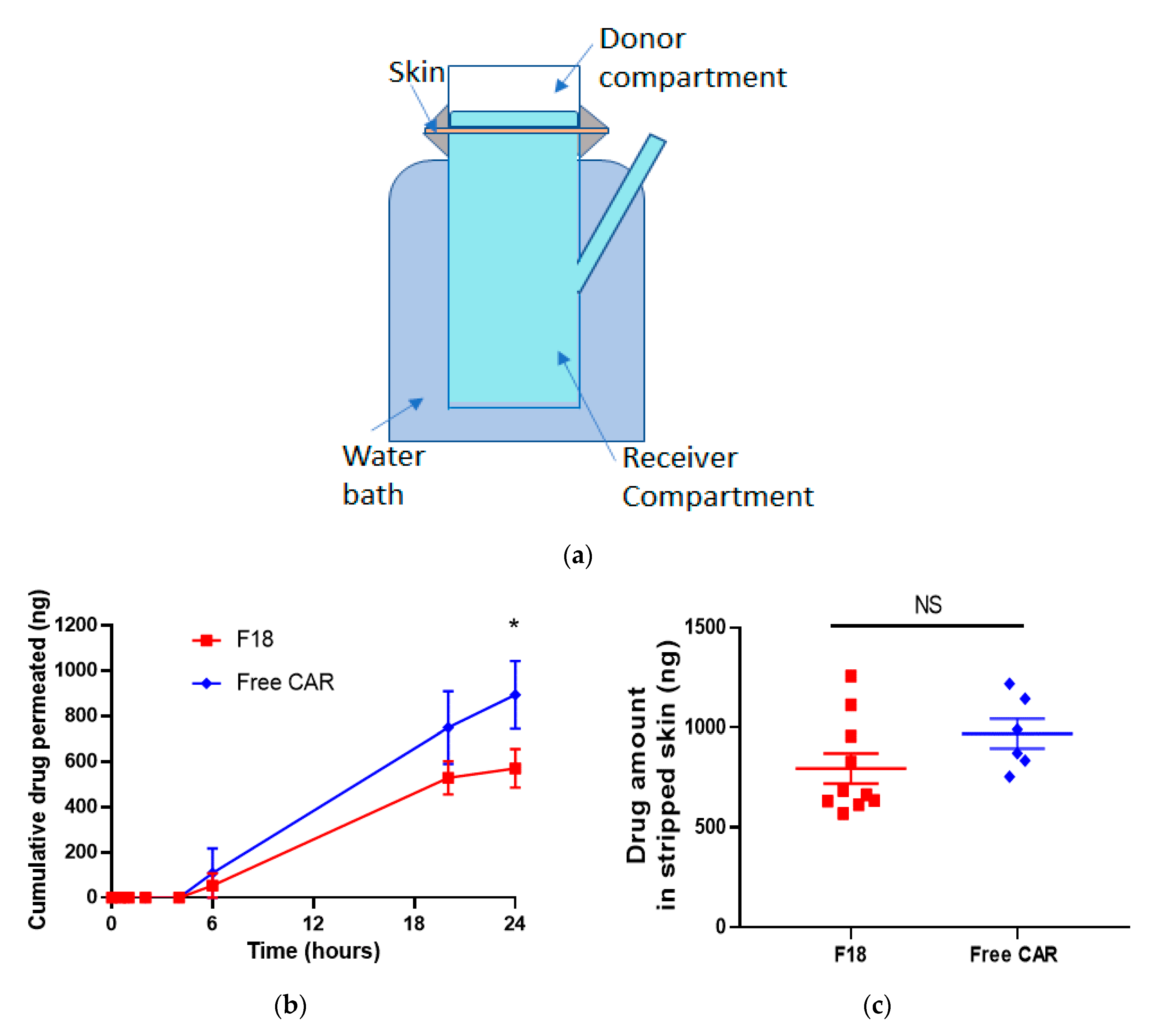
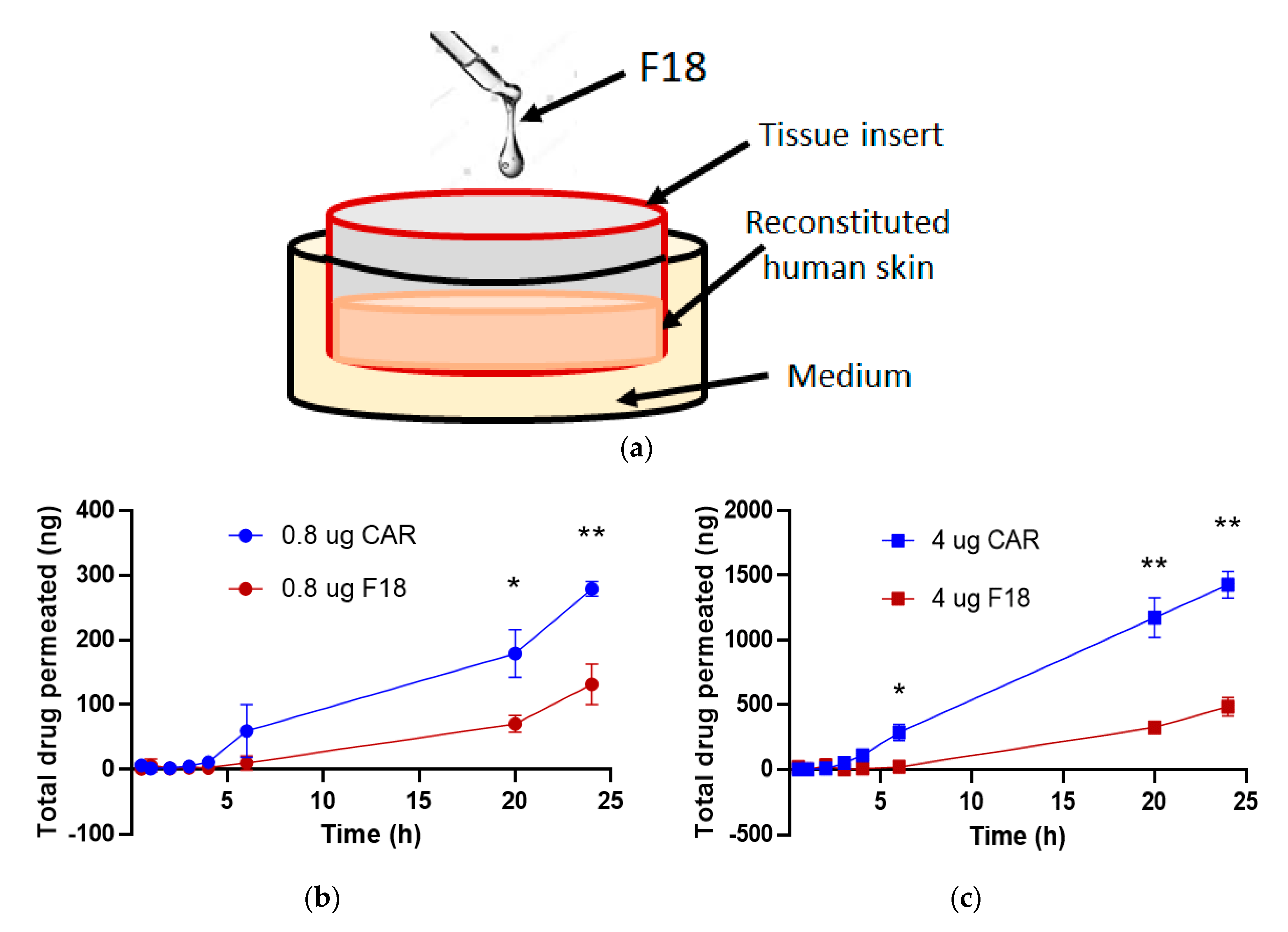
3.3. Ex Vivo Skin Permeation Study
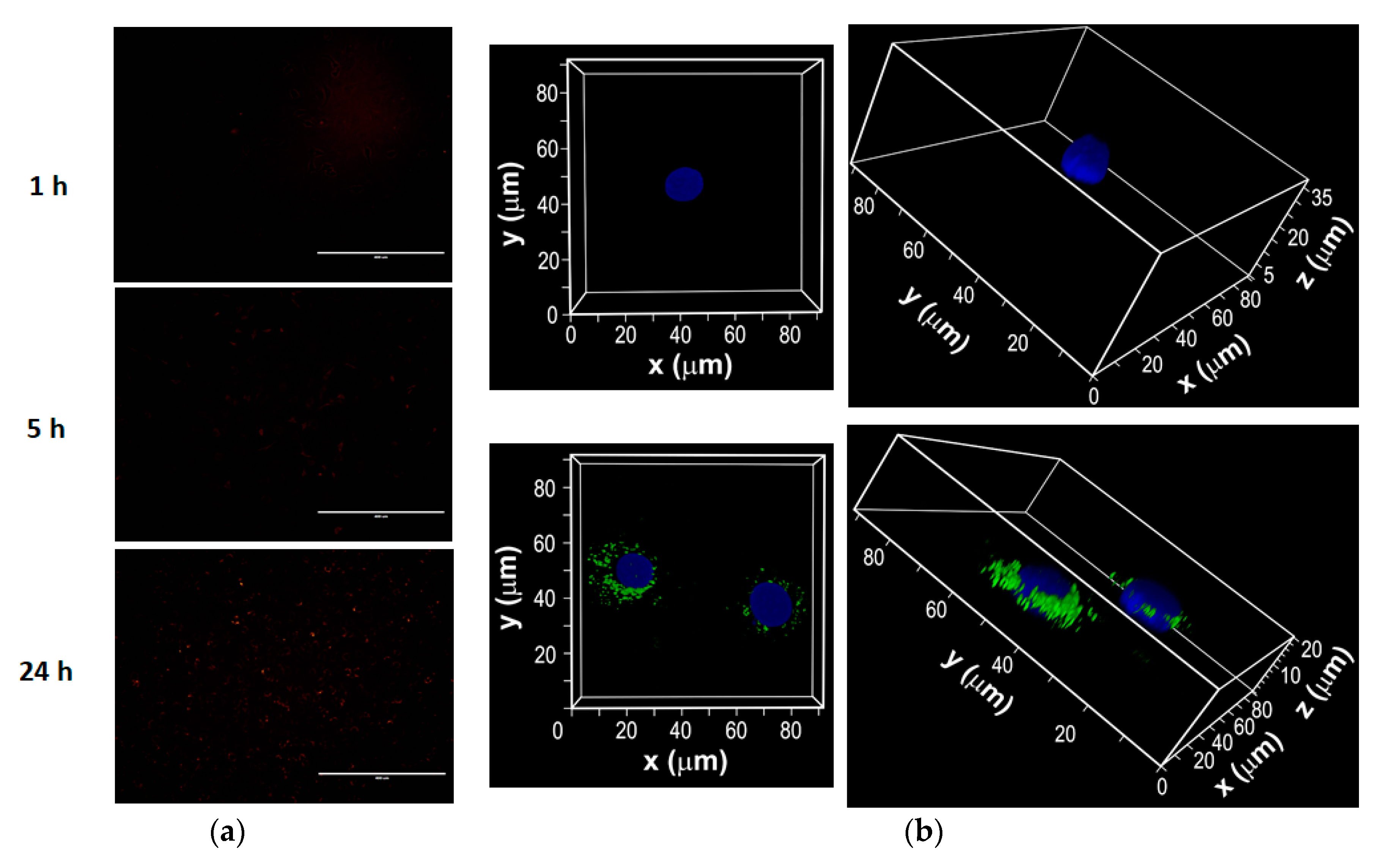
3.4. Intracellular Uptake of Fluorescent Dye-Labeled F18 by JB6 P+ Cells
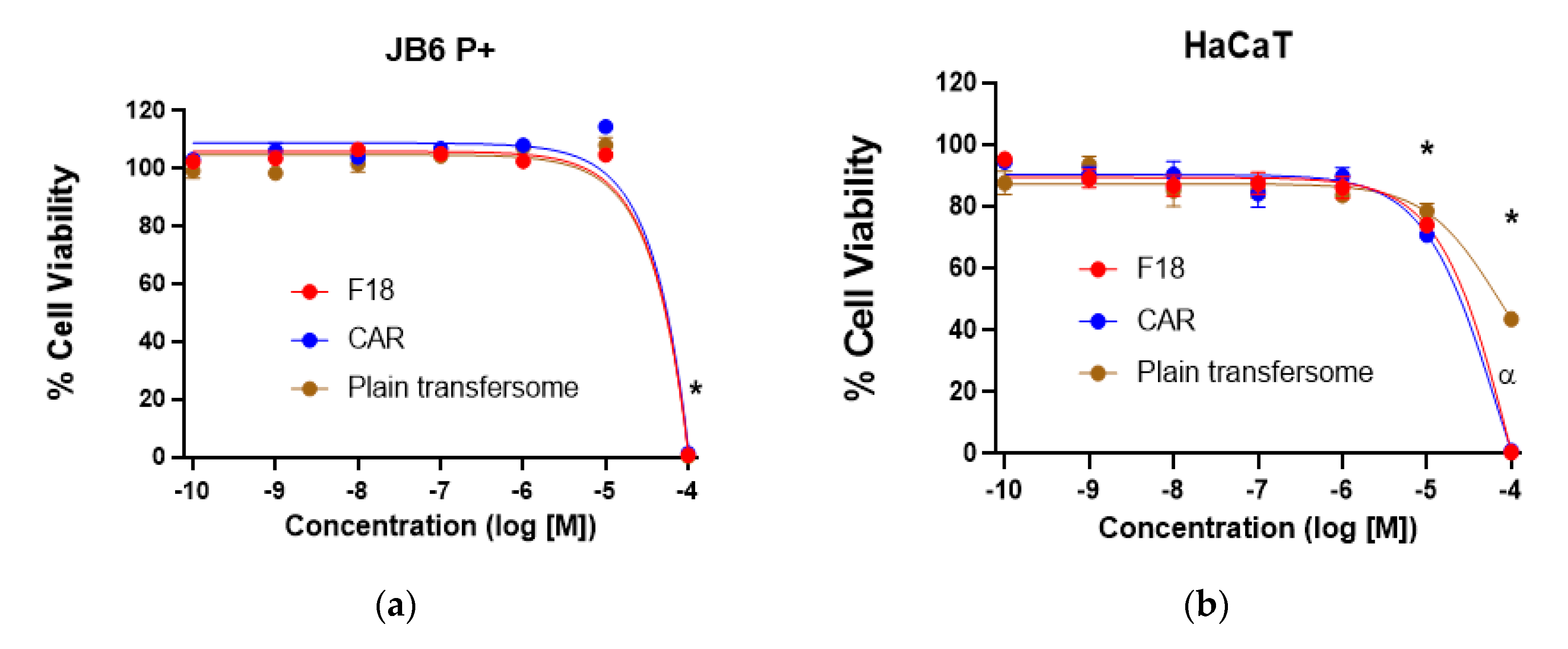
3.5. Cytotoxicity of F18 on JB6 P+ and HaCaT Cells
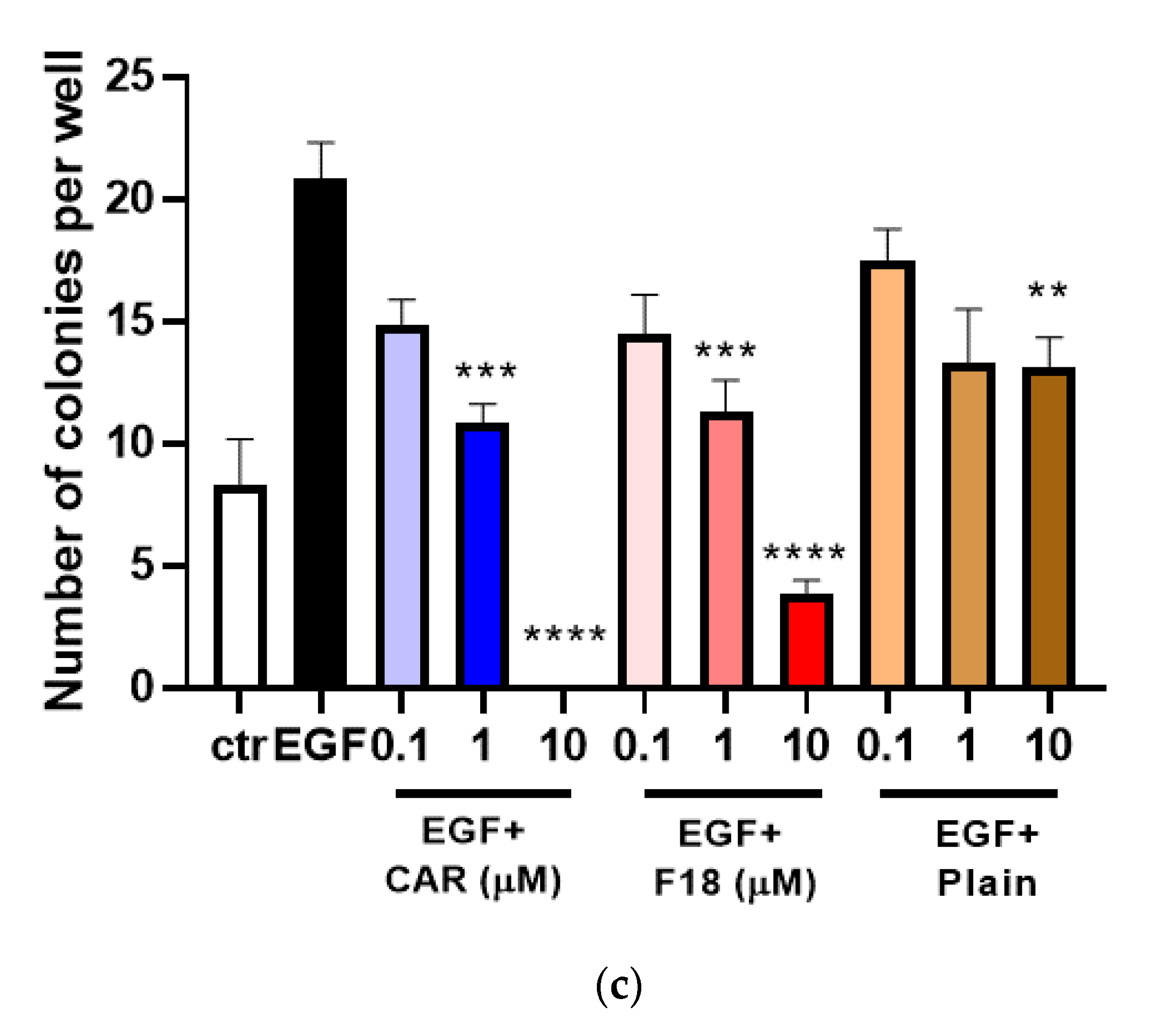
3.6. Effects of F18 on EGF-Induced JB6 P+ Transformation
3.7. Drug Permeation of F18 in Full-Thickness 3D Human Reconstituted Skin Model
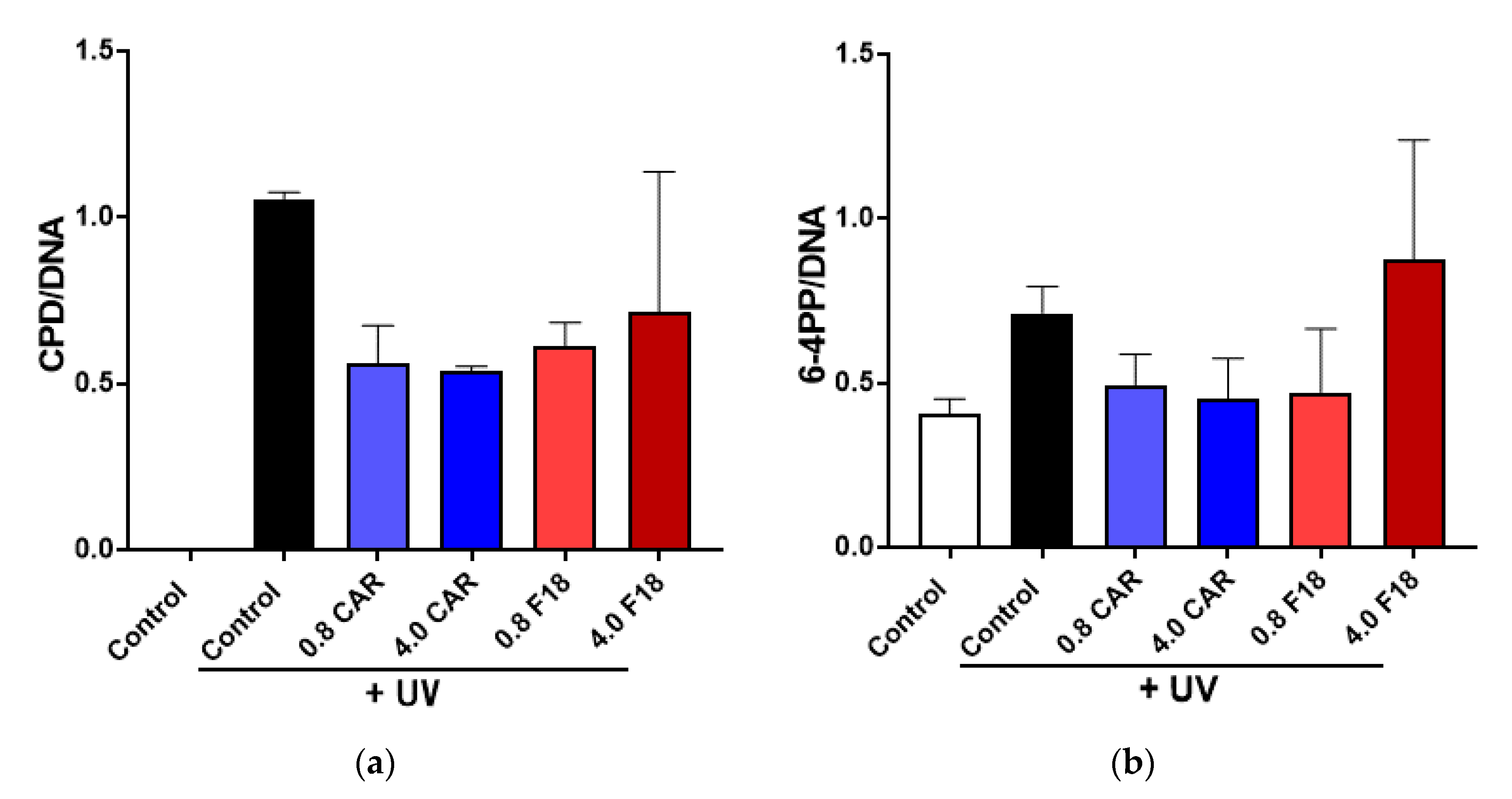
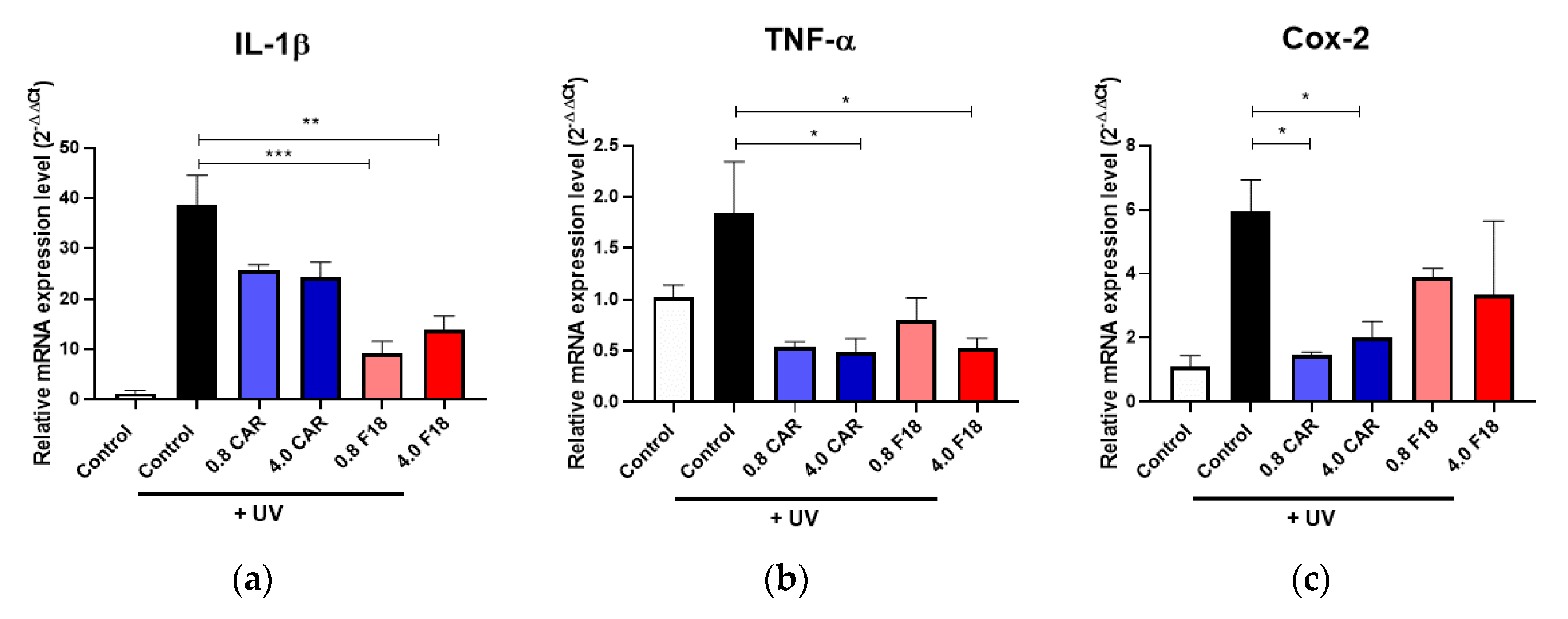
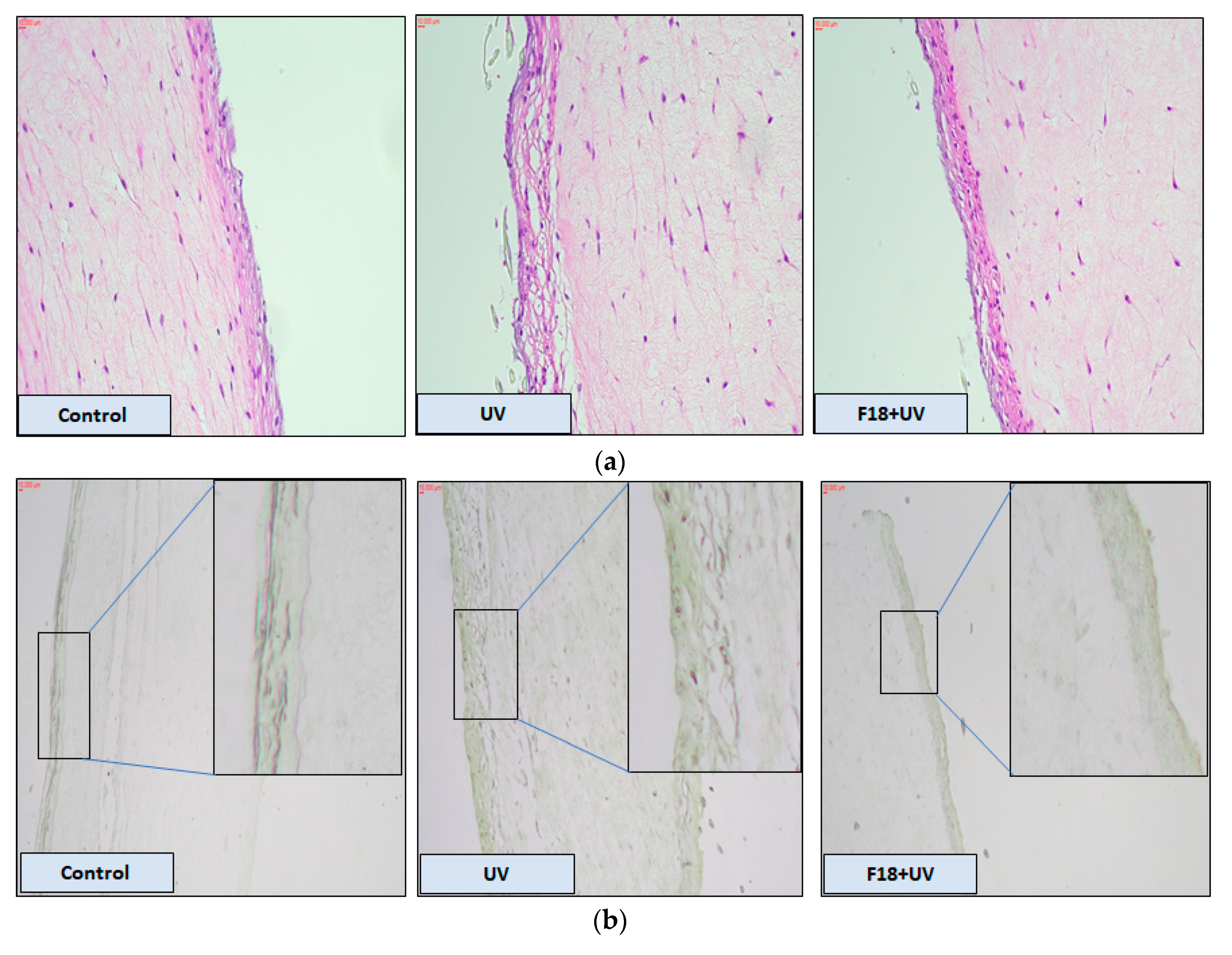
3.8. Effects of F18 on UV-Induced DNA Damage, Inflammation Markers and Apoptosis on Full-Thickness 3D Human Reconstituted Skin
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gruber, P.; Shah, M.; Zito, P.M. Cancer, Skin (Integument). In StatPearls; Treasure Island, Caseel: London, UK, 2020. [Google Scholar]
- Lalotra, A.S.; Singh, V.; Khurana, B.; Agrawal, S.; Shrestha, S.; Arora, D. A Comprehensive Review on Nanotechnology-Based Innovations in Topical Drug Delivery for Treatment of Skin Cancer. Curr. Pharm. Des. 2020. [Google Scholar] [CrossRef]
- Chang, A.; Yeung, S.; Thakkar, A.; Huang, K.M.; Liu, M.M.; Kanassatega, R.S.; Parsa, C.; Orlando, R.; Jackson, E.K.; Andresen, B.T.; et al. Prevention of skin carcinogenesis by the beta-blocker carvedilol. Cancer Prev. Res. 2015, 8, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.M.; Liang, S.; Yeung, S.; Oiyemhonlan, E.; Cleveland, K.H.; Parsa, C.; Orlando, R.; Meyskens, F.L., Jr.; Andresen, B.T.; Huang, Y. Topically Applied Carvedilol Attenuates Solar Ultraviolet Radiation Induced Skin Carcinogenesis. Cancer Prev. Res. 2017, 10, 598–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Liang, S.; Shahid, A.; Andresen, B.T.; Huang, Y. The beta-Blocker Carvedilol Prevented Ultraviolet-Mediated Damage of Murine Epidermal Cells and 3D Human Reconstructed Skin. Int. J. Mol. Sci. 2020, 21, 798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.S.; Lin, W.S.; Lin, C.L.; Kao, C.H. Carvedilol use is associated with reduced cancer risk: A nationwide population-based cohort study. Int. J. Cardiol. 2015, 184, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Rissmann, R.; Oudshoorn, M.H.; Hennink, W.E.; Ponec, M.; Bouwstra, J.A. Skin barrier disruption by acetone: Observations in a hairless mouse skin model. Arch. Dermatol. Res. 2009, 301, 609–613. [Google Scholar] [CrossRef] [Green Version]
- Elsayed, M.M.; Abdallah, O.Y.; Naggar, V.F.; Khalafallah, N.M. Lipid vesicles for skin delivery of drugs: Reviewing three decades of research. Int. J. Pharm. 2007, 332, 1–16. [Google Scholar] [CrossRef]
- Elsayed, M.M.; Abdallah, O.Y.; Naggar, V.F.; Khalafallah, N.M. Deformable liposomes and ethosomes as carriers for skin delivery of ketotifen. Die Pharm. 2007, 62, 133–137. [Google Scholar]
