Co-Encapsulation of Mitoxantrone and β-Elemene in Solid Lipid Nanoparticles to Overcome Multidrug Resistance in Leukemia

 ,
,  ,
,
Abstract
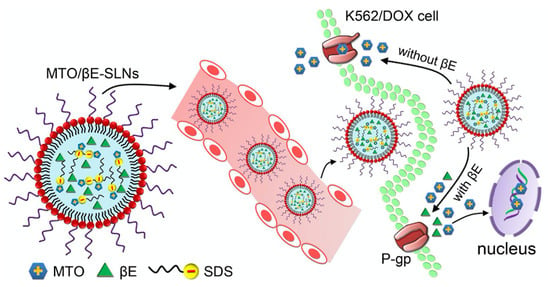
:
1. Introduction
2. Materials
2.1. Reagents
2.2. Cell Cultures and Animals
3. Methods
3.1. Determination of Combinatorial Effects of MTO and βE
3.2. The Preparation of SLNs
3.3. The Characterization of SLNs
3.4. Differential Scanning Calorimetry (DSC) Analysis
3.5. In Vitro Drug Release
3.6. The Cytotoxicity Study of SLNs
3.7. Cellular Uptake and Its Mechanism
3.8. Drug Efflux Study
3.9. Intracellular ATP Production Assay
3.10. Pharmacokinetic and Biodistribution Studies
3.11. In Vivo Antitumor Activity
3.12. Statistical Analysis
4. Results and Discussion
4.1. Combinatorial Effects of MTO and βE
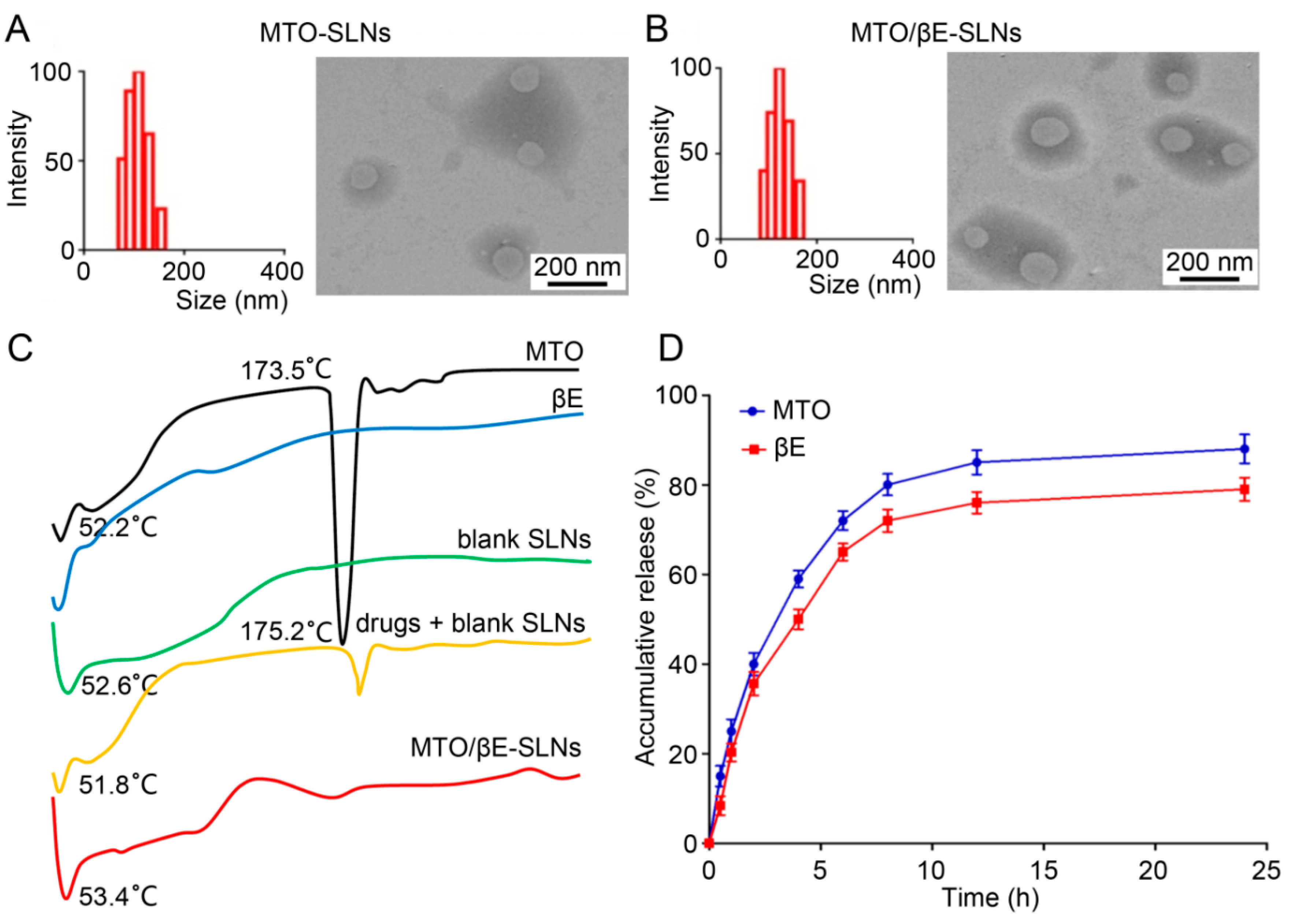
4.2. Characterization of SLNs
4.3. In Vitro Cytotoxicity Studies
4.4. Cellular Uptake Studies
4.5. In Vitro Drug Efflux and Intracellular ATP Production Assay
4.6. The Studies of Pharmacokinetics and Biodistribution of MTO/βE-SLNs
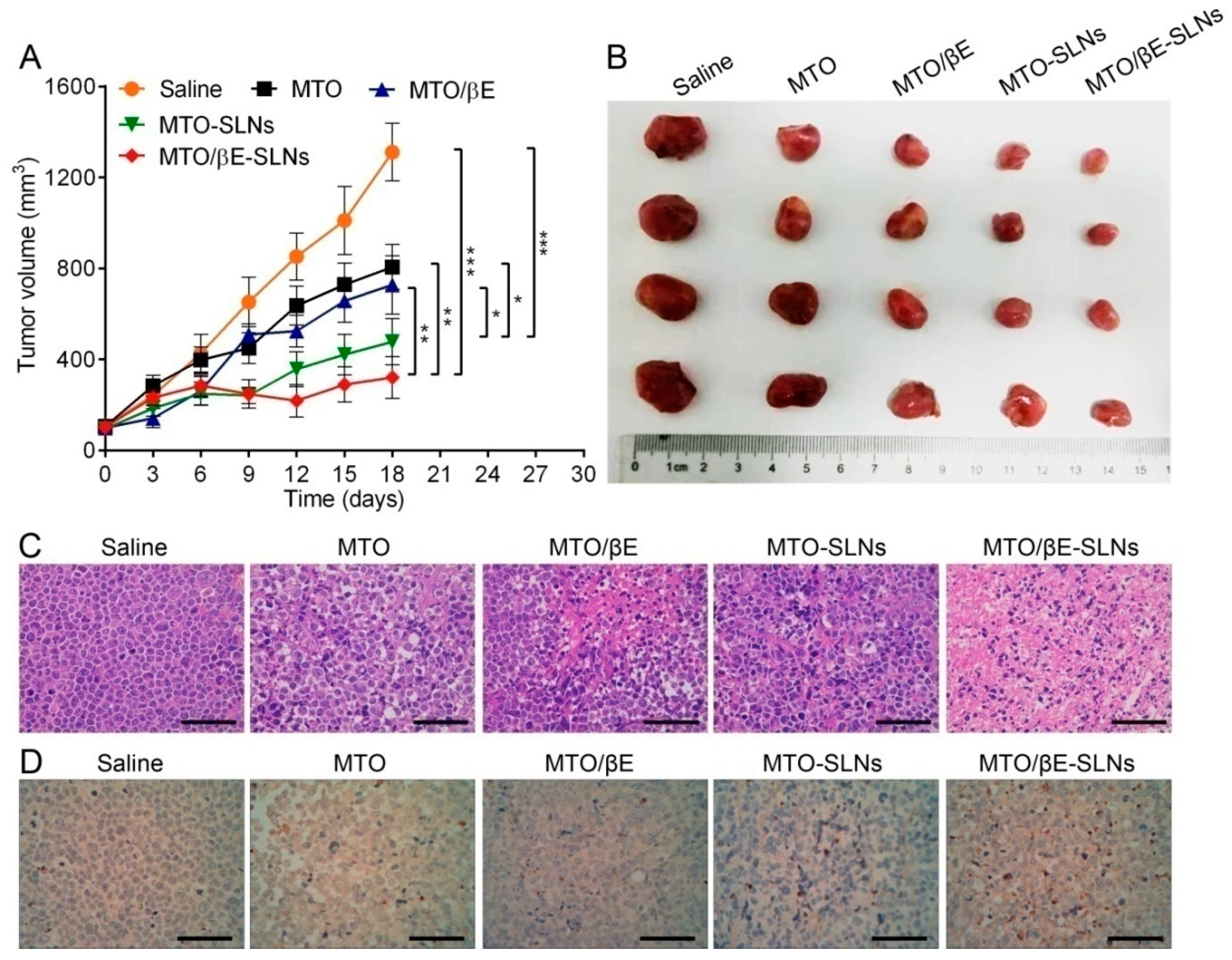
4.7. In Vivo Antitumor Activity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Enache, M.; Toader, A.M.; Enache, M.I. Mitoxantrone-Surfactant Interactions: A Physicochemical Overview. Molecules 2016, 21, 1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipczak, N.; Jaromin, A.; Piwoni, A.; Mahmud, M.; Sarisozen, C.; Torchilin, V.; Gubernator, J. A Triple Co-Delivery Liposomal Carrier That Enhances Apoptosis via an Intrinsic Pathway in Melanoma Cells. Cancers 2019, 11, 1982. [Google Scholar] [CrossRef] [PubMed]
- Im, A.; Amjad, A.; Agha, M.; Raptis, A.; Hou, J.Z.; Farah, R.; Lim, S.; Sehgal, A.; Dorritie, K.A.; Redner, R.L.; et al. Mitoxantrone and Etoposide for the Treatment of Acute Myeloid Leukemia Patients in First Relapse. Oncol. Res. 2016, 24, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Asghar, S.; Gao, S.; Chen, Z.; Huang, L.; Yin, L.; Ping, Q.; Xiao, Y. Polysaccharide-based nanoparticles for co-loading mitoxantrone and verapamil to overcome multidrug resistance in breast tumor. Int. J. Nanomed. 2017, 12, 7337–7350. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, M.; Okusaka, T.; Sato, Y.; Furuse, J.; Mitsunaga, S.; Ueno, H.; Morizane, C.; Inaba, Y.; Kobayashi, T.; Arai, Y. A Phase I/II trial of continuous hepatic intra-arterial infusion of 5-fluorouracil, mitoxantrone and cisplatin for advanced hepatocellular carcinoma. Jpn. J. Clin. Oncol. 2017, 47, 512–519. [Google Scholar] [CrossRef] [Green Version]
