Author Contributions
Conceptualization, N.A.; methodology, N.A., R.A., R.A.A., H.M.A.A., A.S.A.-R.; software, N.A., M.A., M.S.; validation, N.A., R.A., R.A.A., H.M.A.A., A.S.A.-R.; formal analysis, N.A., R.A., R.A.A., H.M.A.A., A.S.A.-R.; investigation, N.A., R.A., R.A.A., H.M.A.A., A.S.A.-R., M.A., M.S.; resources, N.A.; data curation, N.A.; writing—original draft preparation, N.A., R.A., R.A.A., H.M.A.A., A.S.A.-R.; writing—review and editing, N.A., R.A., R.A.A., H.M.A.A., A.S.A.-R.; visualization, N.A., R.A., R.A.A., H.M.A.A., A.S.A.-R., M.A., M.S.; supervision, N.A.; project administration, N.A.; funding acquisition, N.A. All authors have read and agreed to the published version of the manuscript.
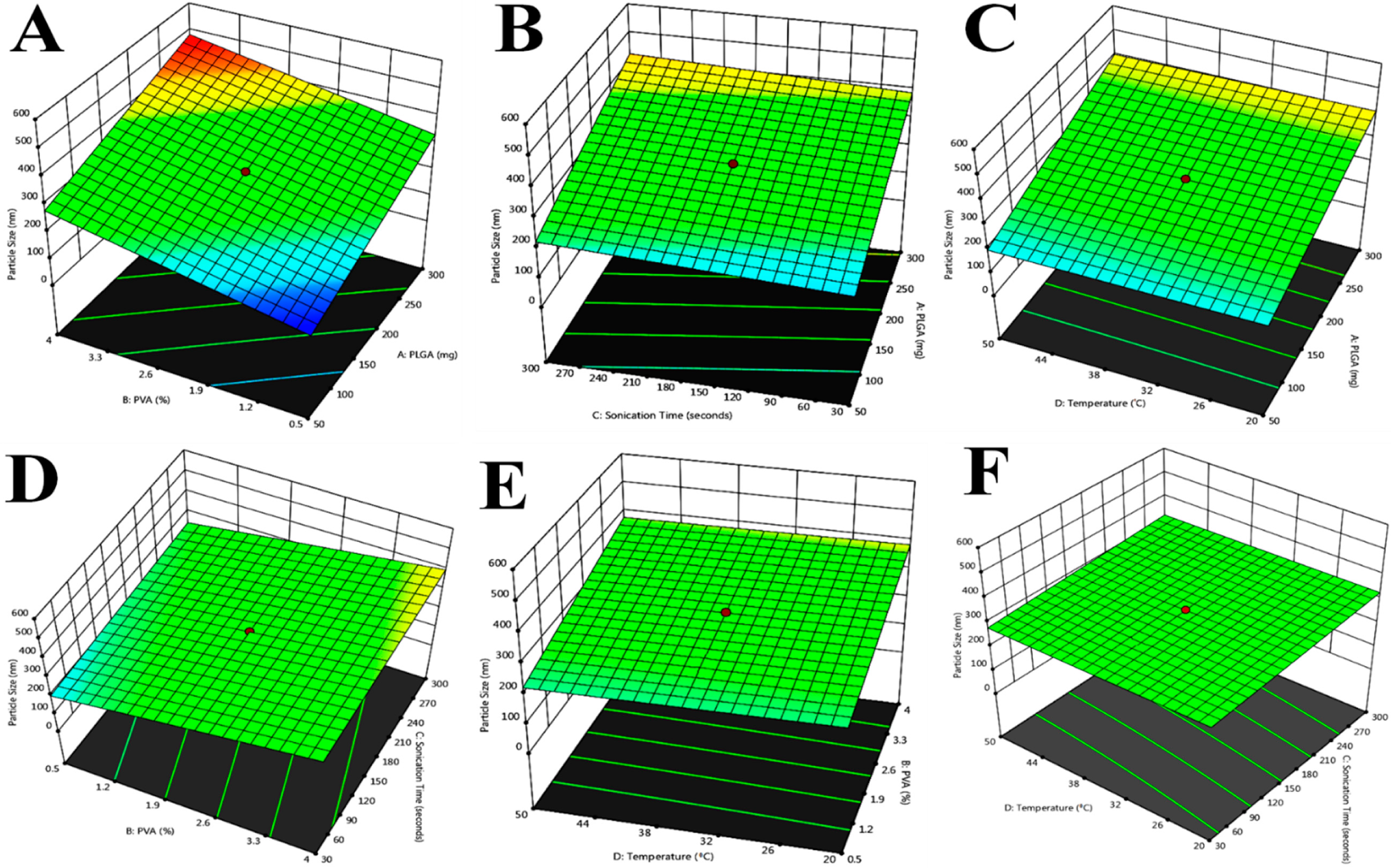
Figure 1.
Response surface plot showing the effect of independent variables on particle size. PVA, PLGA. (A). Particle size, PLGA, sonication time; (B). Particle size, PLGA, temperature; (C). Particle size, PVA (%), sonication time; (D). Particle size, PVA (%), temperature (°C); (E). Particle size, PVA (%), temperature (°C); (F). Particle size, sonication time, temperature (°C).
Figure 1.
Response surface plot showing the effect of independent variables on particle size. PVA, PLGA. (A). Particle size, PLGA, sonication time; (B). Particle size, PLGA, temperature; (C). Particle size, PVA (%), sonication time; (D). Particle size, PVA (%), temperature (°C); (E). Particle size, PVA (%), temperature (°C); (F). Particle size, sonication time, temperature (°C).