- El Zaafarany, G.M.; Awad, G.A.; Holayel, S.M.; Mortada, N.D. Role of edge activators and surface charge in developing ultradeformable vesicles with enhanced skin delivery. Int. J. Pharm. 2010, 397, 164–172. [Google Scholar] [CrossRef]
- El Maghraby, G.M.; Williams, A.C.; Barry, B.W. Skin delivery of 5-fluorouracil from ultradeformable and standard liposomes in-vitro. J. Pharm. Pharmacol. 2001, 53, 1069–1077. [Google Scholar] [CrossRef]
- Arzani, G.; Haeri, A.; Daeihamed, M.; Bakhtiari-Kaboutaraki, H.; Dadashzadeh, S. Niosomal carriers enhance oral bioavailability of carvedilol: Effects of bile salt-enriched vesicles and carrier surface charge. Int. J. Nanomed. 2015, 10, 4797–4813. [Google Scholar] [CrossRef] [Green Version]
- Ghassemi, S.; Haeri, A.; Shahhosseini, S.; Dadashzadeh, S. Labrasol-Enriched Nanoliposomal Formulation: Novel Approach to Improve Oral Absorption of Water-Insoluble Drug, Carvedilol. AAPS PharmSciTech 2018, 19, 2961–2970. [Google Scholar] [CrossRef] [PubMed]
- Gannu, R.; Vishnu, Y.V.; Kishan, V.; Rao, Y.M. In vitro permeation of carvedilol through porcine skin: Effect of vehicles and penetration enhancers. PDA J. Pharm. Sci. Technol. 2008, 62, 256–263. [Google Scholar]
- Lademann, J.; Jacobi, U.; Surber, C.; Weigmann, H.J.; Fluhr, J.W. The tape stripping procedure--evaluation of some critical parameters. Eur. J. Pharm. Biopharm. 2009, 72, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Freitas, J.V.; Praca, F.S.; Bentley, M.V.; Gaspar, L.R. Trans-resveratrol and beta-carotene from sunscreens penetrate viable skin layers and reduce cutaneous penetration of UV-filters. Int. J. Pharm. 