1. Introduction
The vaginal administration of various agents has several advantages over alternatives, such as the presence of rich blood supply and large surface area of the vagina, avoidance of the hepatic first-pass metabolism, possible self-insertion and removal of the dosage form, and the ability to achieve high local drug concentration [
1]. The vagina has been used to deliver drugs for a range of clinical and research applications, including contraception, vaginal infections, and HIV (human immunodeficiency virus) prevention, with many different vaginal formulations, such as gels, creams, pessaries, suppositories, diaphragms, rings, films, tablets, and capsules. Furthermore, a number of these delivery systems have been investigated for the localised delivery of chemotherapeutic drugs to the cervix [
2]. The localized delivery of chemotherapeutic drugs offers a number of advantages over systemic administration, including: (1) direct delivery to the site of action, (2) a lower required dose, (3) limiting systemic drug toxicities, and (4) increased drug stability as it remains in the delivery device until released [
2].
Cervical cancer is a cancer of the cells located in the cervix [
3]. It is the third most common cancer in women globally and is mainly caused by the sexual transmission of the human papillomavirus (HPV) [
2,
3,
4,
5,
6]. The location of the cervix makes it easily accessible through the vagina and allows for a non-invasive localised delivery of chemotherapeutic drugs; adjacent to the cancerous tissue either before resection (neoadjuvant therapy), to reduce tumour size, or after resection (adjuvant therapy) to reduce the risk of recurrence [
7]. The use of local drug delivery systems for the treatment of metastatic cervical tumours may be inefficient when the disease is disseminated in distant organs, such as the lungs, in which case there may be a need for a more systemic approach. However, recent reports show that less than 20% of the cases appear with distant metastasis, which emphasizes that most cervical cancer patients would benefit from localised drug delivery systems [
8].
Each year, approximately 0.6 million women are diagnosed and almost 0.3 million deaths are attributed to cervical cancer [
9,
10]. More than 85% of that burden occurs in women living in low resource countries (developing countries), especially African countries, due to the lack of successful prevention and control programs against this disease [
11]. Alternative screening methods and treatments have been adopted to compensate for the lack of resources. Cervical cancer precursors are screened using visual inspection tests either in combination with 3–5% acetic acid (VIA) or Lugol’s iodine (VILI). The results are instantaneous and women who screen positive for precancerous lesions will be offered a treatment during the same visit [
12]. The most common treatment offered is cryotherapy, which is an ablation (destruction) method involving freezing the affected tissue. This screen and treat strategy can be performed at the primary care level by secondary healthcare workers with minimum supplies and equipment [
12,
13]. Despite the limitations of this concept, it helps to overcome barriers of time, distance, and loss to follow-up [
11].
Our contention is that a vaginal tablet chemotherapeutic formulation can be developed that would complement this current treatment strategy. After surgical treatment the patient will be supplied with a course of these vaginal tablets that can be easily self-administered. Similar to tablets intended for other administration routes, vaginal tablets show advantages including precise dosing, better drug stability, avoidance of antimicrobial agents for preservation, easier handling and storage, and low cost due to large scale production. Furthermore, self-administration is quite easy with no applicators or supervision needed [
14]. We developed a profile for this intervention that consists of a number of key criteria: (1) drug selection, (2) polymer selection, and (3) tablet manufacturing.
(1) Drug selection: A simple criteria for active drug selection was established that would be best suited for deployment, namely being low-cost and off-patent. As an antimetabolite drug, 5-fluorouracil (5-FU) has two mechanisms of action: (i) inhibition of the thymidylate synthase enzyme and (ii) misincorporation into DNA and RNA [
2]. It was developed more than 50 years ago and continues to be widely used in the treatment of cancer [
15,
16]. The topical formulation of 5-FU is used to treat skin cancer and HPV-related warts, lesions, and neoplasia [
2]. Moreover, 5-FU has a relatively short half-life [
17], which requires frequent doses. Dose-related side effects include severe nausea and vomiting, pain, and chronic ulceration. Studies have shown that less frequent or diluted doses have reported favourable side effect profiles [
18]. Therefore, to reduce the amount of 5-FU required, a co-drug was incorporated to produce a synergistic effect and increase safety and the efficacy of the formulation [
19]. One approach to expedite the development of novel formulations is to repurpose pre-existing drugs that have been approved for the treatment of other medical conditions [
20]. Disulfiram (DSF), a drug currently used in the treatment of chronic alcoholism, has been shown to possess anti-tumor activity [
21]. DSF can induce apoptosis in some cell lines and reduce cell growth in certain tumours including prostate cancer, breast cancer, lung cancer, leukaemia and cervical adenocarcinoma [
21,
22,
23]. Furthermore, its chemical structure has a high affinity towards copper, which is essential for the tumour angiogenesis processes, and contributes to making DSF selective to cancerous cells, thus sparing healthy cells [
24].
(2) Polymer selection—a readily available mucoadhesive polymer that can be utilised in low-cost direct compression of vaginal tablets is desirable. Currently available vaginal dosage forms have several limitations, such as leakage, messiness, and low residence time due to the self-cleansing action of the vaginal tract [
25]. Chitosan (CHN) and polyacrylic acid (PAA) are considered mucoadhesive polymers since they have shown an ability to bind to the vaginal mucosa, although remaining attached for different lengths of time [
25]. These polymers are polyelectrolytes that dissociate in aqueous solution into either polycations (CHN) or polyanions (PAA) [
26,
27], which then establish electrostatic interactions in the vaginal environment with the anionic moieties of mucin in the mucus [
14]. This interaction does not weaken over time [
28] and the polymers would degrade into non-toxic, absorbable subunits that can be metabolised once the drug supply is depleted [
8]. CHN produced by the
N-deacetylation of chitin, is composed of ᴅ-glucosamine and
N-acetyl-
d-glucosamine (deacetylated) randomly linked together by β-(1,4) glycosidic bonds [
29]. The percentage of deacetylated glucosamine units (number of ionisable units) along a CHN polymer chain is represented by its degree of deacetylation [
30]. CHN exhibits various degrees of deacetylation that contribute to varying molecular weights [
31], which are directly proportional to physical properties, such as mucoadhesion [
25,
28,
32]. For mucoadhesive formulations, the molecular weight of CHN should be not so high as to impair hydration and chain entanglement with mucins, but not so low as to give poor adhesion [
28]. Thus, a medium molecular weight is a suitable choice [
14]. The amino group in CHN has a pKa value of approximately 6.5, which allows it to dissolve in diluted aqueous acidic solvent with a pH lower than 6 [
32]. As the formulation to be developed is for vaginal delivery where the pH is 4.2–5 [
28], a fast dissolution rate or loss of formulations would be expected from CHN formulations [
33,
34,
35]. PAA exhibits strong hydrogen bonding with the mucin present in the mucosal layer. The hydrophilic nature and cross-linked structure of these polymers make them suitable for controlled drug delivery systems [
26]. Since PAA and CHN are differently charged, polyelectrolyte complex formation may reduce the fast dissolution rate observed in CHN-only formulations [
35].