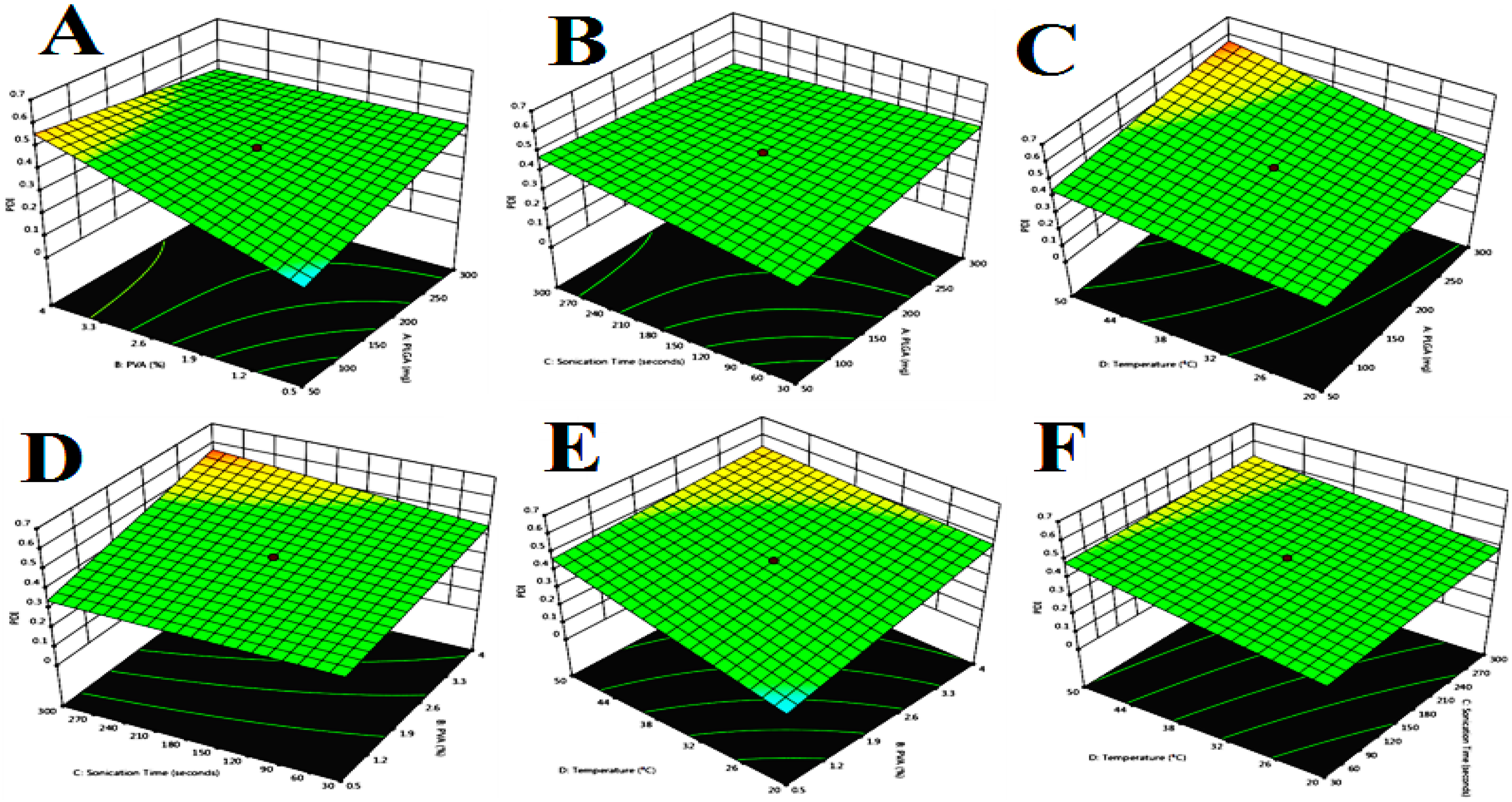
Figure 2.
Response surface plot showing the effect of independent variables on polydispersity index (PDI), PLGA, PVA (%); (A). PDI, PLGA, sonication time; (B). PDI, PLGA, temperature; (C). PDI, PVA (%), sonication time; (D). PDI, PVA (%), temperature; (E). PDI, temperature, sonication time; (F). PDI, temperature, sonication time.
Figure 2.
Response surface plot showing the effect of independent variables on polydispersity index (PDI), PLGA, PVA (%); (A). PDI, PLGA, sonication time; (B). PDI, PLGA, temperature; (C). PDI, PVA (%), sonication time; (D). PDI, PVA (%), temperature; (E). PDI, temperature, sonication time; (F). PDI, temperature, sonication time.
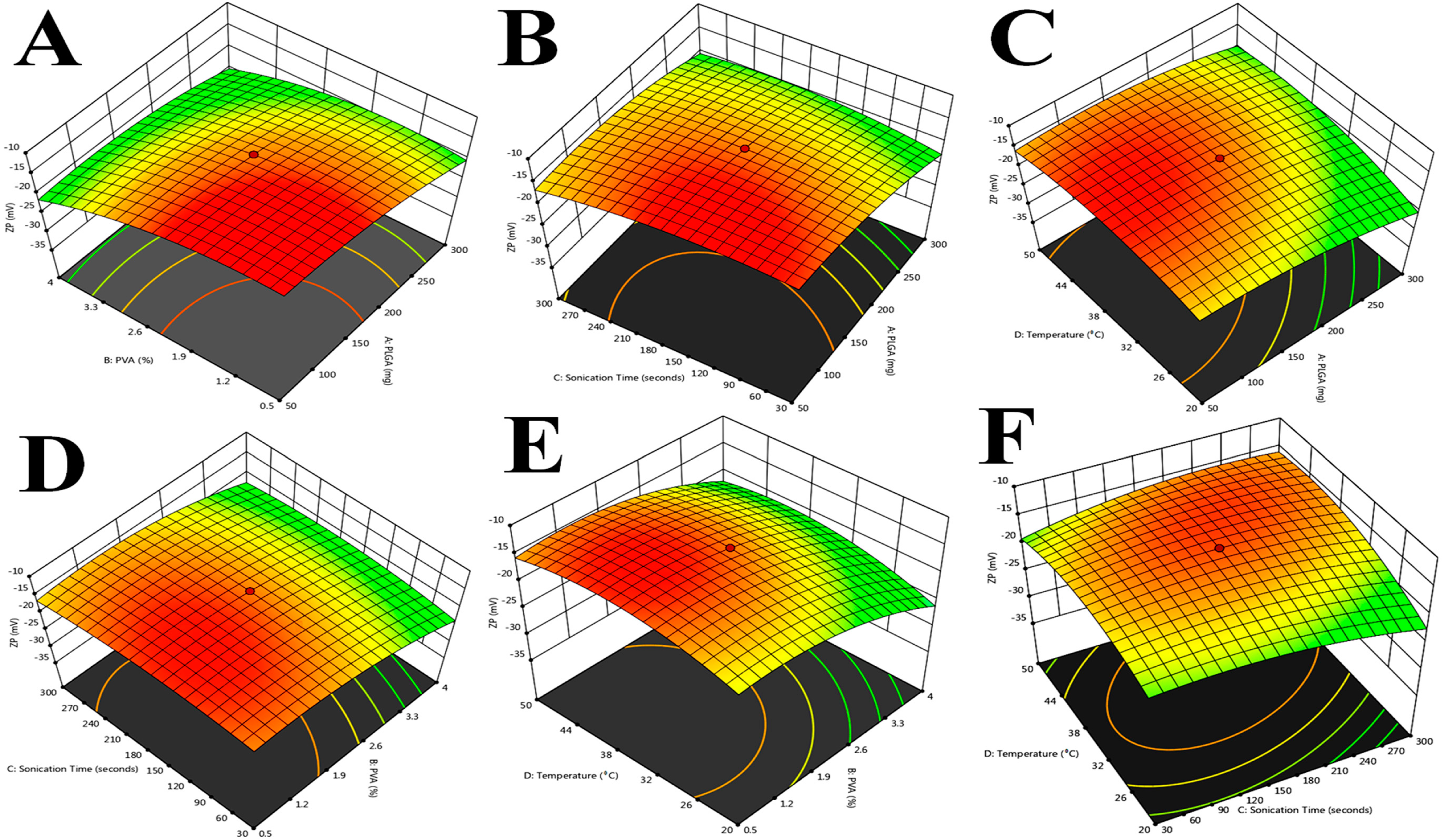
Figure 3.