2015, 484, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Di Sotto, A.; Paolicelli, P.; Nardoni, M.; Abete, L.; Garzoli, S.; Di Giacomo, S.; Mazzanti, G.; Casadei, M.A.; Petralito, S. SPC Liposomes as Possible Delivery Systems for Improving Bioavailability of the Natural Sesquiterpene beta-Caryophyllene: Lamellarity and Drug-Loading as Key Features for a Rational Drug Delivery Design. Pharmaceutics 2018, 10, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kronberg, B.; Dahlman, A.; Carlfors, J.; Karlsson, J.; Artursson, P. Preparation and evaluation of sterically stabilized liposomes: Colloidal stability, serum stability, macrophage uptake, and toxicity. J. Pharm. Sci. 1990, 79, 667–671. [Google Scholar] [CrossRef]
- Lin, S.; Blankschtein, D. Role of the bile salt surfactant sodium cholate in enhancing the aqueous dispersion stability of single-walled carbon nanotubes: A molecular dynamics simulation study. J. Phys. Chem. B 2010, 114, 15616–15625. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Hua, S. Comparison of in vitro dialysis release methods of loperamide-encapsulated liposomal gel for topical drug delivery. Int. J. Nanomed. 2014, 9, 735–744. [Google Scholar] [CrossRef] [Green Version]
