Long-Lasting Exendin-4 Fusion Protein Improves Memory Deficits in High-Fat Diet/Streptozotocin-Induced Diabetic Mice

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Expression and Purification of ExA
2.3. Animal Care
2.4. Pharmacokinetic (PK) Studies
2.5. Induction of T2DM and Experimental Design
2.6. Echo-MRI
2.7. Glucose Tolerance Test (GTT)
2.8. Measurement of Metabolic Parameters
2.9. Tissue Collection and Histological Examination
2.10. Immunofluorescence
2.11. Duolink Immunofluorescence
2.12. Western Blot Analysis
2.13. Morris Water Maze (MWM) Test
2.14. Statistical Analysis
3. Results
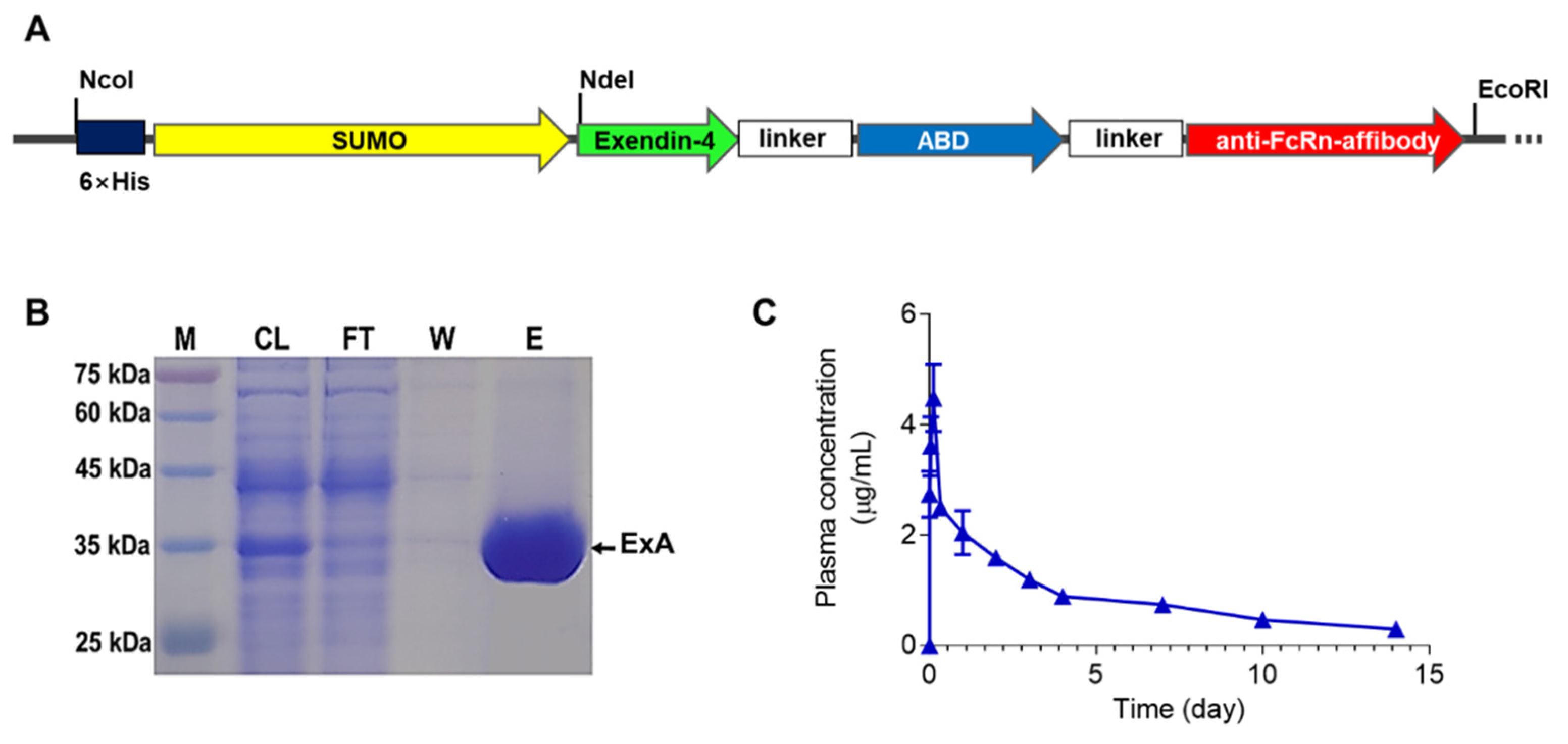
3.1. Expression, Purification, and Pharmacokinetic (PK) Evaluation of ExA
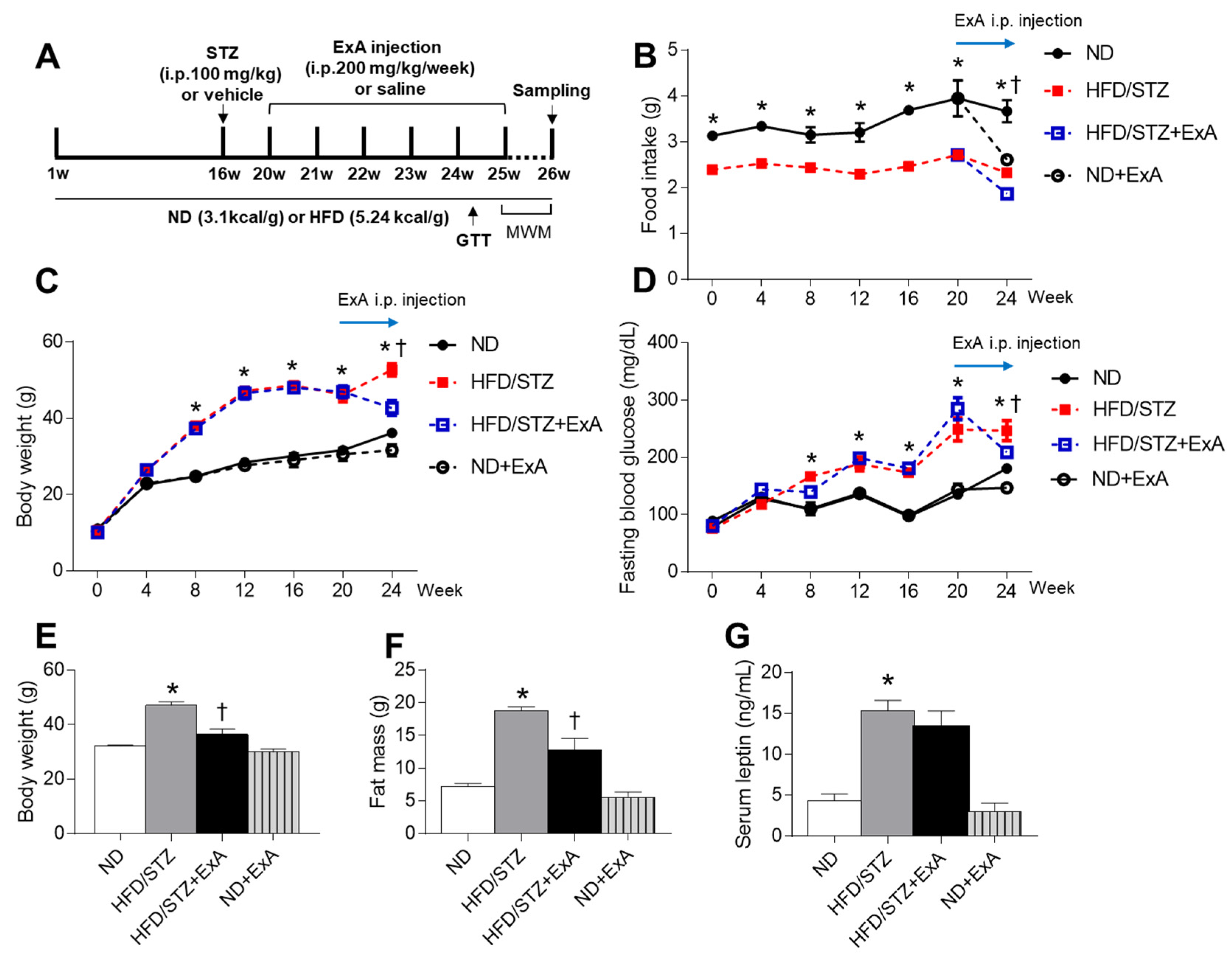
3.2. Effects of ExA Treatment on Food Intake, Body Weight, and Blood Glucose Levels in HFD/STZ Mice
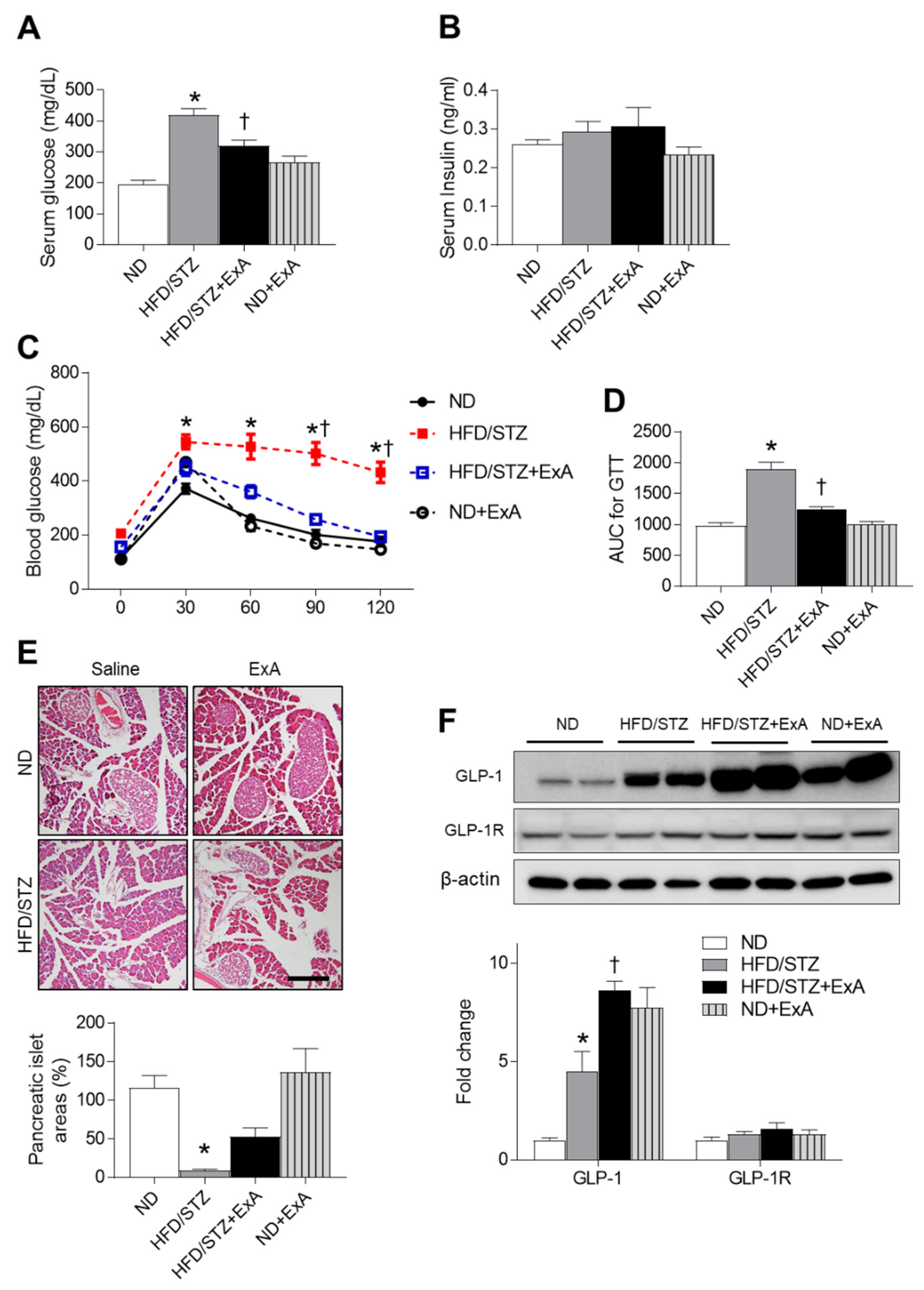
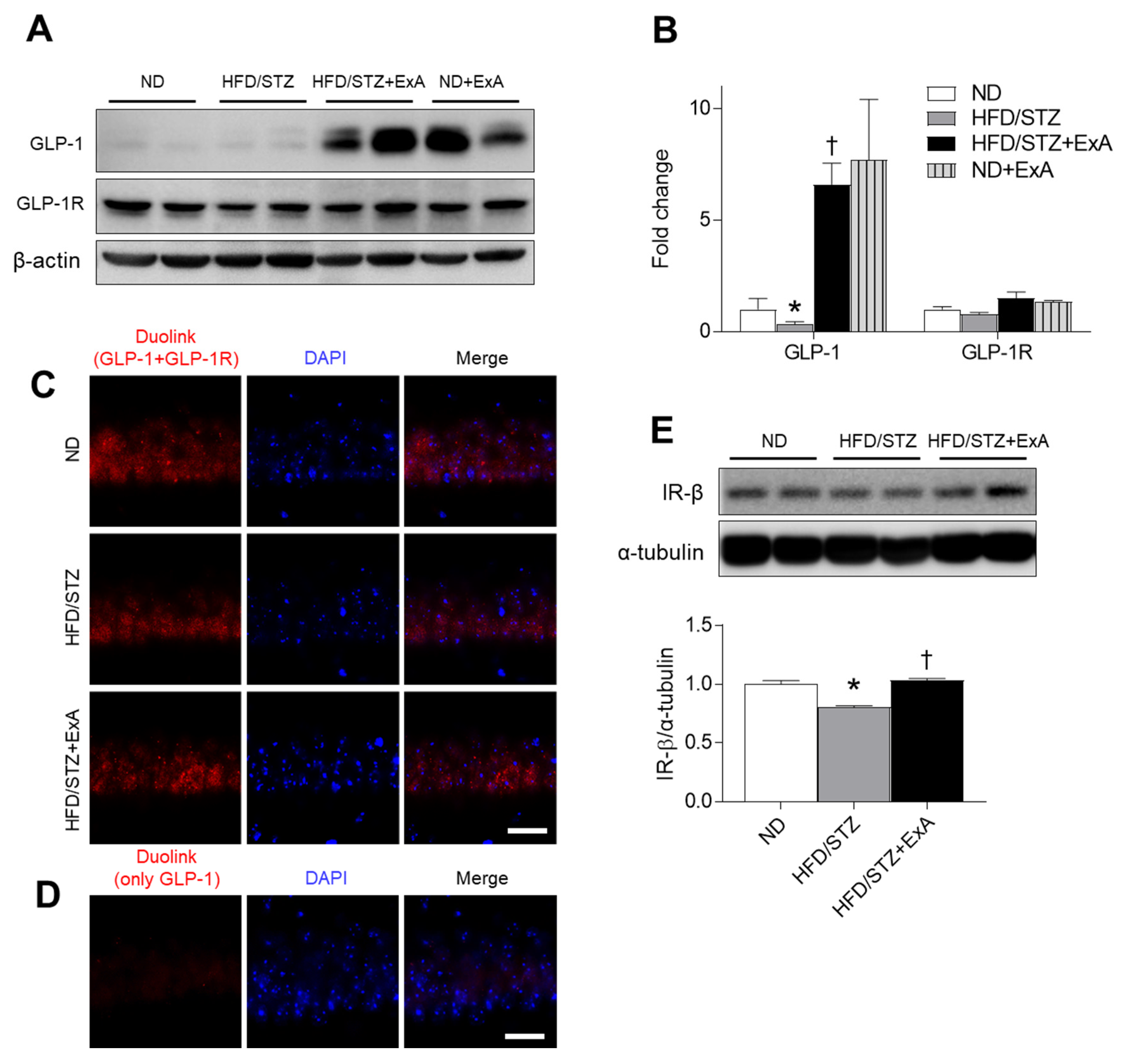
3.3. Effects of ExA Treatment on Insulin Resistance and Pancreatic GLP-1/GLP-1R Expression Levels in HFD/STZ Mice
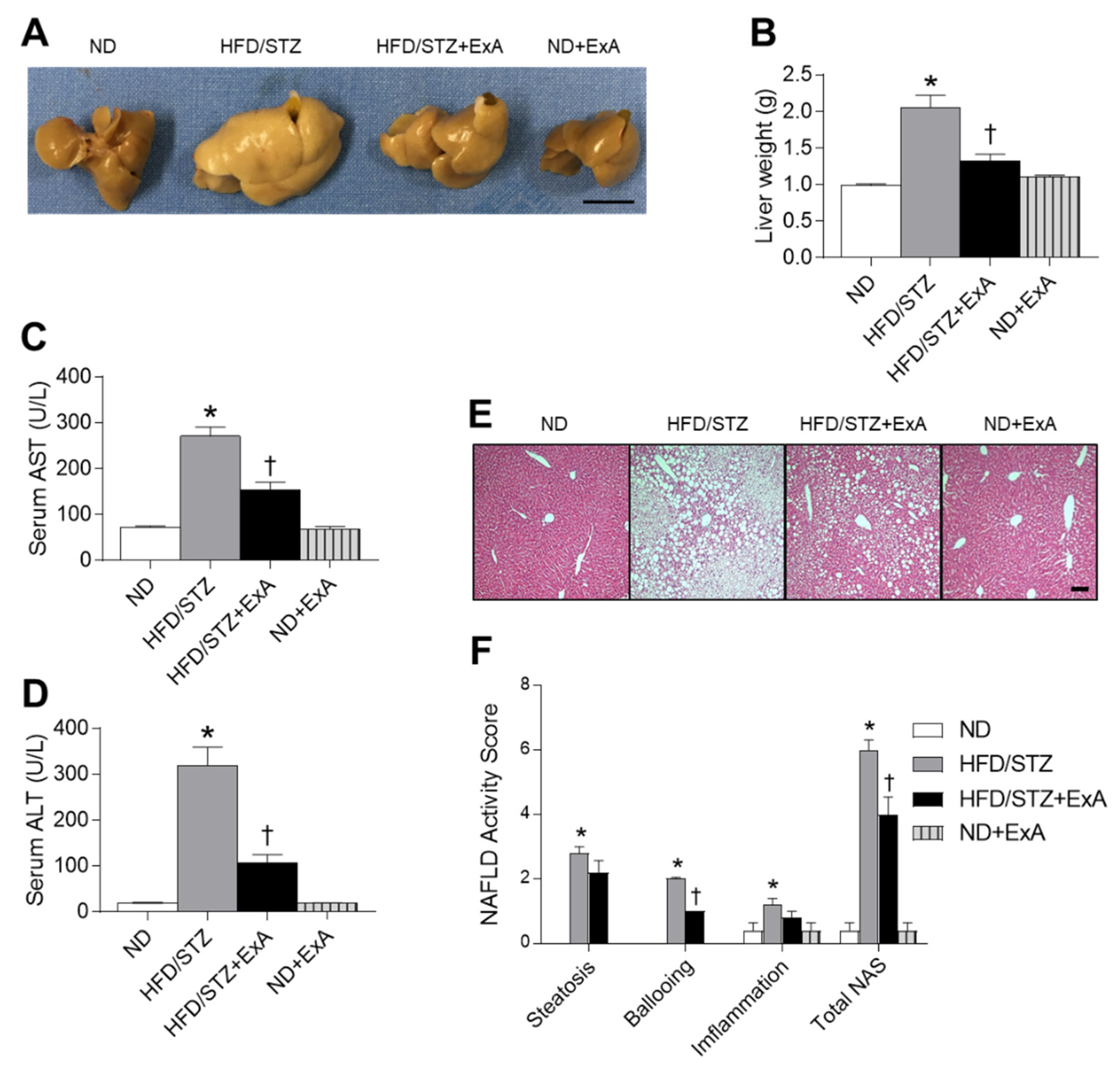
3.4. Effects of ExA Treatment on Hepatic Steatosis in HFD/STZ Mice
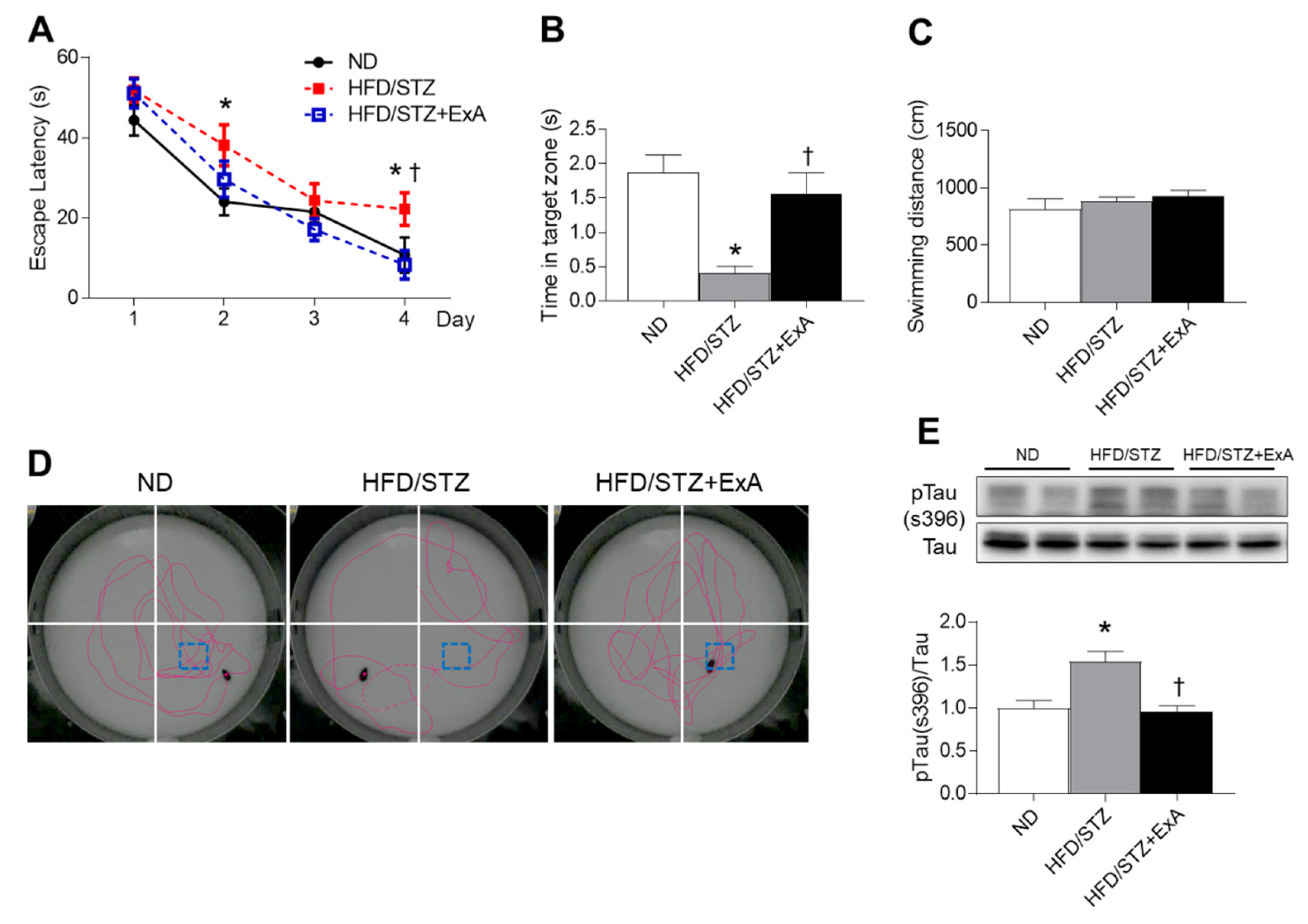
3.5. Effects of ExA Treatment on Memory Deficits and Tau Phosphorylation in HFD/STZ Mice
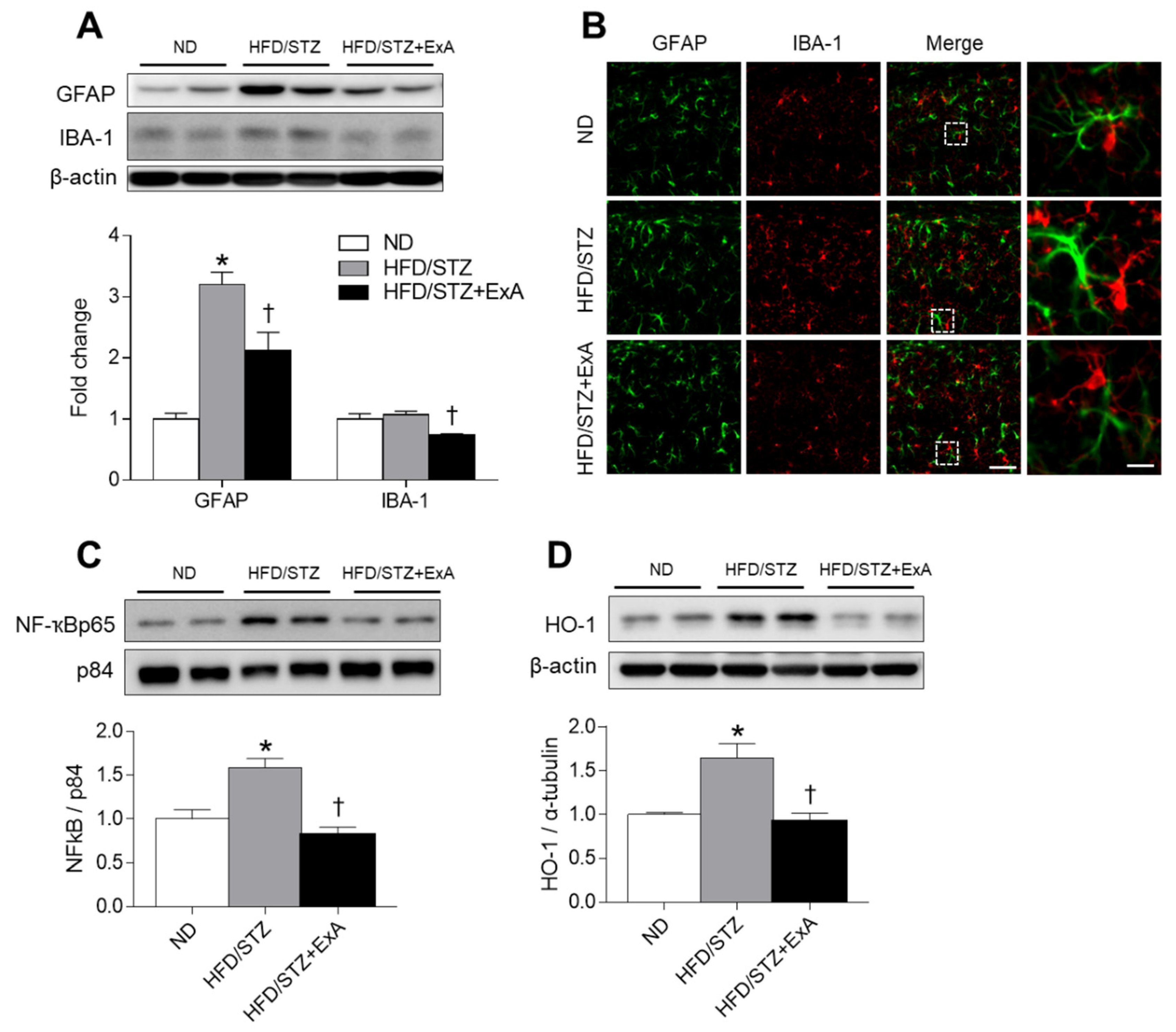
3.6. Effects of ExA Treatment on Neuroinflammation and HO-1 Expression in HFD/STZ Mice
3.7. Effects of ExA Treatment on GLP-1/GLP-1R Activation and Insulin Receptor (IR)-β Expression in HFD/STZ Mice
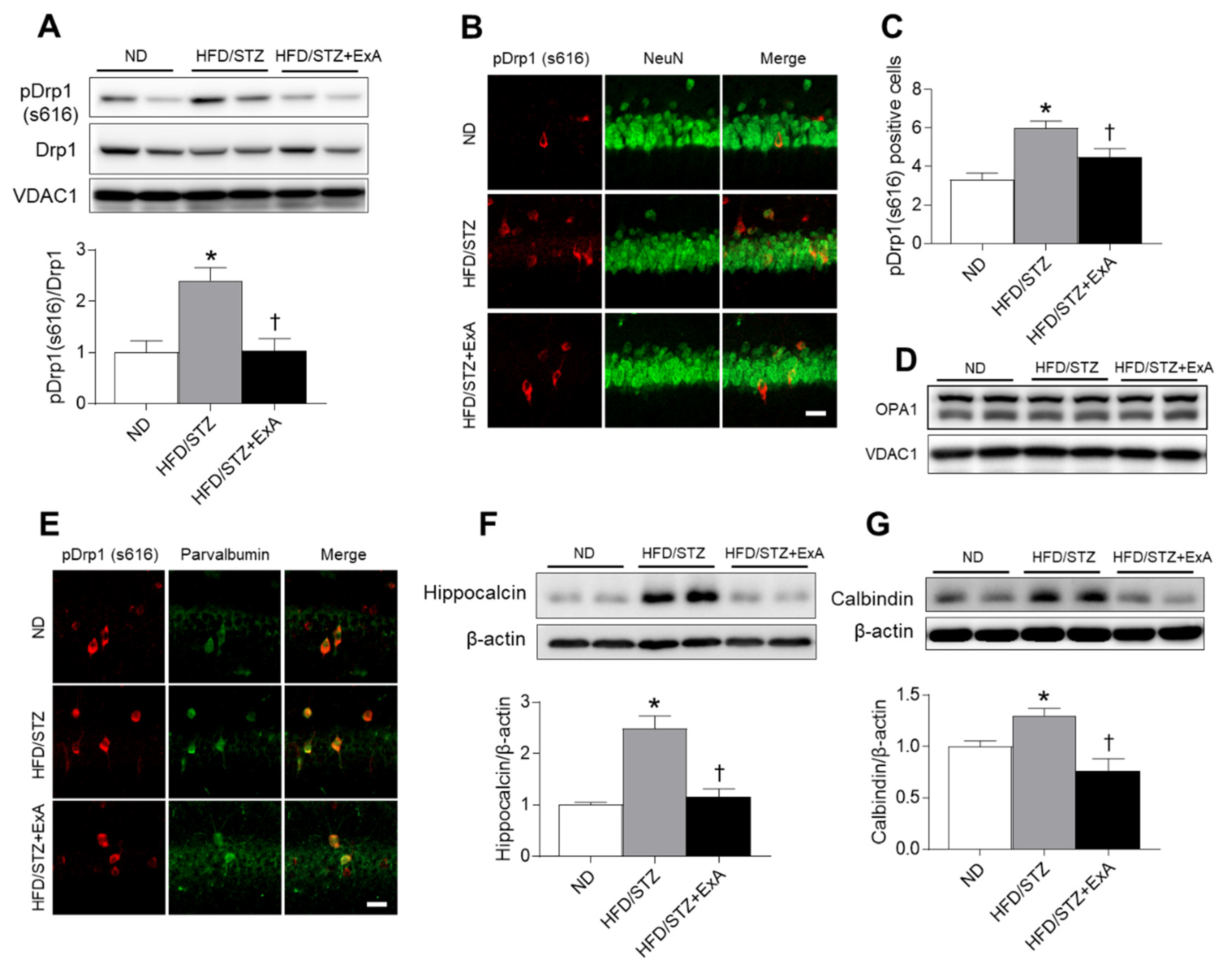
3.8. Effects of ExA Treatment on Hippocampal Mitochondrial Fission and Calcium-Binding Protein (CaBP) Expression in HFD/STZ Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Feinkohl, I.; Price, J.F.; Strachan, M.W.; Frier, B.M. The impact of diabetes on cognitive decline: Potential vascular, metabolic, and psychosocial risk factors. Alzheimers Res. Ther. 2015, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Luchsinger, J.A. Type 2 diabetes and cognitive impairment: Linking mechanisms. J. Alzheimers Dis. 2012, 30, S185–S198. [Google Scholar] [CrossRef] [PubMed]