- Farsani, F.M.; Ganjalikhany, M.R.; Vallian, S. Studies on Non-synonymous Polymorphisms Altering Human DNA Topoisomerase II-Alpha Interaction with Amsacrine and Mitoxantrone: An In Silico Approach. Curr. Cancer Drug Targets 2017, 17, 657–668. [Google Scholar] [CrossRef]
- Consoli, U.; Van, N.T.; Neamati, N.; Mahadevia, R.; Beran, M.; Zhao, S.; Andreeff, M. Cellular pharmacology of mitoxantrone in p-glycoprotein-positive and -negative human myeloid leukemic cell lines. Leukemia 1997, 11, 2066–2074. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, T.; Yamashita, T.; Takemura, H.; Suto, H.; Kishi, S.; Urasaki, Y.; Ueda, T. Effect of PSC 833 on the cytotoxicity and pharmacodynamic of mitoxantrone in multidrug-resistant K562 cells. Leuk. Res. 2000, 24, 249–254. [Google Scholar] [CrossRef]
- Imrichova, D.; Messingerova, L.; Seres, M.; Kavcova, H.; Pavlikova, L.; Coculova, M.; Breier, A.; Sulova, Z. Selection of resistant acute myeloid leukemia SKM-1 and MOLM-13 cells by vincristine-, mitoxantrone- and lenalidomide-induced upregulation of P-glycoprotein activity and downregulation of CD33 cell surface exposure. Eur. J. Pharm. Sci. 2015, 77, 29–39. [Google Scholar] [CrossRef]
- Lv, L.; Liu, C.; Chen, C.; Yu, X.; Chen, G.; Shi, Y.; Qin, F.; Ou, J.; Qiu, K.; Li, G. Quercetin and doxorubicin co-encapsulated biotin receptor-targeting nanoparticles for minimizing drug resistance in breast cancer. Oncotarget 2016, 7, 32184–32199. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, W.; Wu, J.; Zhu, Y. The synergistic reversal effect of multidrug resistance by quercetin and hyperthermia in doxorubicin-resistant human myelogenous leukemia cells. Int. J. Hyperth. 2008, 24, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lin, Z.; Zhang, B.; Guo, L.; Liu, S.; Li, H.; Zhang, J.; Ye, Q. β-elemene sensitizes hepatocellular carcinoma cells to Oxaliplatin by preventing Oxaliplatin-induced degradation of copper transporter 1. Sci. Rep. 2016, 6, 21010. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.J.; Tang, W.; Lu, W.F.; Gao, J.; Kang, H.F.; Ma, X.B.; Min, W.L.; Wang, X.J.; Wu, W.Y. Antiproliferative and apoptotic effects of β-elemene on human hepatoma HepG2 cells. Cancer Cell Int. 2013, 13, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Li, L.; Chen, X.; Zeng, B.; Lin, T. Preliminary evaluation of the potential role of β-elemene in reversing erlotinib-resistant human NSCLC A549/ER cells. Oncol. Lett. 2018, 16, 3380–3388. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Jacob, J.A.; Loganathachetti, D.S.; Nainangu, P.; Chen, B. β-Elemene: Mechanistic Studies on Cancer Cell Interaction and Its Chemosensitization Effect. Front. Pharmacol. 2017, 8, 105. [Google Scholar] [CrossRef] [Green Version]
- Barrero, A.F.; Herrador, M.M.; del Moral, J.F.Q.; Arteaga, P.; Meine, N.; Pérez-Morales, M.C.; Catalán, J.V. Efficient synthesis of the anticancer beta-elemene and other bioactive elemanes from sustainable germacrone. Org. Biomol. Chem. 2011, 9, 1118–1125. [Google Scholar] [CrossRef]
- Zeng, Y.Y.; Zeng, Y.J.; Zhang, N.-N.; Li, C.X.; Xie, T.; Zeng, Z.W. The Preparation, Determination of a Flexible Complex Liposome Co-Loaded with Cabazitaxel and β-Elemene, and Animal Pharmacodynamic on Paclitaxel-Resistant Lung Adenocarcinoma. Molecules 2019, 24, 1697. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.Y.; Zhu, L.X.; Yu, J.D.; Chen, Z.; Gu, M.C.; Mu, C.F.; Liu, Q.; Xiong, Y. Effect of β-elemene on the kinetics of intracellular transport of D-luciferin potassium salt (ABC substrate) in doxorubicin-resistant breast cancer cells and the associated molecular mechanism. Eur. J. Pharm. Sci. 2018, 120, 20–29. [Google Scholar] [CrossRef]
- Yao, C.; Jiang, J.; Tu, Y.; Ye, S.; Du, H.; Zhang, Y. β-elemene reverses the drug resistance of A549/DDP lung cancer cells by activating intracellular redox system, decreasing mitochondrial membrane potential and P-glycoprotein expression, and inducing apoptosis. Thorac. Cancer 2014, 5, 304–312. [Google Scholar] [CrossRef]
- Guo, H.Q.; Zhang, G.N.; Wang, Y.J.; Zhang, Y.K.; Sodani, K.; Talele, T.T.; Ashby, C.R., Jr.; Chen, Z.S. β-Elemene, a compound derived from Rhizoma zedoariae, reverses multidrug resistance mediated by the ABCB1 transporter. Oncol. Rep. 2014, 31, 858–866. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.B.; Li, L.; Fu, J.; Mao, X.P.; Xu, L.Z. Reversion of multidrug resistance in a chemoresistant human breast cancer cell line by β-elemene. Pharmacology 2012, 89, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, H.D.; Chen, L.; Sun, D.W.; Mao, C.F.; Chen, W.; Wu, J.Z.; Zhong, S.L.; Zhao, J.H.; Tang, J.H. β-elemene reverses chemoresistance of breast cancer via regulating MDR-related microRNA expression. Cell Physiol. Biochem. 