(3) Tablet manufacturing—direct compression. Tablets account for nearly 80% of all marketed dosage forms due to economical and stability-related advantages over other dosage forms. Among the tablet-manufacturing processes, direct compression is the simplest and most cost-effective because it only involves blending and compression, as well as many other advantages, including significantly higher manufacturing efficiency and physical and chemical stability [
36]. Tablets kept at a smaller size offer a high drug loading capacity, reduce the risk of an inhomogeneous blend or non-uniform active pharmaceutical ingredient (API) (drug) content in finished tablets, and can also improve patient compliance. Most drug loading in direct compression tablet formulations does not exceed 30% because of poor processability, as most drugs have poor flowability and tabletability properties. These properties are critical to ensure the manufacturability of a direct compression tablet formulation [
36]. Tablet formulations are typically evaluated to comply with the requirement for tabletability, flowability, friability, disintegration time, tablet ejection force, and dissolution performance [
37,
38]. If any of these requirements are not met, the formulation is excluded, and a new formulation is designed and evaluated to address identified deficiencies. This process is repeated until an optimized formulation is identified [
36].
To date there are numerous formulations that combine 5-FU with DSF, and most bilayer tablets are formulated to provide an immediate drug release from one layer and a sustained or controlled drug release from the other. However, in this study we are exploiting the two layers to physically avoid chemical incompatibilities between the two drugs and to use the two layers to release the drugs concurrently for a synergistic effect in situ. The purpose of this present study is to evaluate the first generation of this intervention and to offer advice for future development.
2. Materials and Methods
2.1. Materials
Human cervical epidermoid carcinoma (Ca-Ski) cells were acquired from ATCC TCC® (CRL-1550™). Cell culture media and supplements were purchased from GIBCO™ sourced from Thermo Fisher Scientific (Cork, Ireland). Disulfiram (DSF) was obtained from Sigma Aldrich (Wicklow, Ireland). 5-fluorouracil (5-FU) was obtained from Flurorochem (Glossop, UK). Chitosan (medium molecular weight, ≥90% degree of acetylation) was obtained from Glentham (Corsham, UK). Poly(acrylic) acid was obtained from Lubrizol Advanced Materials (Westerlo, Belgium).
2.2. Evaluation of Powders (Pre-Compression)
A pre-weighed quantity of powder was placed in a measuring cylinder and the volume occupied was recorded as bulk volume. The bulk density was calculated in g/cm
3 using the formula in Equation (1). The powder was then placed in a measuring cylinder on a Copley Scientist (Nottingham, UK) tapped density voltmeter and tapped 1000 times. The tapped volume was measured, and the density was calculated in g/cm
3 using the formula in Equation (2). This procedure was repeated for all the powders used.
The calculated densities were used in computing the compressibility index and Hausner’s ratio using the formulas in Equations (3) and (4) respectively. The flow character of each powder was then evaluated according to
Table 1.
2.3. Preliminary Formulations and Preparation of Single Blends
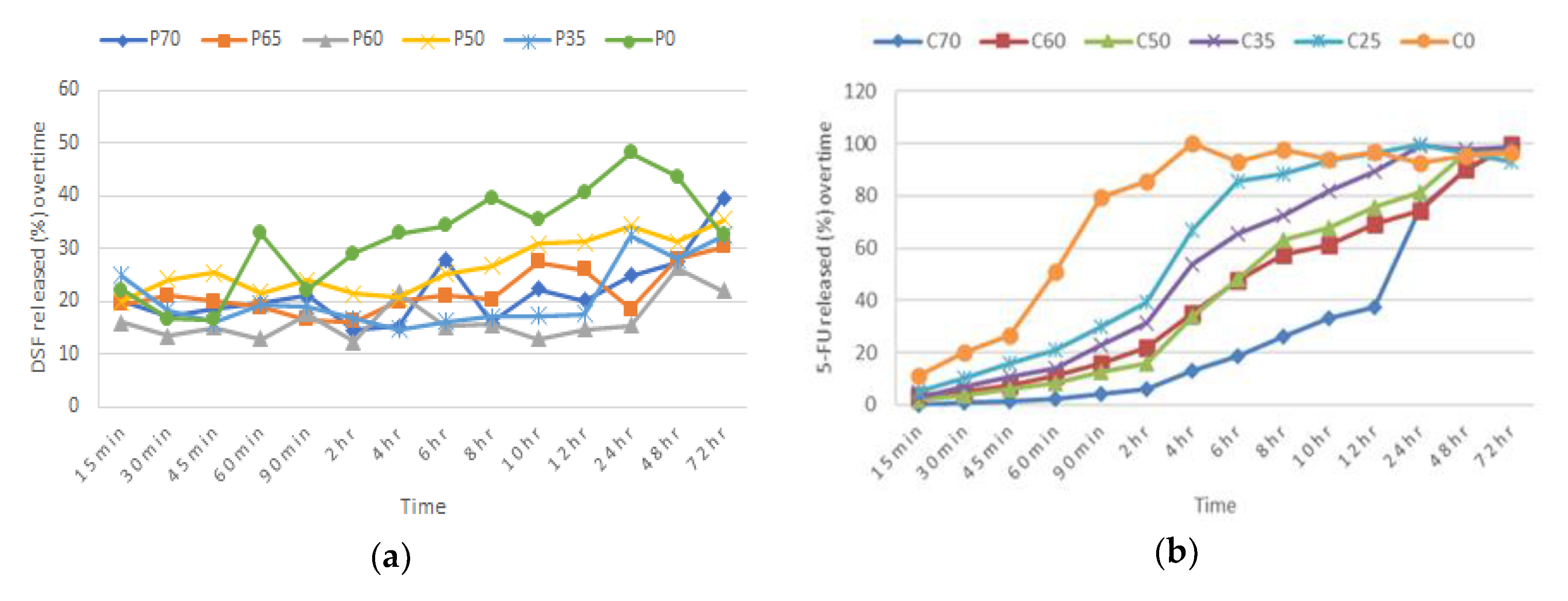
Various batches were prepared by varying the weight ratio of polymers to their respective drug, which was kept at 30%
w/
w, to identify the most effective formulations to make up the bilayer tablet. Each drug and polymer mixture (from this point on will be referred to as Blend 1 (DSF-PAA) and Blend 2 (5-FU-CHN) were separately prepared by homogeneously mixing the drug, polymer, and sorbitol (bulking agent) in a mortar for 15 min (
Table 2 and
Table 3). Each blend (150 mg) was then compressed using a 10-mm-diameter die in a single punch tablet press, to make a single blend tablet. Each tablet for both blends was pressed with a pressure of 3 tons for 30 s. An efficient formulation for each blend was determined according to the percentage of in vitro drug dissolution in simulated vaginal fluid (SVF), over 72 h and measured spectroscopically at 217 nm. Other parameters and conditions of the in vitro drug dissolution test were followed according to
Section 2.10.