- Vagelatos, N.T.; Eslick, G.D. Type 2 diabetes as a risk factor for Alzheimer’s disease: The confounders, interactions, and neuropathology associated with this relationship. Epidemiol. Rev. 2013, 35, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Monte, S.M. Insulin resistance and Alzheimer’s disease. BMB Rep. 2009, 42, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Turton, M.D.; O’Shea, D.; Gunn, I.; Beak, S.A.; Edwards, C.M.; Meeran, K.; Choi, S.J.; Taylor, G.M.; Heath, M.M.; Lambert, P.D.; et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature 1996, 379, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Oka, J.I.; Goto, N.; Kameyama, T. Glucagon-like peptide-1 modulates neuronal activity in the rat’s hippocampus. Neuroreport 1999, 10, 1643–1646. [Google Scholar] [CrossRef]
- Kieffer, T.J.; Habener, J.F. The glucagon-like peptides. Endocr. Rev. 1999, 20, 876–913. [Google Scholar] [CrossRef]
- Yang, Y.; Fang, H.; Xu, G.; Zhen, Y.; Zhang, Y.; Tian, J.; Zhang, D.; Zhang, G.; Xu, J. Liraglutide improves cognitive impairment via the AMPK and PI3K/Akt signaling pathways in type 2 diabetic rats. Mol. Med. Rep. 2018, 18, 2449–2457. [Google Scholar] [CrossRef]
- Heppner, K.M.; Perez-Tilve, D. GLP-1 based therapeutics: Simultaneously combating T2DM and obesity. Front. Neurosci. 2015, 9, 92. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Zhou, Y.; Zhang, M.; Xie, Y.; Ke, S.; Liu, L.; Pan, X.; Chen, Z. Exenatide alleviates mitochondrial dysfunction and cognitive impairment in the 5xFAD mouse model of Alzheimer’s disease. Behav. Brain Res. 2019, 370, 111932. [Google Scholar] [CrossRef]