- Madan, J.R.; Khude, P.A.; Dua, K. Development and evaluation of solid lipid nanoparticles of mometasone furoate for topical delivery. Int. J. Pharm. Investig. 2014, 4, 60–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todorovic, V.S.; Vasovic, M.; Beetge, M.M.; van Zyl, A.W.; Kokovic, V. Stability Development of Immediately Loaded Hybrid Self-Tapping Implants Inserted in the Posterior Maxilla: 1-Year Results of a Randomized Controlled Trial. J. Oral Implantol. 2017, 43, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.O.; Wang, Y.; Stebbins, W.G.; Gao, D.; Zhou, X.; Phelps, R.; Lebwohl, M.; Wei, H. Photoprotective effect of isoflavone genistein on ultraviolet B-induced pyrimidine dimer formation and PCNA expression in human reconstituted skin and its implications in dermatology and prevention of cutaneous carcinogenesis. Carcinogenesis 2006, 27, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Kshirsagar, S.J.; Bhalekar, M.R.; Mohapatra, S.K. Development and evaluation of carvedilol-loaded transdermal drug delivery system: In-vitro and in-vivo characterization study. Drug Dev. Ind. Pharm. 2012, 38, 1530–1537. [Google Scholar] [CrossRef]
- Sapra, B.; Jain, S.; Tiwary, A.K. Effect of Asparagus racemosus extract on transdermal delivery of carvedilol: A mechanistic study. AAPS PharmSciTech 2009, 10, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Aboelwafa, A.A.; El-Setouhy, D.A.; Elmeshad, A.N. Comparative study on the effects of some polyoxyethylene alkyl ether and sorbitan fatty acid ester surfactants on the performance of transdermal carvedilol proniosomal gel using experimental design. AAPS PharmSciTech 2010, 11, 1591–1602. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipid | SA | Ratio | Size (nm) | PDI | Zeta Potential (mV) | EE (%) | ||