Response surface plot showing the effect of independent variables on Zeta potential (ZP). (A). Zeta potential, PVA (%), PLGA; (B). Zeta potential, sonication time, PLGA; (C). Zeta potential, temperature (°C), PLGA; (D). Zeta potential, sonication time, PVA (%); (E). Zeta potential, temperature, PVA (%); (F). Zeta potential, temperature, sonication time.
Figure 3.
Response surface plot showing the effect of independent variables on Zeta potential (ZP). (A). Zeta potential, PVA (%), PLGA; (B). Zeta potential, sonication time, PLGA; (C). Zeta potential, temperature (°C), PLGA; (D). Zeta potential, sonication time, PVA (%); (E). Zeta potential, temperature, PVA (%); (F). Zeta potential, temperature, sonication time.
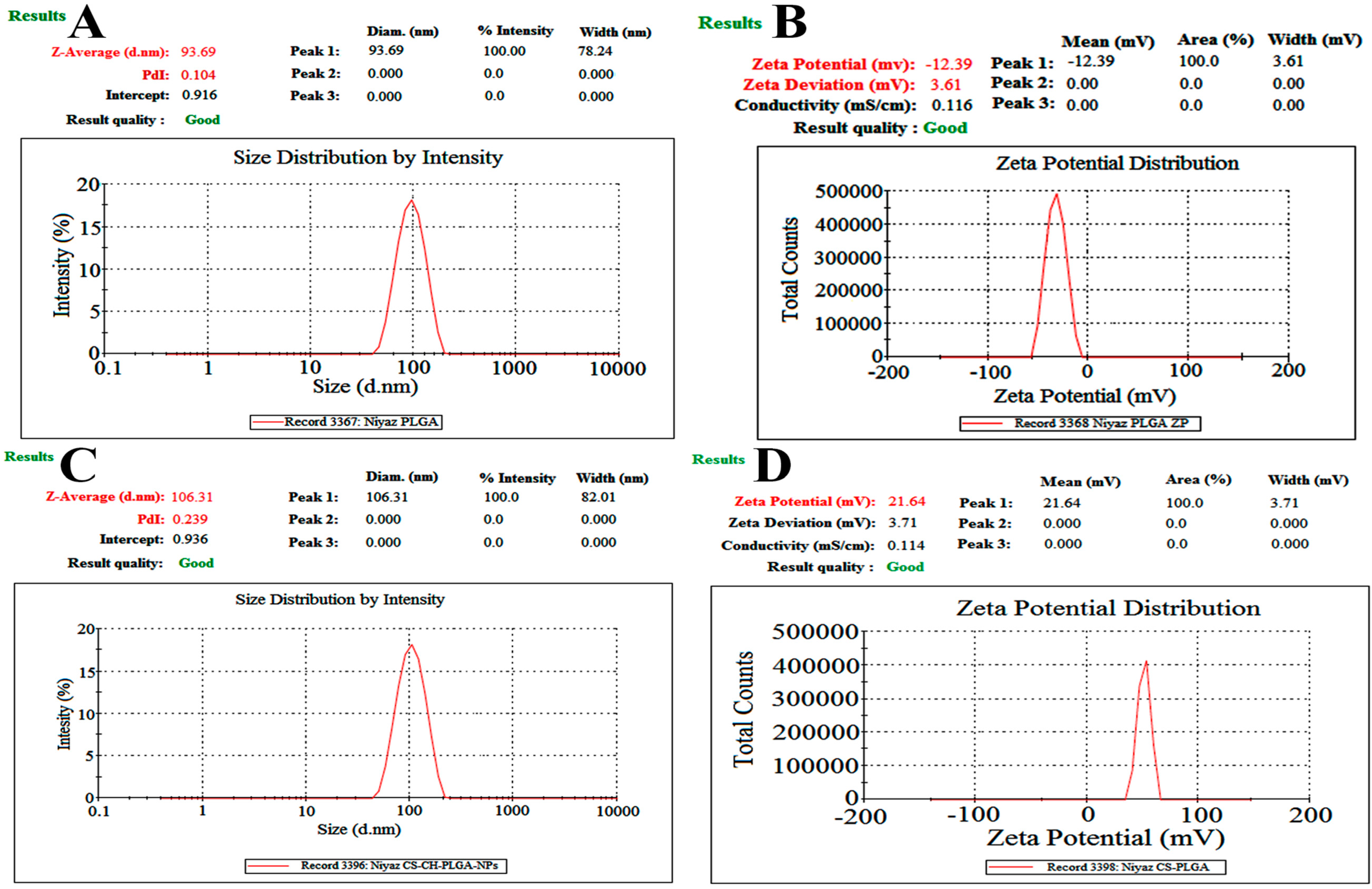
Figure 4.