- Duarte, A.I.; Candeias, E.; Correia, S.C.; Santos, R.X.; Carvalho, C.; Cardoso, S.; Placido, A.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Crosstalk between diabetes and brain: Glucagon-like peptide-1 mimetics as a promising therapy against neurodegeneration. Biochim. Biophys. Acta 2013, 1832, 527–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gault, V.A.; Porter, W.D.; Flatt, P.R.; Holscher, C. Actions of exendin-4 therapy on cognitive function and hippocampal synaptic plasticity in mice fed a high-fat diet. Int. J. Obes. (Lond) 2010, 34, 1341–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onoviran, O.F.; Li, D.; Toombs Smith, S.; Raji, M.A. Effects of glucagon-like peptide 1 receptor agonists on comorbidities in older patients with diabetes mellitus. Adv. Chronic. Dis. 2019, 10, 2040622319862691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastin, A.J.; Akerstrom, V.; Pan, W. Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier. J. Mol. Neurosci. 2002, 18, 7–14. [Google Scholar] [CrossRef]
- Seijsing, J.; Lindborg, M.; Hoiden-Guthenberg, I.; Bonisch, H.; Guneriusson, E.; Frejd, F.Y.; Abrahmsen, L.; Ekblad, C.; Lofblom, J.; Uhlen, M.; et al. An engineered affibody molecule with pH-dependent binding to FcRn mediates extended circulatory half-life of a fusion protein. Proc. Natl. Acad Sci. USA 2014, 111, 17110–17115. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Jeong, E.A.; Kim, K.E.; Yi, C.O.; Jin, Z.; Lee, J.E.; Lee, D.H.; Kim, H.J.; Kang, S.S.; Cho, G.J.; et al. TonEBP/NFAT5 haploinsufficiency attenuates hippocampal inflammation in high-fat diet/streptozotocin-induced diabetic mice. Sci. Rep. 2017, 7, 7837. [Google Scholar] [CrossRef] [Green Version]
- Jeon, B.T.; Jeong, E.A.; Shin, H.J.; Lee, Y.; Lee, D.H.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Roh, G.S. Resveratrol attenuates obesity-associated peripheral and central inflammation and improves memory deficit in mice fed a high-fat diet. Diabetes 2012, 61, 1444–1454. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Tian, H.; Wu, J.; Cheng, J.; Luo, C.; Sai, W.; Song, X.; Gao, X.; Yao, W. C-terminal site-specific PEGylated Exendin-4 analog: A long-acting glucagon like Peptide-1 receptor agonist, on glycemic control and beta cell function in diabetic db/db mice. J. Pharm. Sci. 2018, 138, 23–30. [Google Scholar] [CrossRef]