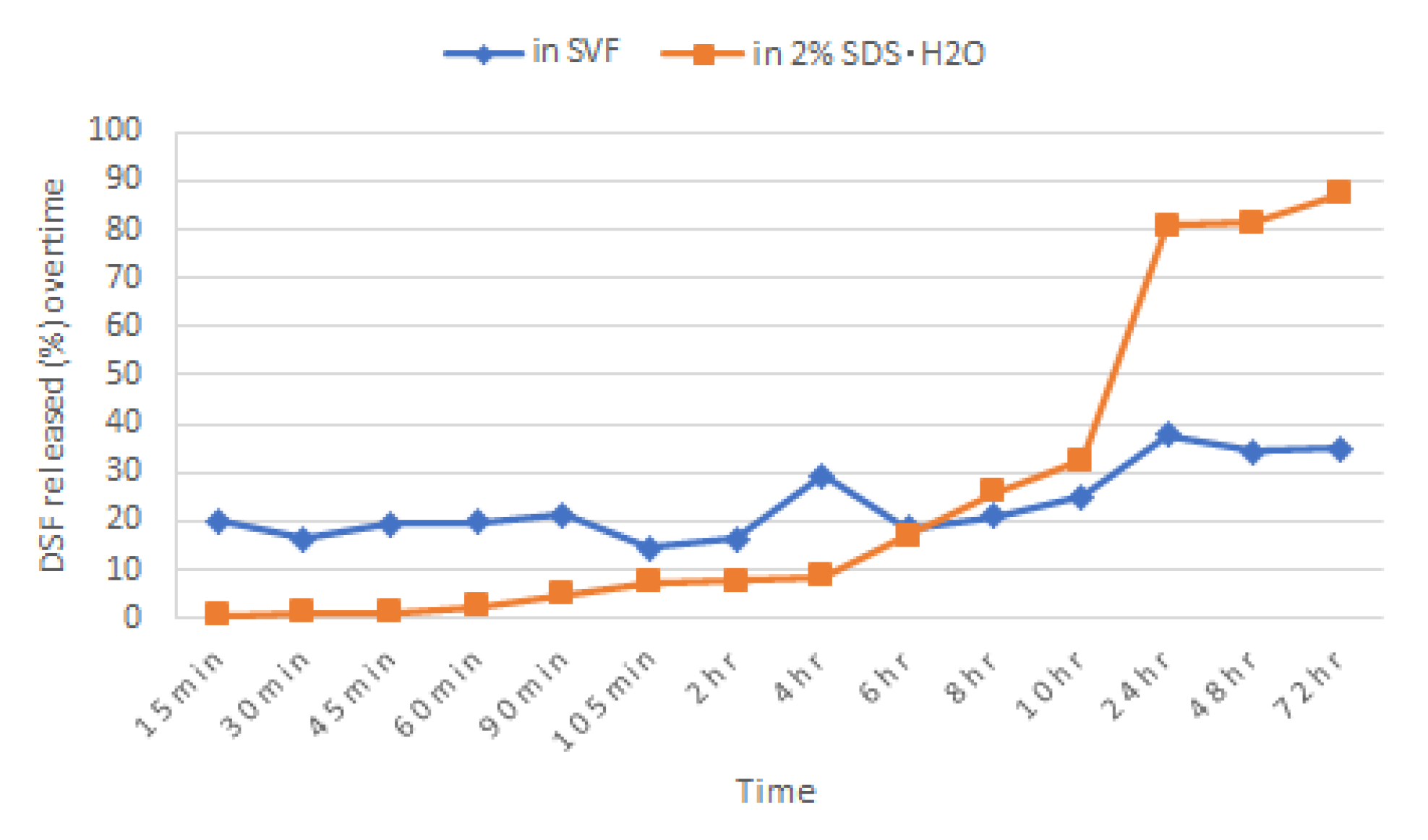
2.4. Dissolution of DSF Single Blend Tablet in 2% Sodium Dodecyl Sulphate (SDS) Aqueous Solution
Briefly, 2% (
w/
v) of sodium dodecyl sulphate (SDS) was added to distilled water and maintained at pH 4.2 by acetic acid to mimic the vaginal pH. A tablet of Blend 1 was immersed in 900 mL of this dissolution medium and other parameters and conditions of the in vitro drug dissolution test were followed according to
Section 2.10. Samples were taken and measured spectroscopically at 217 nm.
2.5. Preparation of Bilayer Tablet
Bilayer tablets were prepared by a direct compression procedure involving 2 steps. The formulation of blend 1 (150 mg) was compressed using a 10-mm-diameter die in a single punch machine press, with a pressure of 2 tons for 20 s. The upper punch was raised, and blend 2 formulation (150 mg) was then placed on top of the pressed layer; the 2 layers were then compressed into a bilayer tablet with a pressure of 3 tons for 30 s. In order to ensure good cohesion, the two layers were compressed with different pressures.
2.6. Physical Evaluation of Single Blend and Bilayer Tablets
2.6.1. Tablet Uniformity
Twenty tablets were randomly selected and weighed individually using an electric balance, and the thickness was measured using vernier calipers.
2.6.2. Hardness
Ten bilayer tablets were selected randomly and crushed individually between the anvils of the Pharmatron Schleuniger (Thun, Switzerland) Model 6D hardness tester. The energy required to break the individual tablets was recorded and the mean was calculated.
2.6.3. Friability
Twenty tablets were randomly selected, dusted and weighed (
W0) together using an electronic balance. The tablets were then placed into the drum of the friabilator and were rotated at 25 rpm for 4 min. The tablets were dusted and reweighed (
W). The degree of friability was calculated as percentage of weight loss using the formula in Equation (5), requiring that it be less than or equal to 1% to pass the test.