|---|---|---|---|---|---|---|---|---|
| Group 1 | F1 | DSPC | T-80 | 1:1:0.25 | 313.13 ± 3.26 | 0.28 ± 0.03 | −5.54 ± 1.45 | 99.8 |
| F2 | DSPC | T-80 | 1:2:0.25 | 341.65 ± 18.27 | 0.29 ± 0.02 | −3.20 ± 0.19 | 99.4 | |
| F3 | DSPC | T-80 | 1:3:0.25 | 341.40 ± 19.77 | 0.23 ± 0.00 | −2.35 ± 0.60 | 99.2 | |
| F4 | DSPC | T-80 | 1:4:0.25 | 619.14 ± 54.66 | 0.30 ± 0.02 | −2.16 ± 0.19 | 99.6 | |
| F5 | DSPC | T-80 | 1:5:0.25 | 639.15 ± 73.71 | 0.26 ± 0.02 | −1.89 ± 0.24 | 99.5 | |
| F6 | DSPC | T-80 | 1:1:0.5 | 370.11 ± 6.63 | 0.30 ± 0.01 | −5.10 ± 0.56 | 100.8 | |
| F7 | DSPC | T-80 | 1:2:0.5 | 319.73 ± 10.37 | 0.28 ± 0.02 | −2.06 ± 0.19 | 99.1 | |
| F8 | DSPC | T-80 | 1:3:0.5 | 246.26 ± 10.78 | 0.28 ± 0.01 | −1.85 ± 0.06 | 98.5 | |
| F9 | DSPC | T-80 | 1:4:0.5 | 364.77 ± 18.35 | 0.29 ± 0.01 | −1.86 ± 0.39 | 101.1 | |
| F10 | DSPC | T-80 | 1:5:0.5 | 365.96 ± 16.11 | 0.25 ± 0.01 | −1.61 ± 0.30 | 100.7 | |
| Group 2 | F11 | SPC | T-80 | 1:1:0.25 | 286.92 ± 1.52 | 0.29 ± 0.01 | 11.60 ± 0.93 | 99.6 |
| F12 | SPC | T-80 | 1:2:0.25 | 234.56 ± 3.37 | 0.27 ± 0.00 | 10.58 ± 1.42 | 97.2 | |
| F13 | SPC | T-80 | 1:3:0.25 | 213.46 ± 1.46 | 0.28 ± 0.01 | 12.53 ± 1.15 | 91.6 | |
| F14 | SPC | T-80 | 1:4:0.25 | 238.50 ± 3.14 | 0.28 ± 0.01 | 13.3 ± 0.96 | 100.3 | |
| F15 | SPC | T-80 | 1:5:0.25 | 240.41 ± 3.63 | 0.27 ± 0.01 | 13.07 ± 0.53 | 91.9 | |
| F16 | SPC | T-80 | 1:1:0.5 | 243.73 ± 37.47 | 0.30 ± 0.05 | 8.74 ± 0.24 | 99.5 | |