- Raufman, J.P.; Singh, L.; Singh, G.; Eng, J. Truncated glucagon-like peptide-1 interacts with exendin receptors on dispersed acini from guinea pig pancreas. Identification of a mammalian analogue of the reptilian peptide exendin-4. J. Biol. Chem. 1992, 267, 21432–21437. [Google Scholar]
- Tourrel, C.; Bailbe, D.; Meile, M.J.; Kergoat, M.; Portha, B. Glucagon-like peptide-1 and exendin-4 stimulate beta-cell neogenesis in streptozotocin-treated newborn rats resulting in persistently improved glucose homeostasis at adult age. Diabetes 2001, 50, 1562–1570. [Google Scholar] [CrossRef]
- Buse, J.B.; Klonoff, D.C.; Nielsen, L.L.; Guan, X.; Bowlus, C.L.; Holcombe, J.H.; Maggs, D.G.; Wintle, M.E. Metabolic effects of two years of exenatide treatment on diabetes, obesity, and hepatic biomarkers in patients with type 2 diabetes: An interim analysis of data from the open-label, uncontrolled extension of three double-blind, placebo-controlled trials. Clin. Ther. 2007, 29, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Gumuslu, E.; Mutlu, O.; Celikyurt, I.K.; Ulak, G.; Akar, F.; Erden, F.; Ertan, M. Exenatide enhances cognitive performance and upregulates neurotrophic factor gene expression levels in diabetic mice. Fundam. Clin. Pharm. 2016, 30, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Gault, V.A.; Holscher, C. GLP-1 agonists facilitate hippocampal LTP and reverse the impairment of LTP induced by beta-amyloid. Eur. J. Pharm. 2008, 587, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.L.; Quattrini, A.; Lentz, S.I.; Figueroa-Romero, C.; Cerri, F.; Backus, C.; Hong, Y.; Feldman, E.L. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia 2010, 53, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Gao, P.; Wei, C.; Li, H.; Zhang, L.; Zhao, Y.; Wu, B.; Tian, Y.; Zhang, W.; Wu, L.; et al. Calcium sensing receptor protects high glucose-induced energy metabolism disorder via blocking gp78-ubiquitin proteasome pathway. Cell Death Dis. 2017, 8, e2799. [Google Scholar] [CrossRef] [Green Version]