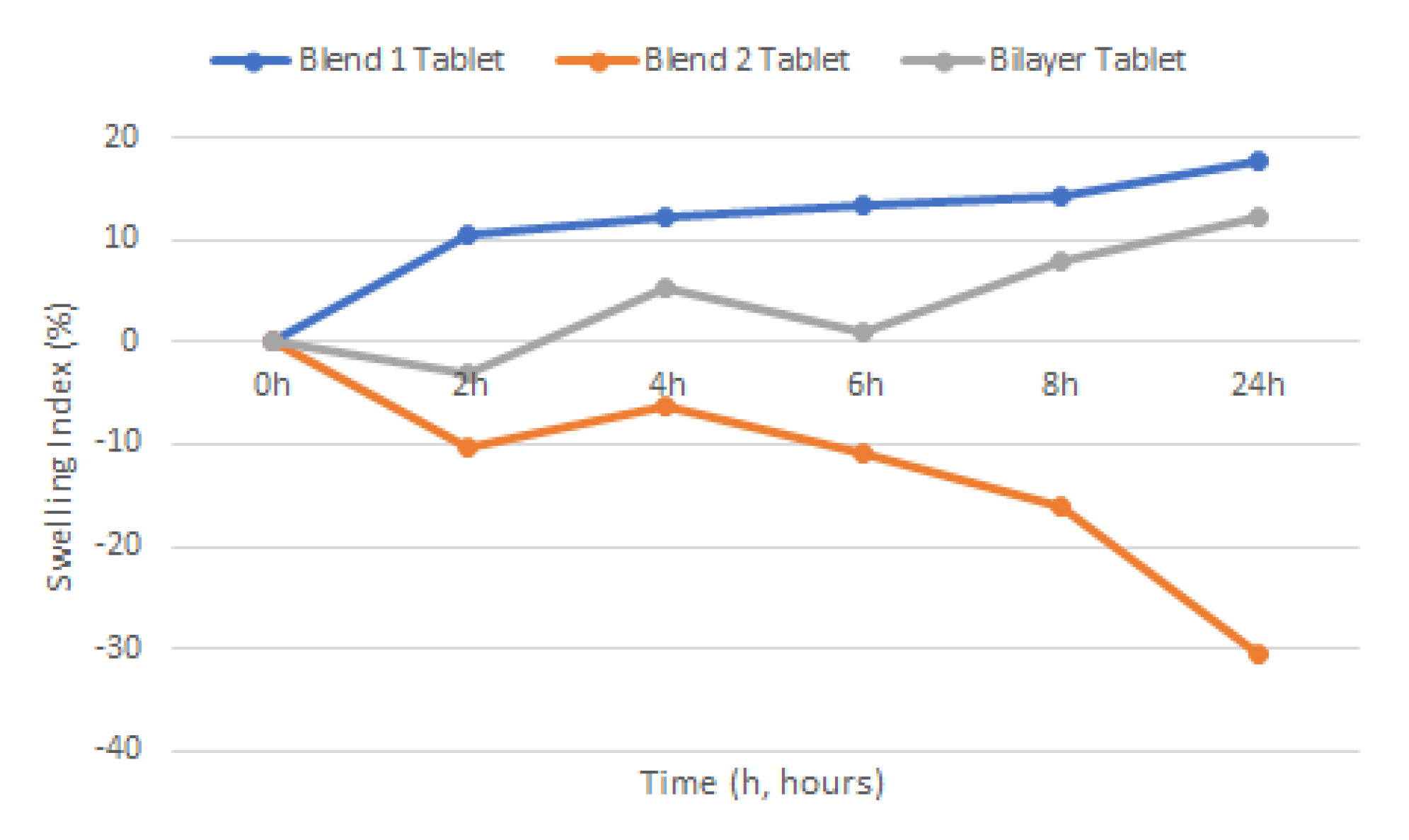
2.7. Swelling
Swelling study of the various tablets was carried out by submerging tablets in a steel basket into 25 mL of 2% SDS solution (pH 4.2) medium with the temperature maintained at 37 ± 1 °C. Weight of individual tablets was taken prior to the swelling study,
w1. Individual tablets were taken out at time intervals of 2, 4, 6, 8, and 24 h and left to dry in an oven at 60 °C for 24 h before being re-weighed (
w2). Swelling would be indicated by the weight gained by the tablets. Percent hydration (swelling index) was calculated using Equation (6).
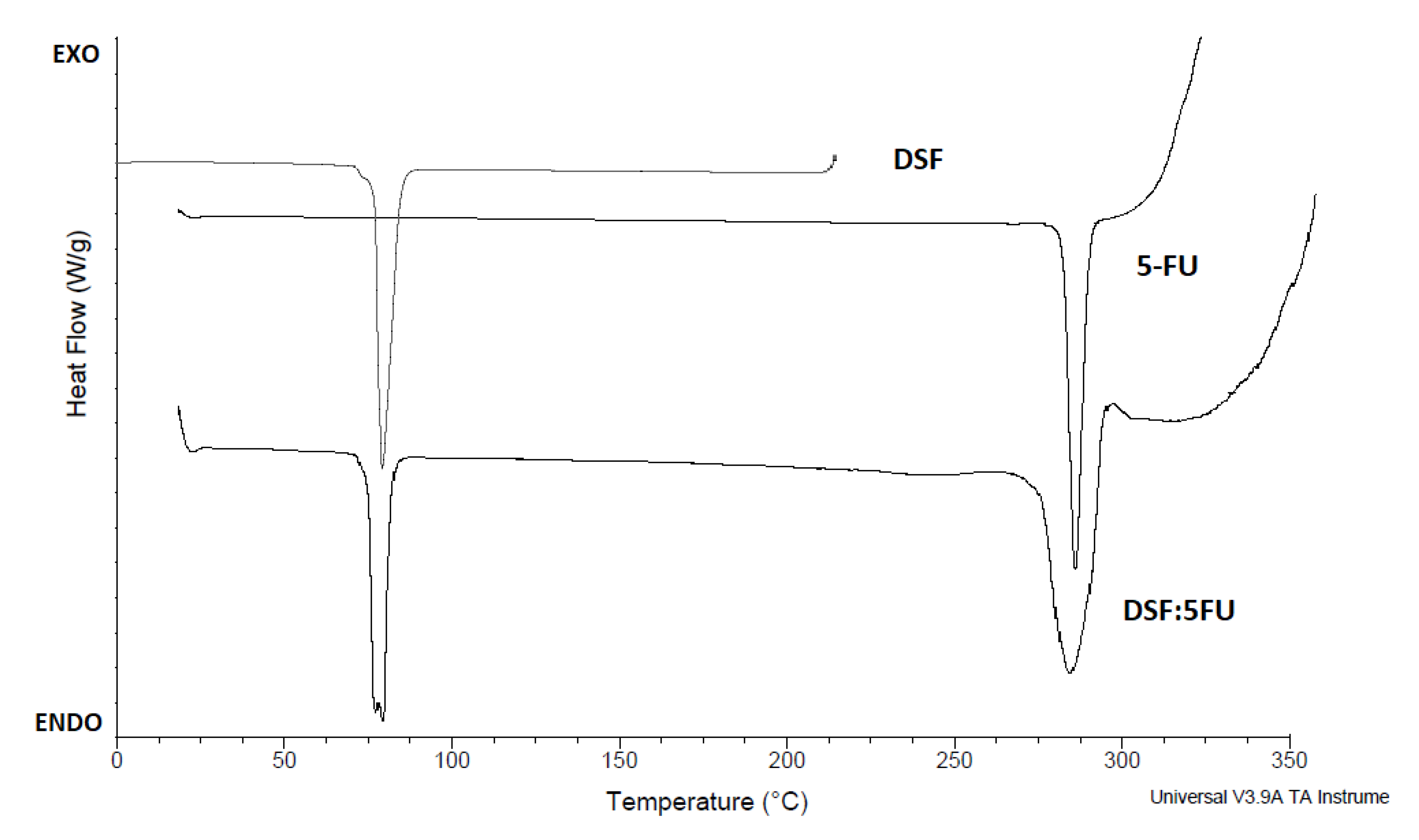
2.8. Differential Scanning Calorimetry (DSC)
Differential scanning calorimetry (DSC) thermograms were performed using a differential scanning calorimeter (Universal V3.9A TA Instruments, New Castle, DE, USA). Samples (8 mg) were placed into the covered aluminum pans and heated from 25 to 330 °C at a heating rate of 20 °C per minute. The scans were taken under a nitrogen atmosphere. An empty covered aluminum pan was used as the reference.