2014, 34, 2027–2037. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mu, X.D.; Li, E.Z.; Luo, Y.; Song, N.; Qu, X.J.; Hu, X.J.; Liu, Y.P. The role of E3 ubiquitin ligase Cbl proteins in β-elemene reversing multi-drug resistance of human gastric adenocarcinoma cells. Int. J. Mol. Sci. 2013, 14, 10075–10089. [Google Scholar] [CrossRef] [PubMed]
- Zhai, B.; Zeng, Y.; Zeng, Z.; Zhang, N.; Li, C.; Zeng, Y.; You, Y.; Wang, S.; Chen, X.; Sui, X.; et al. Drug delivery systems for elemene, its main active ingredient β-elemene, and its derivatives in cancer therapy. Int. J. Nanomed. 2018, 13, 6279–6296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, F.; Vicent, M.J. Combination therapy: Opportunities and challenges for polymer-drug conjugate as anticancer nanomedicines. Adv. Drug Deliv. Rev. 2009, 61, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Yhee, J.Y.; Son, S.; Lee, H.; Kim, K. Nanoparticle-Based Combination Therapy for Cancer Treatment. Curr. Pharm. Des. 2015, 21, 3158–3166. [Google Scholar] [CrossRef]
- Wang, J.; Seebacher, N.; Shi, H.; Kan, Q.; Duan, Z. Novel strategies to prevent the development of multidrug resistance (MDR) in cancer. Oncotarget 2017, 8, 84559–84571. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, I.S.; He, W.; Yin, L. Understanding of human ATP binding cassette super family and novel multidrug resistance modulators to overcome MDR. Biomed. Pharmacother. 2018, 100, 335–348. [Google Scholar] [CrossRef]
- Shao, Y.; Luo, W.; Guo, Q.; Li, X.; Zhang, Q.; Li, J. In vitro and in vivo effect of hyaluronic acid modified, doxorubicin and gallic acid co-delivered lipid-polymeric hybrid nano-system for leukemia therapy. Drug Des. Dev. Ther. 2019, 13, 2043–2055. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Su, Z.; Li, S.; Chen, Y.; Chen, X.; Xiao, Y.; Sun, M.; Ping, Q.; Zong, L. Multistep targeted nano drug delivery system aiming at leukemic stem cells and minimal residual disease. Mol. Pharm. 2013, 10, 2479–2489. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Asghar, S.; Yang, L.; Gao, S.; Chen, Z.; Huang, L.; Zong, L.; Ping, Q.; Xiao, Y. Chitosan hydrochloride/hyaluronic acid nanoparticles coated by mPEG as long-circulating nanocarriers for systemic delivery of mitoxantrone. Int. J. Biol. Macromol. 