- Biessels, G.J.; ter Laak, M.P.; Hamers, F.P.; Gispen, W.H. Neuronal Ca2+ disregulation in diabetes mellitus. Eur. J. Pharm. 2002, 447, 201–209. [Google Scholar] [CrossRef]
- McQuail, J.A.; Frazier, C.J.; Bizon, J.L. Molecular aspects of age-related cognitive decline: The role of GABA signaling. Trends Mol. Med. 2015, 21, 450–460. [Google Scholar] [CrossRef] [Green Version]
- Kann, O. The interneuron energy hypothesis: Implications for brain disease. Neurobiol. Dis. 2016, 90, 75–85. [Google Scholar] [CrossRef]
- Kreymann, B.; Williams, G.; Ghatei, M.A.; Bloom, S.R. Glucagon-like peptide-1 7-36: A physiological incretin in man. Lancet 1987, 2, 1300–1304. [Google Scholar] [CrossRef]
- Seo, S.; Ju, S.; Chung, H.; Lee, D.; Park, S. Acute effects of glucagon-like peptide-1 on hypothalamic neuropeptide and AMP activated kinase expression in fasted rats. Endocr. J. 2008, 55, 867–874. [Google Scholar] [CrossRef] [Green Version]
- Hayes, M.R.; Leichner, T.M.; Zhao, S.; Lee, G.S.; Chowansky, A.; Zimmer, D.; De Jonghe, B.C.; Kanoski, S.E.; Grill, H.J.; Bence, K.K. Intracellular signals mediating the food intake-suppressive effects of hindbrain glucagon-like peptide-1 receptor activation. Cell Metab. 2011, 13, 320–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanoski, S.E.; Fortin, S.M.; Arnold, M.; Grill, H.J.; Hayes, M.R. Peripheral and central GLP-1 receptor populations mediate the anorectic effects of peripherally administered GLP-1 receptor agonists, liraglutide and exendin-4. Endocrinology 2011, 152, 3103–3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Jung, J.; Kim, H.; Heo, R.W.; Yi, C.O.; Lee, J.E.; Jeon, B.T.; Kim, W.H.; Hahm, J.R.; Roh, G.S. Exendin-4 Improves Nonalcoholic Fatty Liver Disease by Regulating Glucose Transporter 4 Expression in ob/ob Mice. Korean J. Physiol. Pharm. 