2.9. Scanning Electron Microscopy (SEM)
Scanning electron microscopy (SEM) was performed on a Tescan Mira SEM (Oxford Instruments, Abingdon, UK) using a range of magnifications to evaluate the surface morphology of the tablets and drug using the secondary electrons function. Tablets were snap broken through the transversal plane and cross-sectional areas placed under the microscope. This process was repeated for tablets submerged in liquid nitrogen for 10 min. As a first step, the samples were placed on an aluminum stub and were gold coated using Baltec SCD 005 sputter coater (BAL-TEC Gmbh, Pfäffikon, Switzerland) for 110 s at 0.1 mBar vacuum before observation.
2.10. Content Uniformity
Randomly chosen tablets of each formulation batch were weighed accurately, powdered, and 100 mg were dissolved in 100 mL of methanol. Each mixture was shaken at 100 rpm in an incubator overnight and kept at 37 ± 1 °C. Samples (1 mL) were suitably diluted with methanol and analyzed for drug content by a Shimadzu (Kyoto, Japan) UV spectrophotometer at 217 nm and 266 nm.
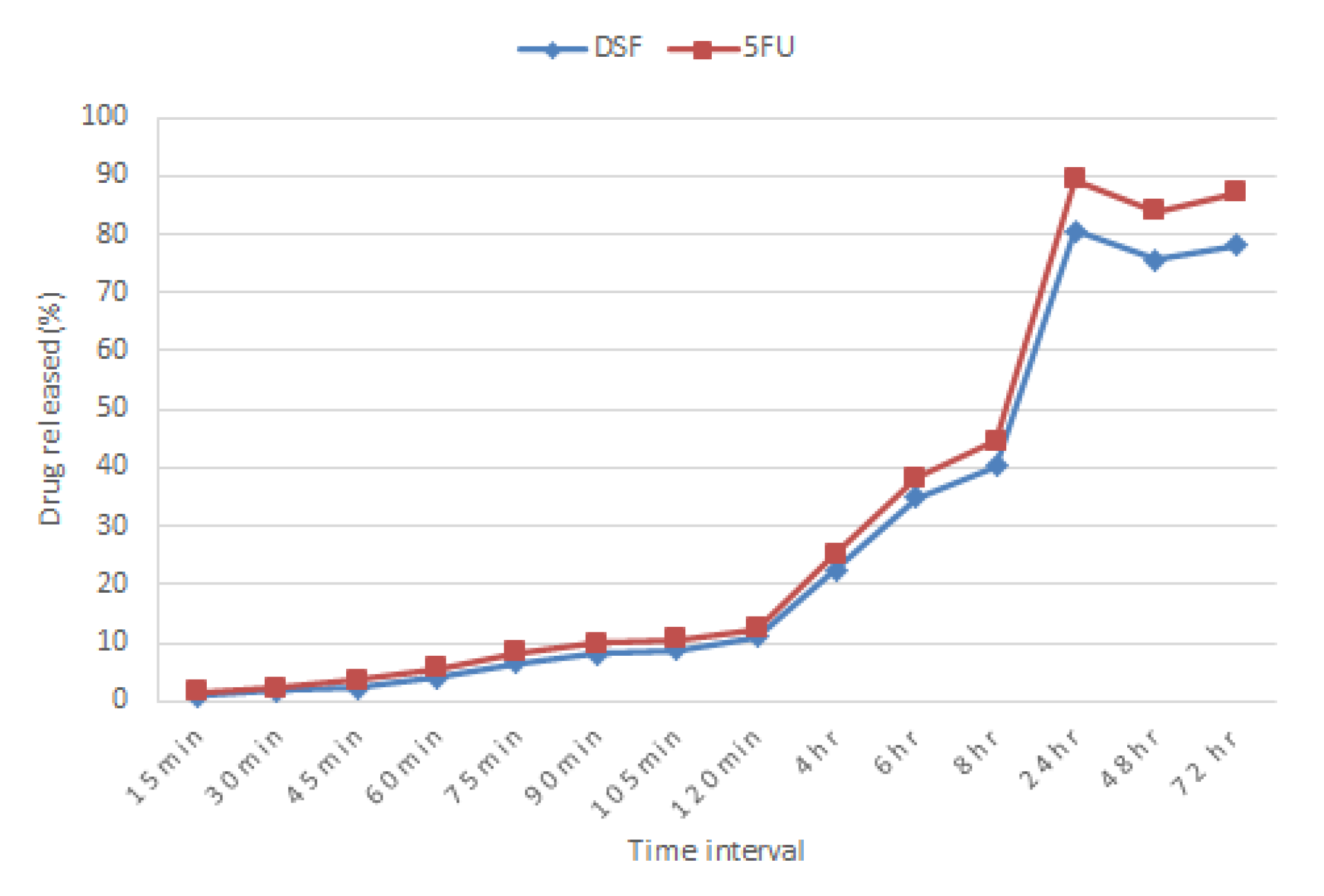
2.11. In Vitro Release Study
Using the paddle method (USP II method) with the rotation speed of 100 rpm and maintained at 37 ± 1 °C, tablets were immersed in 900 mL of 2% SDS solution (pH 4.2). Acetic acid was added into the medium solution to replicate the pH of the cervix at a reproductive age. Samples (5 mL) were withdrawn at 15, 30, 45-, 60-, 90-, and 105-min intervals, then at 2-, 4-, 6-, 8-, 24-, 48-, and 72-h intervals. Samples were assayed using a Shimadzu (Kyoto, Japan) UV spectrophotometer at 217 nm and 266 nm for DSF and 5-FU respectively.