2018, 113, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Zhao, X.L.; Li, J.Z.; Kang, S.M.; Yang, C.R.; Jin, Y.H.; Liu, D.; Chen, D.W. Preparation and characterization of β-elemene-loaded microemulsion. Drug Dev. Ind. Pharm. 2011, 37, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Xia, X.; Yang, Y.; Ye, J.; Dong, W.; Ma, P.; Jin, Y.; Liu, Y. Co-encapsulation of paclitaxel and baicalein in nanoemulsions to overcome multidrug resistance via oxidative stress augmentation and P-glycoprotein inhibition. Int. J. Pharm. 2016, 513, 8–16. [Google Scholar] [CrossRef]
- Guissi, N.E.I.; Li, H.; Xu, Y.; Semcheddine, F.; Chen, M.; Su, Z.; Ping, Q. Mitoxantrone- and Folate-TPGS2k Conjugate Hybrid MicellarAggregates To Circumvent Toxicity and Enhance Efficiency for BreastCancer Therapy. Mol. Pharm. 2017, 14, 1082–1094. [Google Scholar] [CrossRef]
- Tang, J.; Zhang, L.; Gao, H.; Liu, Y.; Zhang, Q.; Ran, R.; Zhang, Z.; He, Q. Co-delivery of doxorubicin and P-gp inhibitor by a reduction-sensitive liposome to overcome multidrug resistance, enhance anti-tumor efficiency and reduce toxicity. Drug Deliv. 2016, 23, 1130–1143. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, Y.; Wang, D.; Gao, Y.; Li, J.; Ma, S.; Zhao, L.; Zhang, C.; Liu, Y.; Li, X. pH-responsive polymeric micelles based on poly(2-ethyl-2-oxazoline)-poly(D,L-lactide) for tumor-targeting and controlled delivery of doxorubicin and P-glycoprotein inhibitor. Acta Biomater. 2015, 17, 182–192. [Google Scholar] [CrossRef]
- Mohammad, I.S.; Teng, C.; Chaurasiya, B.; Yin, L.; Wu, C.; He, W. Drug-delivering-drug approach-based codelivery of paclitaxel and disulfiram for treating multidrug-resistant cancer. Int. J. Pharm. 2019, 557, 304–313. [Google Scholar] [CrossRef]
- Shi, C.; Zhang, Z.; Shi, J.; Wang, F.; Luan, Y. Co-delivery of docetaxel and chloroquine via PEO-PPO-PCL/TPGS micelles for overcoming multidrug resistance. Int. J. Pharm. 2015, 495, 932–939. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Sun, X.; Yan, X.; Jacobson, O.; Sun, W.; Wang, Z.; Tong, X.; Xia, Y.; Ling, D.; Chen, X. Improved Tumor Uptake by Optimizing Liposome Based RES Blockade Strategy. Theranostics 2017, 7, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.M.; Malherbe, F.; Eldridge, D.; Palombo, E.A.; Harding, I.H. Physicochemical characterization of solid lipid nanoparticles (SLNs) prepared by a novel microemulsion technique. J. Colloid Interface Sci. 2014, 428, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Eldridge, D.; Palombo, E.; Harding, I. Lipid Nanoparticles: Production, Characterization and Stability; Springer International Publishing: New York, NY, USA, 2015; pp. 11–22. [Google Scholar]