2014, 18, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drucker, D.J.; Buse, J.B.; Taylor, K.; Kendall, D.M.; Trautmann, M.; Zhuang, D.; Porter, L.; Group, D.-S. Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: A randomised, open-label, non-inferiority study. Lancet 2008, 372, 1240–1250. [Google Scholar] [CrossRef]
- Diamant, M.; Van Gaal, L.; Stranks, S.; Northrup, J.; Cao, D.; Taylor, K.; Trautmann, M. Once weekly exenatide compared with insulin glargine titrated to target in patients with type 2 diabetes (DURATION-3): An open-label randomised trial. Lancet 2010, 375, 2234–2243. [Google Scholar] [CrossRef]
- Bergenstal, R.M.; Wysham, C.; Macconell, L.; Malloy, J.; Walsh, B.; Yan, P.; Wilhelm, K.; Malone, J.; Porter, L.E.; Group, D.-S. Efficacy and safety of exenatide once weekly versus sitagliptin or pioglitazone as an adjunct to metformin for treatment of type 2 diabetes (DURATION-2): A randomised trial. Lancet 2010, 376, 431–439. [Google Scholar] [CrossRef]
- Ohki, T.; Isogawa, A.; Iwamoto, M.; Ohsugi, M.; Yoshida, H.; Toda, N.; Tagawa, K.; Omata, M.; Koike, K. The effectiveness of liraglutide in nonalcoholic fatty liver disease patients with type 2 diabetes mellitus compared to sitagliptin and pioglitazone. Sci. World J. 2012, 2012, 496453. [Google Scholar] [CrossRef]
- Cuthbertson, D.J.; Irwin, A.; Gardner, C.J.; Daousi, C.; Purewal, T.; Furlong, N.; Goenka, N.; Thomas, E.L.; Adams, V.L.; Pushpakom, S.P.; et al. Improved glycaemia correlates with liver fat reduction in obese, type 2 diabetes, patients given glucagon-like peptide-1 (GLP-1) receptor agonists. PLoS ONE 2012, 7, e50117. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; De Minicis, S.; Candelaresi, C.; Faraci, G.; Pacetti, D.; Vivarelli, M.; Nicolini, D.; et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011, 31, 1285–1297. [Google Scholar] [CrossRef] [Green Version]