| F17 | SPC | T-80 | 1:2:0.5 | 204.55 ± 41.88 | 0.24 ± 0.03 | 15.13 ± 1.06 | 98.9 | |
| F18 | SPC | T-80 | 1:3:0.5 | 162.92 ± 9.57 | 0.26 ± 0.01 | 17.13 ± 0.29 | 99.8 | |
| Group 3 | F19 | HEPC | T-80 | 1:1:0.25 | 301.47 ± 7.33 | 0.25 ± 0.03 | −3.54 ± 0.88 | 98.8 |
| F20 | HEPC | T-80 | 1:2:0.25 | 380.62 ± 32.01 | 0.25 ± 0.03 | −1.01 ± 0.30 | 98.8 | |
| F21 | HEPC | T-80 | 1:3:0.25 | 214.38 ± 5.31 | 0.26 ± 0.01 | −2.85 ± 1.11 | 98.8 | |
| F22 | HEPC | T-80 | 1:1:0.5 | 477.29 ± 57.66 | 0.32 ± 0.02 | −1.81 ± 0.28 | 98.1 | |
| F23 | HEPC | T-80 | 1:2:0.5 | 272.68 ± 6.52 | 0.32 ± 0.01 | −1.11 ± 0.40 | 97.7 | |
| F24 | HEPC | T-80 | 1:3:0.5 | 381.12 ± 70.36 | 0.28 ± 0.02 | −1.20 ± 0.34 | 99.7 | |
| Group 4 | F25 | SPC | SC | 1:1:0.25 | 303.63 ± 2.90 | 0.27 ± 0.01 | 8.14 ± 0.81 | 97.6 |
| F26 | SPC | SC | 1:2:0.25 | 224.96 ± 2.62 | 0.26 ± 0.01 | 12.13 ± 0.24 | 98.8 | |
| F27 | SPC | SC | 1:3:0.25 | 213.14 ± 1.49 | 0.26 ± 0.01 | 12.33 ± 0.47 | 94.1 | |
| F28 | SPC | SC | 1:1:0.5 | 206.41 ± 5.51 | 0.30 ± 0.01 | 9.62 ± 0.19 | 96.9 | |
| F29 | SPC | SC | 1:2:0.5 | 217.44 ± 5.13 | 0.27 ± 0.01 | 12.53 ± 0.44 | 98.2 | |
| F30 | SPC | SC | 1:3:0.5 | 189.12 ± 2.23 | 0.27 ± 0.01 | 12.43 ± 0.28 | 91.9 |
| Lipid | SA | Ratio | Size (nm) | PDI | Zeta Potential (mV) | EE% | |
|---|---|---|---|---|---|---|---|
| F8 | DSPC | T-80 | 1:3:0.5 | 549.97 ± 25.11 | 0.31 ± 0.01 | 0.9 ± 0.1 | 53.9 |
| F18 | SPC | T-80 | 1:3:0.5 | 197.11 ± 4.68 | 0.3 ± 0.01 | 15.7 ± 0.7 | 69.7 |