- Kovačević, A.B.; Müller, R.H.; Savić, S.D.; Vuleta, G.M.; Keck, C.M. Solid lipid nanoparticles (SLN) stabilized with polyhydroxy surfactants: Preparation, characterization and physical stability investigation. Colloids Surf. A 2014, 444, 15–25. [Google Scholar] [CrossRef]
- Leonardi, D.; Salomon, C.J. Unexpected performance of physical mixtures over solid dispersions on the dissolution behavior of benznidazole from tablets. J. Pharm. Sci. 2013, 102, 1016–1023. [Google Scholar] [CrossRef]
- Sui, Z.-H.; Xu, H.; Wang, H.; Jiang, S.; Chi, H.; Sun, L. Intracellular Trafficking Pathways of Edwardsiella tarda: From Clathrin- and Calveolin-Mediated Endocytosis to Endosome and Lysosome. Front. Cell. Infect. Microbiol. 2017, 7, 400. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.P.; Singla, B.; Ghoshal, P.; Faulkner, J.L.; Cherian-Shaw, M.; O’Connor, P.M.; She, J.X.; de Chantemele, E.J.B.; Csányi, G. Identification of novel macropinocytosis inhibitors using a rational screen of Food and Drug Administration-approved drugs. Br. J. Pharmacol. 2018, 175, 3640–3655. [Google Scholar] [CrossRef] [Green Version]
- Chai, G.H.; Hu, F.Q.; Sun, J.; Du, Y.Z.; You, J.; Yuan, H. Transport pathways of solid lipid nanoparticles across Madin-Darby canine kidney epithelial cell monolayer. Mol. Pharm. 2014, 11, 3716–3726. [Google Scholar] [CrossRef]
- Desai, P.V.; Sawada, G.A.; Watson, I.A.; Raub, T.J. Integration of in silico and in vitro tools for scaffold optimization during drug discovery: Predicting P-glycoprotein efflux. Mol. Pharm. 2013, 10, 1249–1261. [Google Scholar] [CrossRef]
- Zhang, G.N.; Ashby, C.R.; Zhang, Y.K.; Chen, Z.S.; Guo, H. The reversal of antineoplastic drug resistance in cancer cells by β-elemene. Chin. J. Cancer 2015, 34, 45. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | IC50 (µg/mL) | CI | |
|---|---|---|---|
| MTO | βE | ||
| Free MTO | 48.19 | ------- | ------- |
| Free βE | ------- | 140.30 | ------- |
| MTO/βE (10/1) | 38.60 | 0.39 | 0.83 |
| MTO/βE (5/1) | 32.63 | 6.53 | 0.72 |
| MTO/βE (2/1) | 24.07 | 12.04 | 0.59 |
| MTO/βE (1/1) | 15.99 | 15.99 | 0.45 |