- Lund, A.; Knop, F.K.; Vilsboll, T. Emerging GLP-1 receptor agonists. Expert Opin. Emerg. Drugs 2011, 16, 607–618. [Google Scholar] [CrossRef]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Duffy, K.B.; Ottinger, M.A.; Ray, B.; Bailey, J.A.; Holloway, H.W.; Tweedie, D.; Perry, T.; Mattson, M.P.; Kapogiannis, D.; et al. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J. Alzheimers Dis. 2010, 19, 1205–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Li, D.; Feng, P.; Li, L.; Xue, G.F.; Li, G.; Holscher, C. A novel dual GLP-1 and GIP incretin receptor agonist is neuroprotective in a mouse model of Parkinson’s disease by reducing chronic inflammation in the brain. Neuroreport 2016, 27, 384–391. [Google Scholar] [CrossRef]
- Iwai, T.; Ito, S.; Tanimitsu, K.; Udagawa, S.; Oka, J. Glucagon-like peptide-1 inhibits LPS-induced IL-1beta production in cultured rat astrocytes. Neurosci. Res. 2006, 55, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, T.; Tanaka, R.; Shimada, Y.; Yamashiro, K.; Ueno, Y.; Shimura, H.; Urabe, T.; Hattori, N. Exendin-4 Inhibits Matrix Metalloproteinase-9 Activation and Reduces Infarct Growth After Focal Cerebral Ischemia in Hyperglycemic Mice. Stroke 2016, 47, 1328–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, S.B.; Pineda, F.J.; Hardwick, J.M. Mitochondrial fission and fusion dynamics: The long and short of it. Cell Death Differ. 2008, 15, 1147–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Okamoto, K.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Jeon, B.T.; Heo, R.W.; Jeong, E.A.; Yi, C.O.; Lee, J.Y.; Kim, K.E.; Kim, H.; Roh, G.S. Effects of caloric restriction on O-GlcNAcylation, Ca2+ signaling, and learning impairment in the hippocampus of ob/ob mice. Neurobiol. Aging 2016, 44, 127–137. [Google Scholar] [CrossRef]
- Lietzau, G.; Nystrom, T.; Ostenson, C.G.; Darsalia, V.; Patrone, C. Type 2 diabetes-induced neuronal pathology in the piriform cortex of the rat is reversed by the GLP-1 receptor agonist exendin-4. Oncotarget 2016, 7, 5865–5876. [Google Scholar] [CrossRef] [Green Version]