Figure S1 shows a scan of UV spectrum of 5-FU, DSF and as a blend from 200 to 400 nm. An equal amount of sample withdrawn was replaced with fresh medium kept at the same temperature to maintain sink conditions. The cumulative percentage of drug released was calculated.
2.12. Ex Vivo Mucoadhesion Assessment
The assessment was performed as described by Cazorla-Luna et al. (2019, 2020) with modifications [
25,
39]. Ewe vaginal mucosa was obtained from Gilligan’s Farm (Four Mile House, Roscommon, Ireland) farming-and-butcher operations and used as a model for ex vivo adhesion. The mucosa was cut into fragments of 75 × 25 mm and fixed on glass slides with cyanoacrylate adhesive. Then, each tablet was placed in the center of the mucosa and pressed with a contact force of 500 g for 10 s. The slides were positioned at an angle of 60° and immersed in 45 mL of simulated vaginal fluid (
Table 4), prepared according to Owen and Katz (1999) [
40] and, posteriorly incubated at 36.5 °C at 30 rpm until total detachment. The adhesion time was determined by observation of the samples. All assays were performed in triplicate.
2.13. Cell Culture Studies
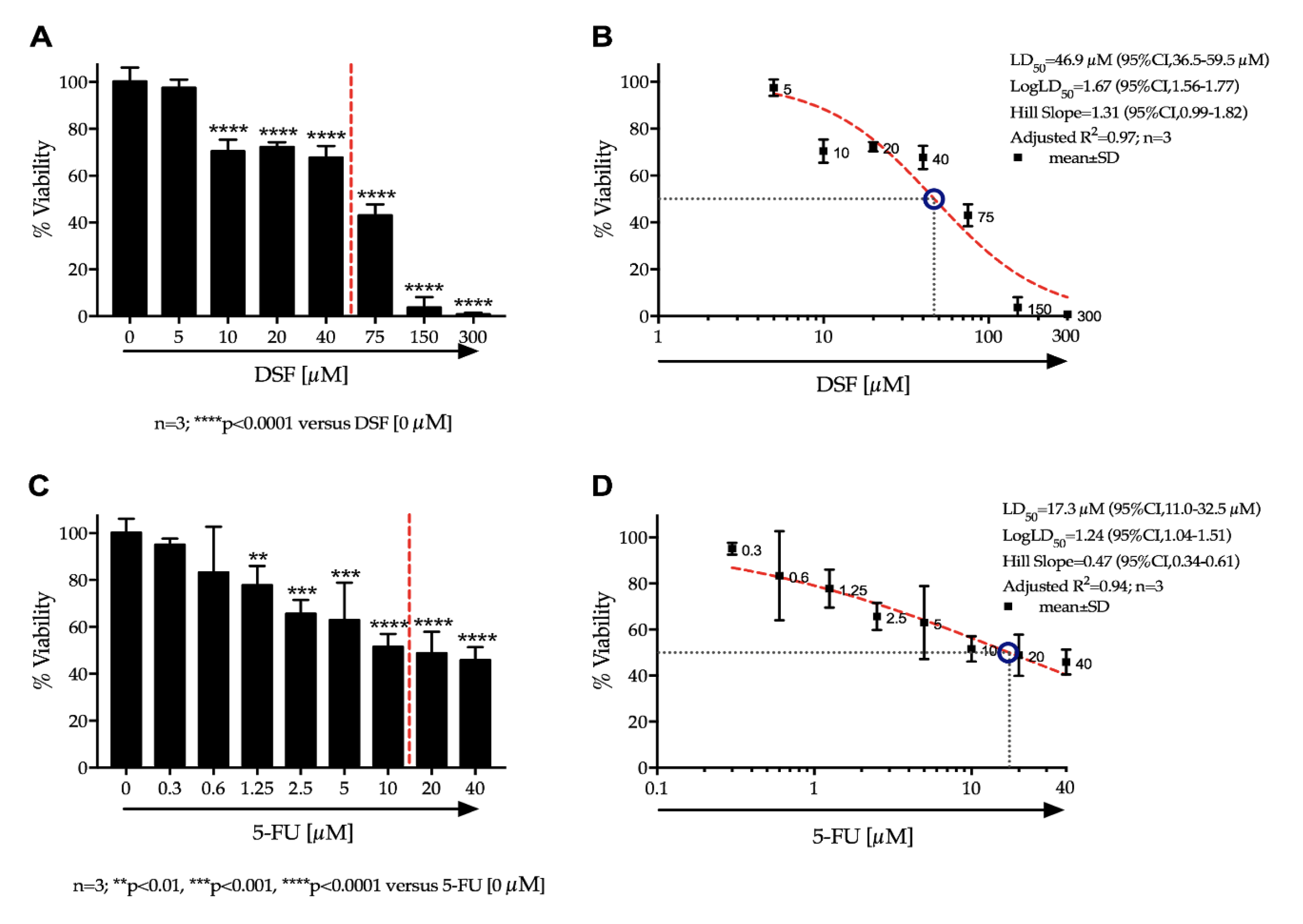
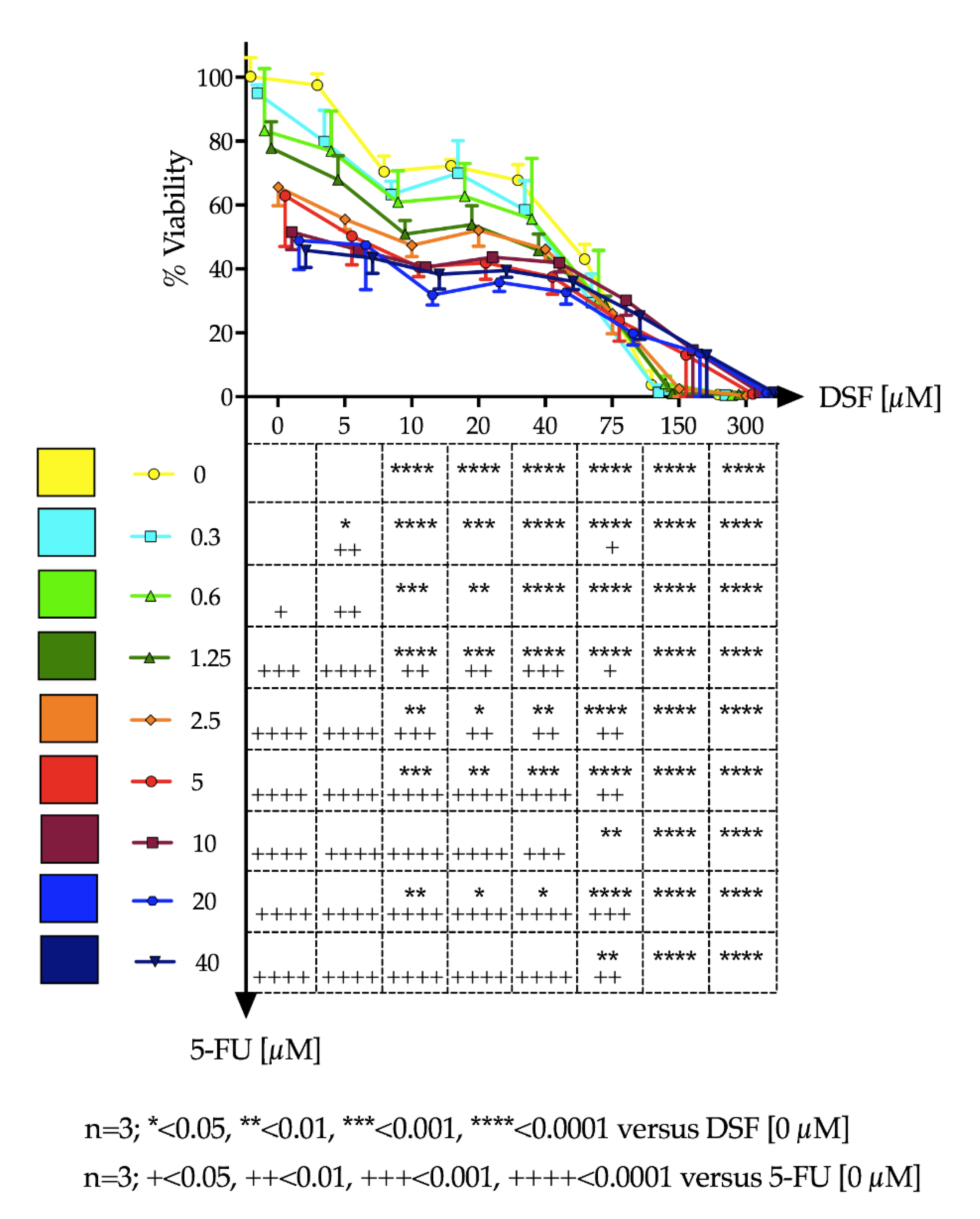
Ca-Ski cells were cultured in humidified incubators (37 °C, 5% CO2) in RPMI media supplemented with 10% fetal calf serum (FCS), 1% L-glutamine and 1% penicillin/streptomycin. For experiments, Ca-Ski cells were seeded onto 96 well plates at a density of 7 × 103 cells/well per well and cultured until 60–70% confluence was reached. Media was replaced and cells were treated with varying drug concentrations of DSF and 5-FU independently and in combination. 