| MTO/βE (1/2) | 11.01 | 22.02 | 0.38 |
| MTO/βE (1/5) | 8.98 | 44.90 | 0.51 |
| MTO/βE (1/10) | 8.21 | 82.08 | 0.76 |
| Time (Months) | Formulation | Particle Size (nm) | PDI | Zeta Potential (mv) | EE (%) | |
|---|---|---|---|---|---|---|
| MTO | Be | |||||
| 0 | MTO-SLNs | 121.5 ± 1.8 | 0.171 ± 0.016 | −20.86 ± 1.8 | 99.61 | — |
| MTO/βE-SLNs | 124.6 ± 1.4 | 0.162 ± 0.011 | −16.47 ± 0.9 | 97.98 | 94.42 | |
| 3 | MTO-SLNs | 125.4 ± 2.1 | 0.159 ± 0.014 | −22.15 ± 1.1 | 96.32 | — |
| MTO/βE-SLNs | 128.8 ± 1.2 | 0.195 ± 0.009 | −18.33 ± 0.8 | 94.85 | 90.22 | |
| Formulations | IC50 | IDR | IRDR | |
|---|---|---|---|---|
| K562 | K562/DOX | |||
| βE | 122.2 | 140.3 | ------- | ------ |
| MTO | 5.025 | 48.19 | 9.59 | ------ |
| MTO/βE | 1.82 | 11.01 | 6.05 | 1.58 |
| MTO-SLNs | 0.98 | 5.61 | 5.72 | 1.67 |
| MTO/βE-SLNs | 0.51 | 2.25 | 4.41 | 2.17 |
| Formulation | MTO | βE | MTO in MTO/βE-SLNs | βE in MTO/βE-SLNs |
|---|---|---|---|---|
| AUC0–24 (mg·h·L−1) | 1.18 ± 0.15 | 4.86 ± 1.76 | 51.55 ± 2.19 ** | 64.46 ± 2.95 ** |
| T1/2 (h) | 1.53 ± 0.33 | 2.9 ± 0.45 | 13.13 ± 1.15 * | 16.53 ± 1.22 * |
| Cmax (mg/L) | 3.45 ± 0.58 | 6.68 ± 1.21 | 67.78 ± 2.89 ** | 90.15 ± 3.59 ** |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amerigos Daddy J.C., K.; Chen, M.; Raza, F.; Xiao, Y.; Su, Z.; Ping, Q. Co-Encapsulation of Mitoxantrone and β-Elemene in Solid Lipid Nanoparticles to Overcome Multidrug Resistance in Leukemia. Pharmaceutics 2020, 12, 191. https://doi.org/10.3390/pharmaceutics12020191
Amerigos Daddy J.C. K, Chen M, Raza F, Xiao Y, Su Z, Ping Q. Co-Encapsulation of Mitoxantrone and β-Elemene in Solid Lipid Nanoparticles to Overcome Multidrug Resistance in Leukemia. Pharmaceutics. 2020; 12(2):191. https://doi.org/10.3390/pharmaceutics12020191
Chicago/Turabian StyleAmerigos Daddy J.C., Kambere, Minglei Chen, Faisal Raza, Yanyu Xiao, Zhigui Su, and Qineng Ping. 2020. "Co-Encapsulation of Mitoxantrone and β-Elemene in Solid Lipid Nanoparticles to Overcome Multidrug Resistance in Leukemia" Pharmaceutics 12, no. 2: 191. https://doi.org/10.3390/pharmaceutics12020191
APA StyleAmerigos Daddy J.C., K., Chen, M., Raza, F., Xiao, Y., Su, Z., & Ping, Q. (2020). Co-Encapsulation of Mitoxantrone and β-Elemene in Solid Lipid Nanoparticles to Overcome Multidrug Resistance in Leukemia. Pharmaceutics, 12(2), 191. https://doi.org/10.3390/pharmaceutics12020191