Dynamic light scattering techniques for determining the particle size distribution of CH-loaded–PLGA–NPs’ globule size (A) and Zeta potential (B), as well as CS-coated–CH-loaded–PLGA–NPs’ particle size (C) and zeta potential (D) images.
Figure 4.
Dynamic light scattering techniques for determining the particle size distribution of CH-loaded–PLGA–NPs’ globule size (A) and Zeta potential (B), as well as CS-coated–CH-loaded–PLGA–NPs’ particle size (C) and zeta potential (D) images.
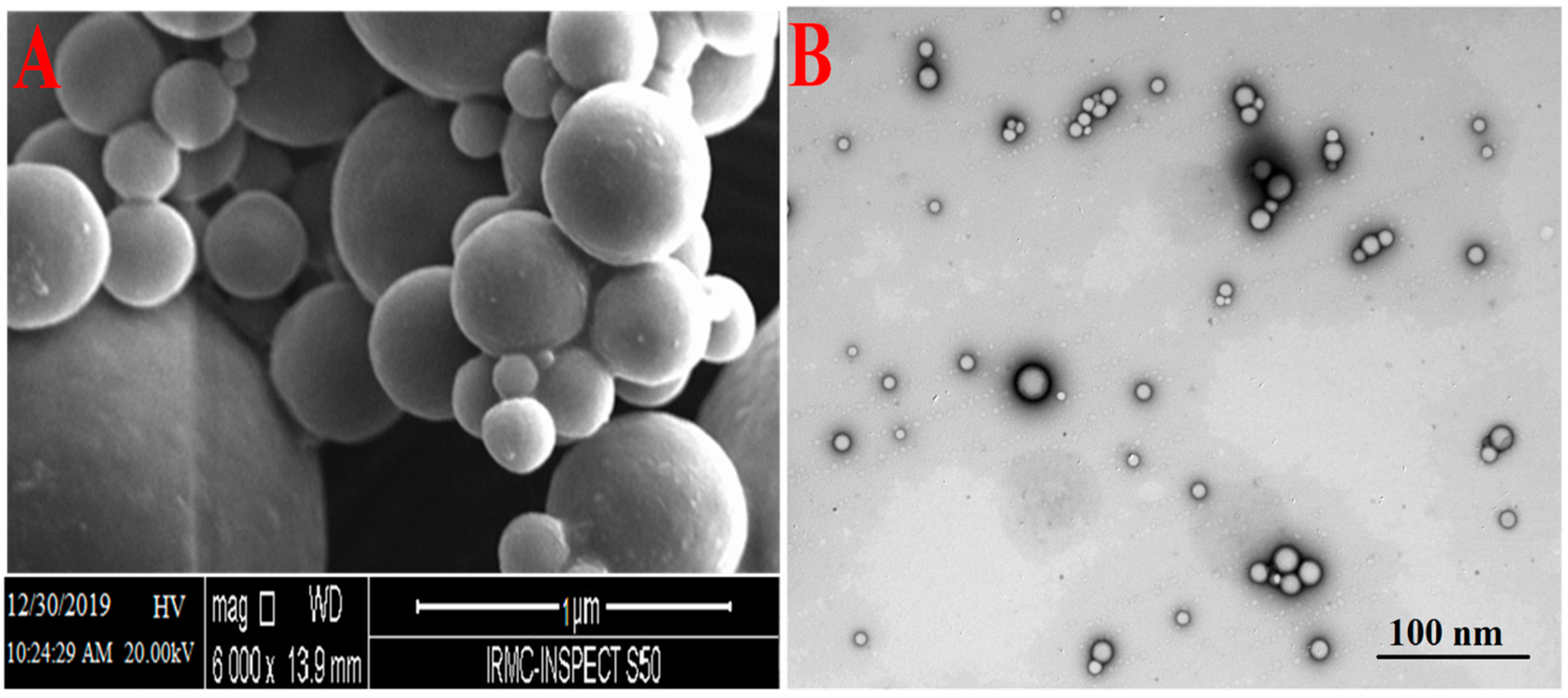
Figure 5.
Scanning electron microscopy (SEM) (A) and transmission electron microscopy (TEM) (B) images of CS-coated–CH-loaded–PLGA–NPs.
Figure 5.
Scanning electron microscopy (SEM) (A) and transmission electron microscopy (TEM) (B) images of CS-coated–CH-loaded–PLGA–NPs.