5-FU and DSF solutions were prepared in dimethyl sulfoxide (DMSO). Stock preparations were made and diluted down at least 100 times in growth media before addition to cells, in order to prevent cytotoxic effects of DMSO. After 48 h, test solutions were removed, and cells were gently washed three times with Dulbecco’s phosphate-buffered saline (DPBS). Cells were incubated with 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) dissolved in DPBS at a final concentration of 0.5 mg/mL. After 3 h the MTT solution was removed. 0.1 mL DMSO was added to each well to dissolve the formazan created by viable cells. Absorbance was read using a Synergy Multi-plate reader at 540 nm. Percentage cell-viability (% Viability) was calculated using absorbance readings of treatment wells against that of the negative control wells.
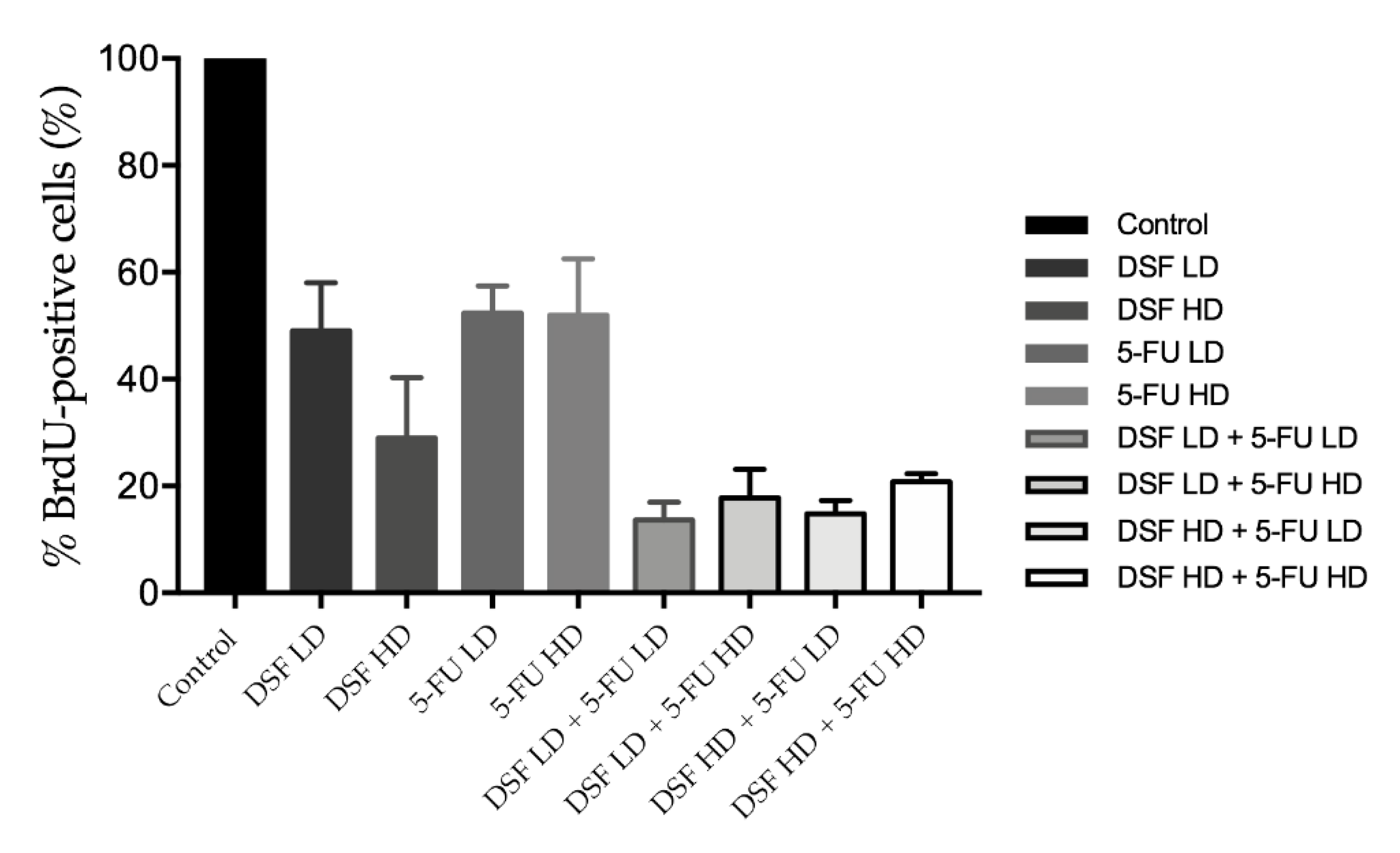
2.14. BrdU Assay
Cells were seeded in 96 well plates at a density of 7 × 103 cells/well (0.1 mL final volume) for 24 h. Cells were then treated with low (40 µM) or high (75 µM) dose DSF; low (10 µM) or high (20 µM) dose 5-FU. To determine additive effect cells were also treated with drug combinations which included: (1) low dose DSF combined with low dose 5-FU; (2) high dose DSF with high dose 5-FU; (3) low dose DSF with high dose 5-FU; and (4) high dose DSF with low dose 5-FU). As a control, a group of cells was kept untreated. Cells were treated for 24 h and subjected to the Cell Proliferation ELISA, BrdU (colorimetric) assay (Roche, Switzerland), following the manufacturer’s protocol. Briefly, cells were incubated for two hours with the BrdU labeling solution, fixed and treated with anti-BrdU-POD solution for 90 min, washed three times with phosphate saline buffer, and incubated with substrate solution for 30 min for the development of color. Absorbance was read at 370 nm in a Synergy™ HTX (BioTek®, Winooski, VT, USA) plate reader. Results were plotted as % BrdU positive cells which represents the population of proliferating cells.