| F21 | HEPC | T-80 | 1:3:0.25 | 2164.57 ± 162.45 | 0.34 ± 0.02 | −0.5 ± 0.8 | 93.1 |
| F30 | SPC | SC | 1:3:0.5 | 217.69 ± 3.8 | 0.3 ± 0.01 | 21 ± 0.9 | 46.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.; Shamim, M.A.; Shahid, A.; Yeung, S.; Andresen, B.T.; Wang, J.; Nekkanti, V.; Meyskens, F.L., Jr.; Kelly, K.M.; Huang, Y. Topical Delivery of Carvedilol Loaded Nano-Transfersomes for Skin Cancer Chemoprevention. Pharmaceutics 2020, 12, 1151. https://doi.org/10.3390/pharmaceutics12121151
Chen M, Shamim MA, Shahid A, Yeung S, Andresen BT, Wang J, Nekkanti V, Meyskens FL Jr., Kelly KM, Huang Y. Topical Delivery of Carvedilol Loaded Nano-Transfersomes for Skin Cancer Chemoprevention. Pharmaceutics. 2020; 12(12):1151. https://doi.org/10.3390/pharmaceutics12121151
Chicago/Turabian StyleChen, Mengbing, Md Abdullah Shamim, Ayaz Shahid, Steven Yeung, Bradley T. Andresen, Jeffrey Wang, Vijaykumar Nekkanti, Frank L. Meyskens, Jr., Kristen M. Kelly, and Ying Huang. 2020. "Topical Delivery of Carvedilol Loaded Nano-Transfersomes for Skin Cancer Chemoprevention" Pharmaceutics 12, no. 12: 1151. https://doi.org/10.3390/pharmaceutics12121151
APA StyleChen, M., Shamim, M. A., Shahid, A., Yeung, S., Andresen, B. T., Wang, J., Nekkanti, V., Meyskens, F. L., Jr., Kelly, K. M., & Huang, Y. (2020). Topical Delivery of Carvedilol Loaded Nano-Transfersomes for Skin Cancer Chemoprevention. Pharmaceutics, 12(12), 1151. https://doi.org/10.3390/pharmaceutics12121151