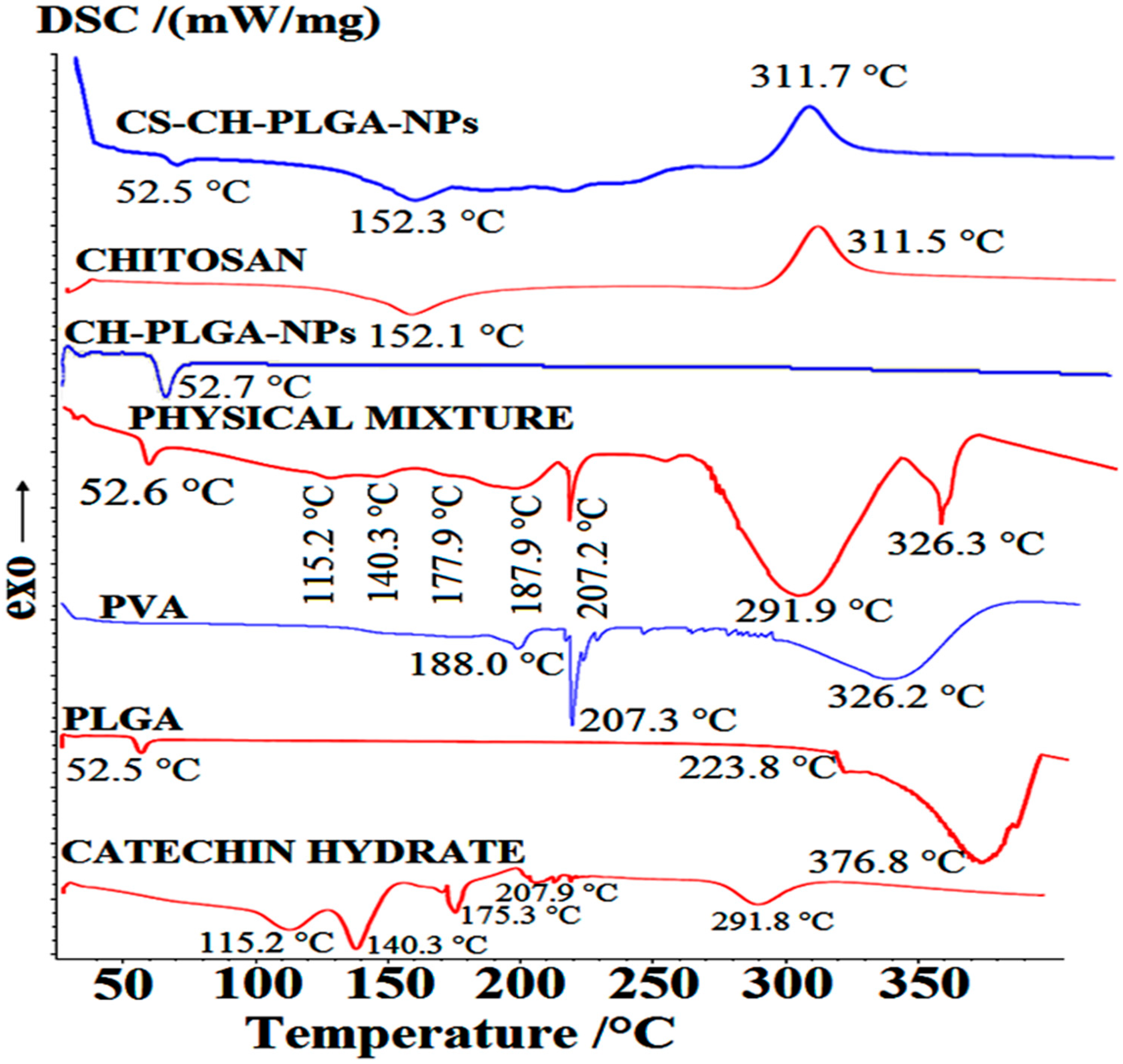
Figure 6.
The differential scanning calorimetry (DSC) thermograms of pure catechin (CH); polymer (PLGA); polyvinyl alcohol (PVA); a physical mixture of CH, PVA, and PLGA; freeze-dried CH-loaded–optimized polymeric (PLGA)–NPs; pure chitosan; and CS-coated–CH-loaded–optimized polymeric PLGA–NPs.
Figure 6.
The differential scanning calorimetry (DSC) thermograms of pure catechin (CH); polymer (PLGA); polyvinyl alcohol (PVA); a physical mixture of CH, PVA, and PLGA; freeze-dried CH-loaded–optimized polymeric (PLGA)–NPs; pure chitosan; and CS-coated–CH-loaded–optimized polymeric PLGA–NPs.
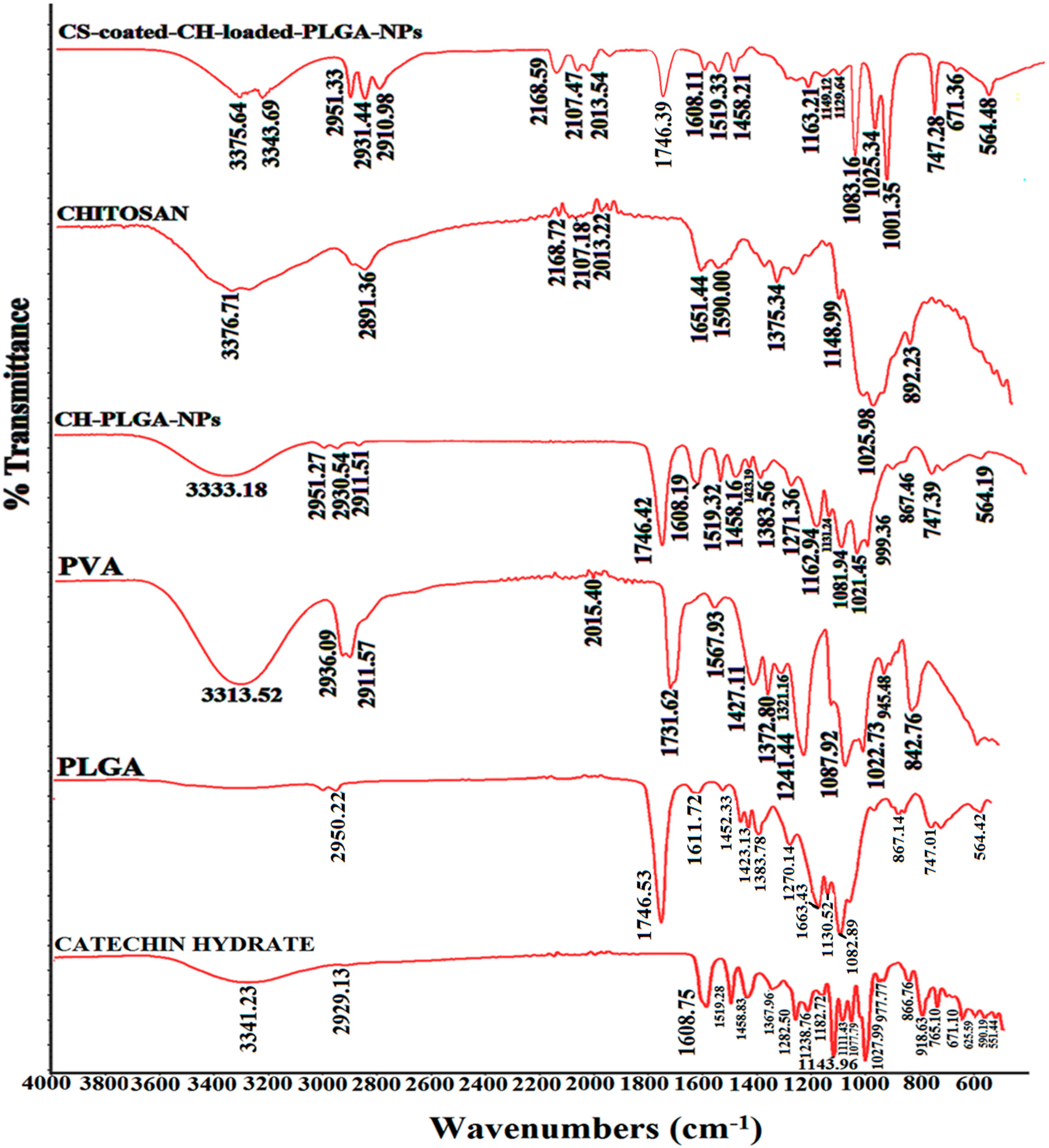
Figure 7.