- Druga, R. Neocortical inhibitory system. Folia. Biol. (Praha) 2009, 55, 201–217. [Google Scholar]
- Larsson, M.; Lietzau, G.; Nathanson, D.; Ostenson, C.G.; Mallard, C.; Johansson, M.E.; Nystrom, T.; Patrone, C.; Darsalia, V. Diabetes negatively affects cortical and striatal GABAergic neurons: An effect that is partially counteracted by exendin-4. Biosci. Rep. 2016, 36, e00421. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Papageorgiou, I.E.; Draguhn, A. Highly energized inhibitory interneurons are a central element for information processing in cortical networks. J. Cereb. Blood Flow Metab. 2014, 34, 1270–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, K.-A.; Jin, Z.; Lee, J.Y.; An, H.S.; Choi, E.B.; Kim, K.E.; Shin, H.J.; Jeong, E.A.; Min, K.A.; Shin, M.C.; et al. Long-Lasting Exendin-4 Fusion Protein Improves Memory Deficits in High-Fat Diet/Streptozotocin-Induced Diabetic Mice. Pharmaceutics 2020, 12, 159. https://doi.org/10.3390/pharmaceutics12020159
Park K-A, Jin Z, Lee JY, An HS, Choi EB, Kim KE, Shin HJ, Jeong EA, Min KA, Shin MC, et al. Long-Lasting Exendin-4 Fusion Protein Improves Memory Deficits in High-Fat Diet/Streptozotocin-Induced Diabetic Mice. Pharmaceutics. 2020; 12(2):159. https://doi.org/10.3390/pharmaceutics12020159
Chicago/Turabian StylePark, Kyung-Ah, Zhen Jin, Jong Youl Lee, Hyeong Seok An, Eun Bee Choi, Kyung Eun Kim, Hyun Joo Shin, Eun Ae Jeong, Kyoung Ah Min, Meong Cheol Shin, and et al. 2020. "Long-Lasting Exendin-4 Fusion Protein Improves Memory Deficits in High-Fat Diet/Streptozotocin-Induced Diabetic Mice" Pharmaceutics 12, no. 2: 159. https://doi.org/10.3390/pharmaceutics12020159
APA StylePark, K. -A., Jin, Z., Lee, J. Y., An, H. S., Choi, E. B., Kim, K. E., Shin, H. J., Jeong, E. A., Min, K. A., Shin, M. C., & Roh, G. S. (2020). Long-Lasting Exendin-4 Fusion Protein Improves Memory Deficits in High-Fat Diet/Streptozotocin-Induced Diabetic Mice. Pharmaceutics, 12(2), 159. https://doi.org/10.3390/pharmaceutics12020159