2.15. Statistical Data Analysis
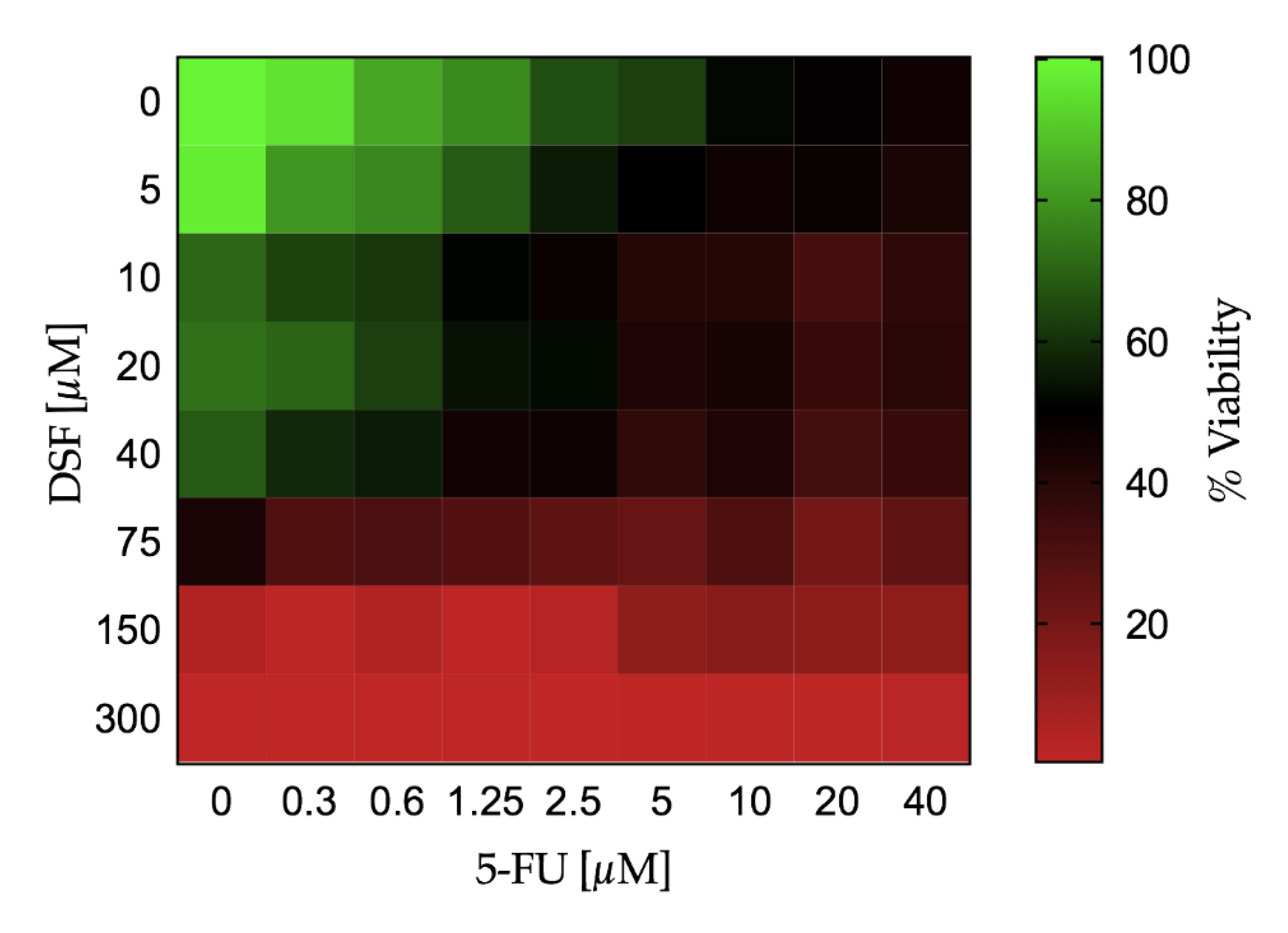
Continuous variables were expressed as mean ± standard deviation (SD). The change in cell viability and cell proliferation over different doses of DSF or 5-FU was tested by a one-way analysis of variance (ANOVA). If the one-way ANOVA reached statistical significance, pairwise comparisons were performed among different doses of the study drug versus no drug administration (i.e., PBS) by the Fisher’s least significant difference (LSD) test. The effect on cell viability and the BrdU uptake assay combining both DSF and 5-FU at different doses was evaluated by a two-way ANOVA. If the two-way ANOVA reached statistical significance, pairwise comparisons were performed among different dose combination of the study drug versus no drug administration (i.e., PBS) by the Fisher’s LSD test. The effect of dose increase of 5-FU in combination with DSF on the cell viability was depicted using a Karnaugh color map and a heat map. The LD50 with 95% confidence interval for DSF and 5-FU was estimated using a non-linear regression by least squares ordinary fit. Goodness of fit was represented by adjusted R square. A p-value < 0.05 (two-tailed) was deemed statistically significant. Statistical analyses were performed using STATA-14/MP (StataCorp LP, College Station, TX, USA), GraphPad Prism 7a (GraphPad Software, San Diego, CA, USA), and Microsoft Excel for Mac 2017, Version 15.32 (Microsoft, Redmond, WA, USA).

 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}