Different Fourier transform infrared spectroscopy (FTIR) spectra with an attenuated total reflection (ATR) attachment of pure catechin hydrate (CH), PVA, PLGA–NPs, CH-loaded–PLGA–NPs, chitosan, and chitosan-coated–CH-loaded–PLGA–NPs.
Figure 7.
Different Fourier transform infrared spectroscopy (FTIR) spectra with an attenuated total reflection (ATR) attachment of pure catechin hydrate (CH), PVA, PLGA–NPs, CH-loaded–PLGA–NPs, chitosan, and chitosan-coated–CH-loaded–PLGA–NPs.
Figure 8.
(A) In vitro release profile of CS–S, CH-loaded–PLGA–NPs, and chitosan-coated–CH-loaded–PLGA–NPs performed using the dialysis bag method, revealing the sustained release pattern of CH-loaded–PLGA–NPs and Chitosan-coated–CH-loaded–PLGA–NPs (mean ± SD, n = 3). (B) Ex vivo permeation profiles of developed chitosan-coated–CH-loaded–PLGA–NPs as compared with CH-loaded–PLGA–NPs and pure CH–S through goat nasal mucosa.
Figure 8.
(A) In vitro release profile of CS–S, CH-loaded–PLGA–NPs, and chitosan-coated–CH-loaded–PLGA–NPs performed using the dialysis bag method, revealing the sustained release pattern of CH-loaded–PLGA–NPs and Chitosan-coated–CH-loaded–PLGA–NPs (mean ± SD, n = 3). (B) Ex vivo permeation profiles of developed chitosan-coated–CH-loaded–PLGA–NPs as compared with CH-loaded–PLGA–NPs and pure CH–S through goat nasal mucosa.
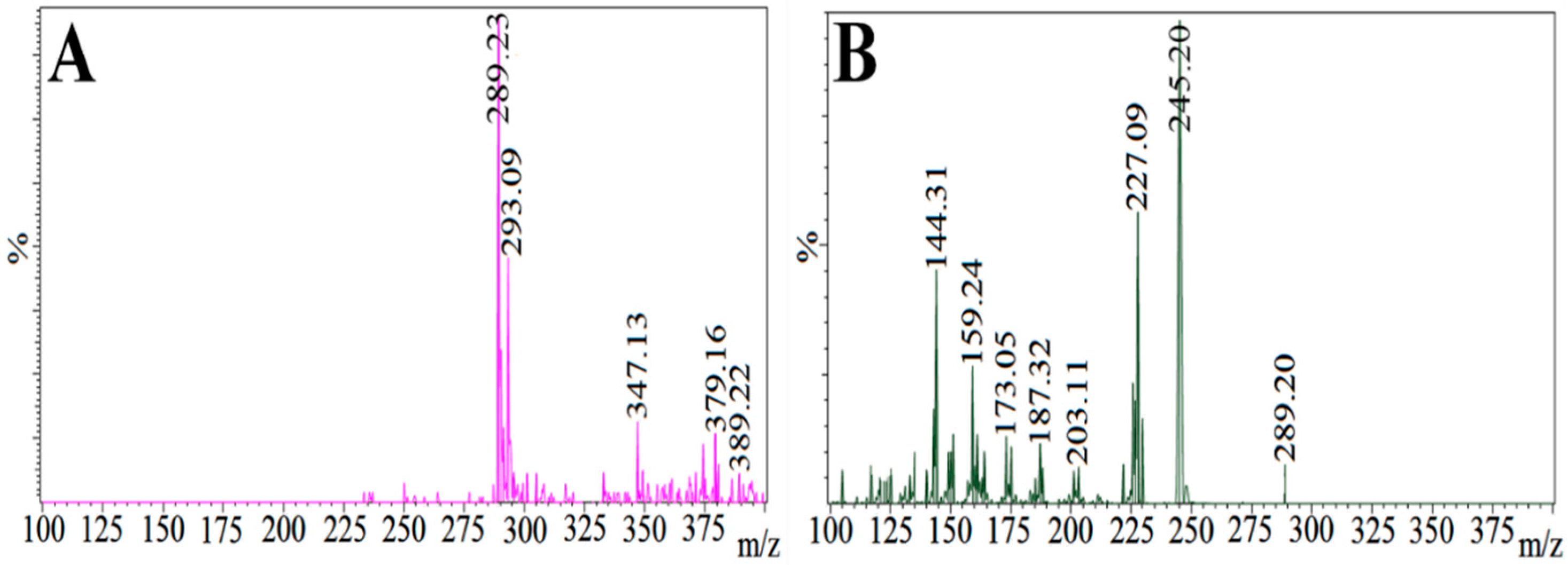
Figure 9.
Mass spectrum of (A) catechin hydrate (CH) parent ion (protonated precursor [M–H]− ions at m/z 289.23) and (B) catechin hydrate (CH) product ion (major fragmented product ion at m/z 245.20) showing fragmentation transitions.
Figure 9.
Mass spectrum of (A) catechin hydrate (CH) parent ion (protonated precursor [M–H]− ions at m/z 289.23) and (B) catechin hydrate (CH) product ion (major fragmented product ion at m/z 245.20) showing fragmentation transitions.
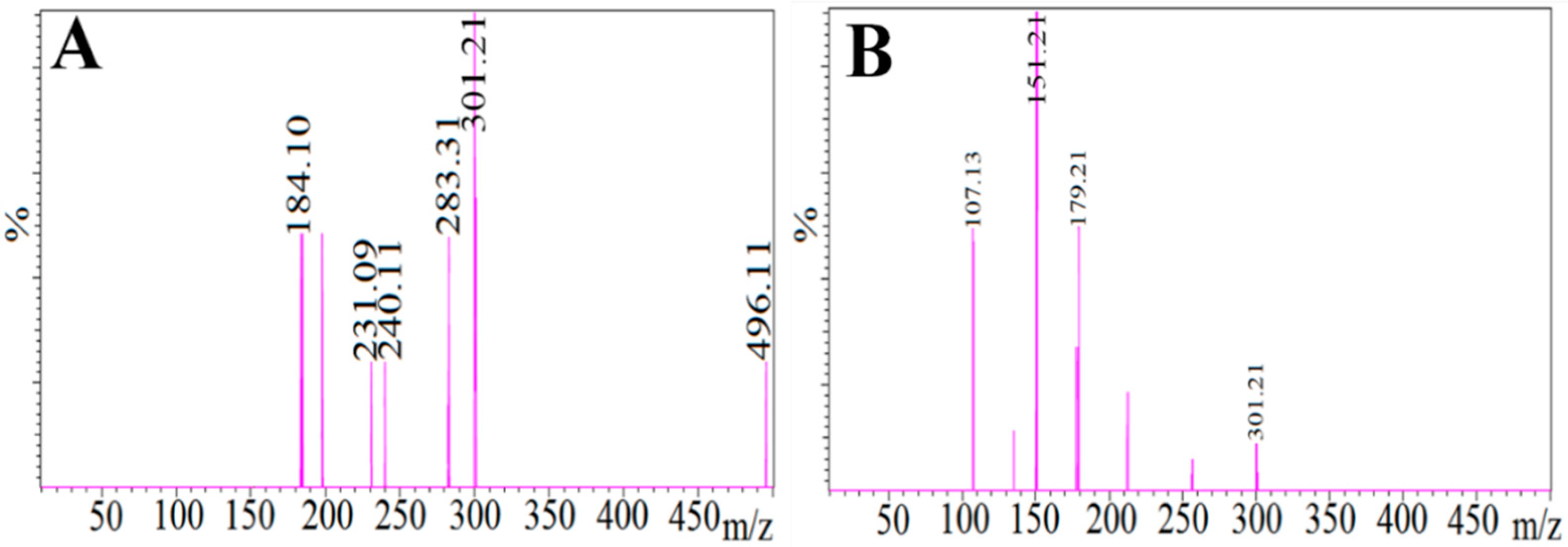
Figure 10.
Mass spectrum of, (A) quercetin (IS) precursor ion (protonated precursor [M–H]− ions at m/z 301.21 and (B) internal standard (IS) product ion (major fragmented product ions at m/z 151.21) showing fragmentation transitions.
Figure 10.
Mass spectrum of, (A) quercetin (IS) precursor ion (protonated precursor [M–H]− ions at m/z 301.21 and (B) internal standard (IS) product ion (major fragmented product ions at m/z 151.21) showing fragmentation transitions.
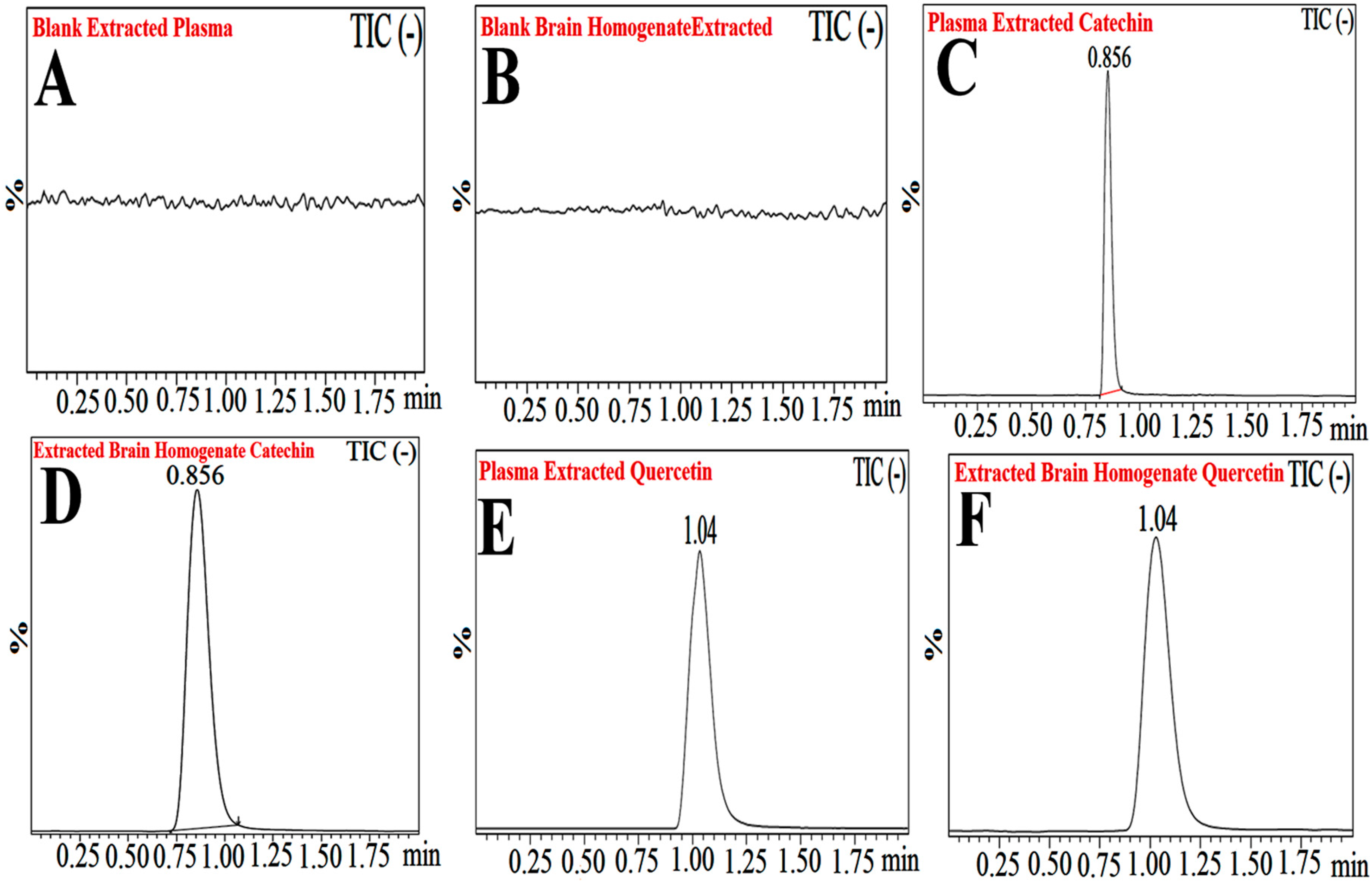
Figure 11.
Typical chromatograms of extracted blank plasma (A), extracted blank brain homogenate (B), plasma extracted catechin hydrate (CH) (C), extracted brain homogenate catechin hydrate (CH) (D), plasma extracted quercetin (IS) (E), and extracted brain homogenate quercetin (IS) (F).
Figure 11.
Typical chromatograms of extracted blank plasma (A), extracted blank brain homogenate (B), plasma extracted catechin hydrate (CH) (C), extracted brain homogenate catechin hydrate (CH) (D), plasma extracted quercetin (IS) (E), and extracted brain homogenate quercetin (IS) (F).
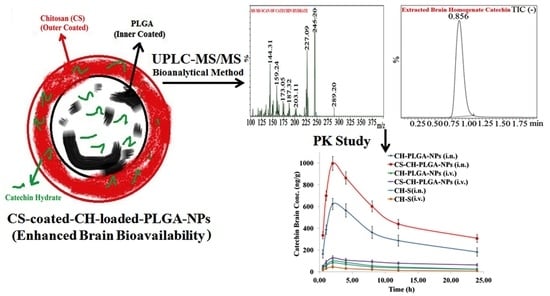
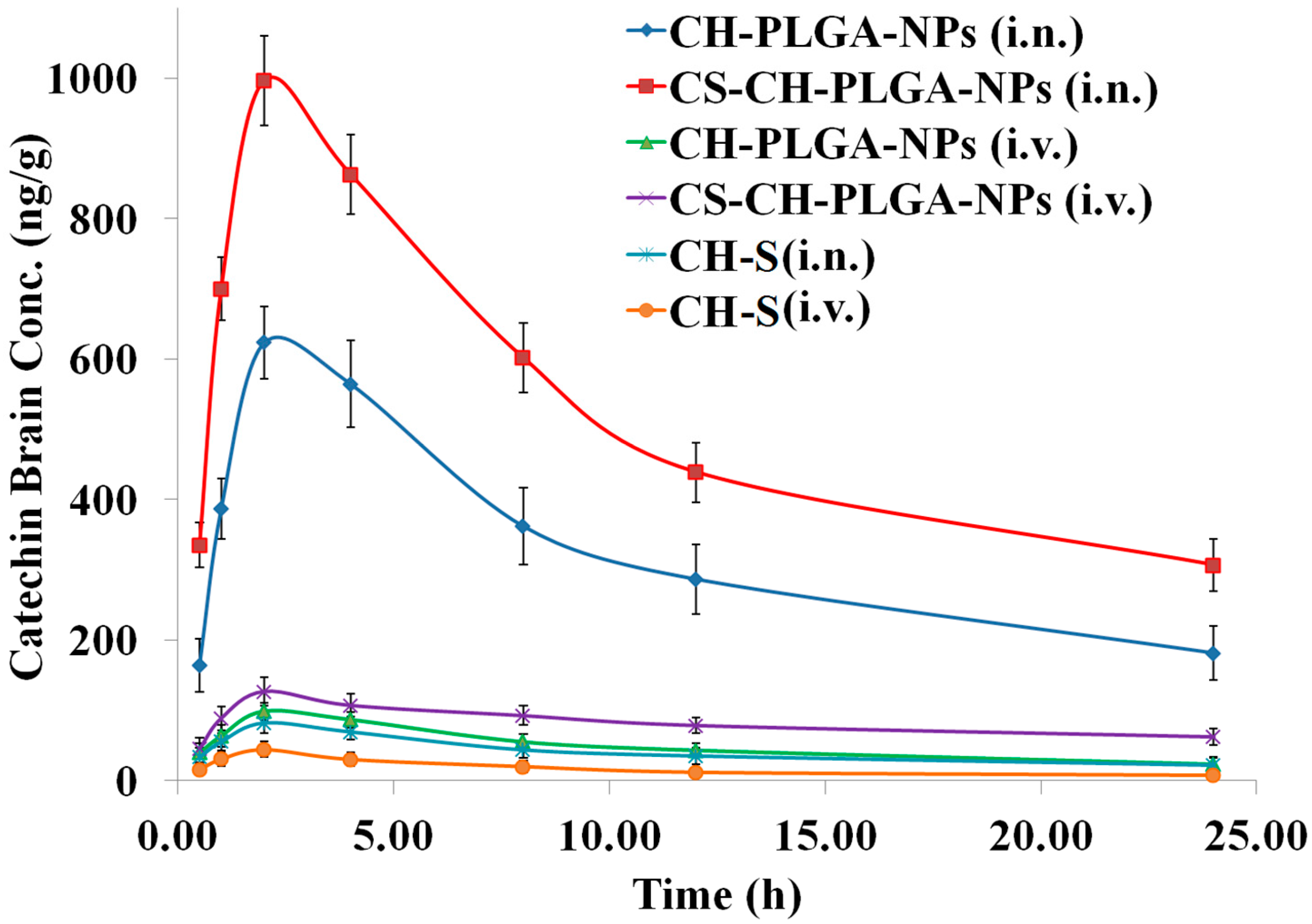
Figure 12.
Pharmacokinetic profiles of catechin hydrate (CH) concentration in the brain at different time intervals after administration of optimized different nanoformulations compared with pure catechin hydrate (CH).
Figure 12.
Pharmacokinetic profiles of catechin hydrate (CH) concentration in the brain at different time intervals after administration of optimized different nanoformulations compared with pure catechin hydrate (CH).
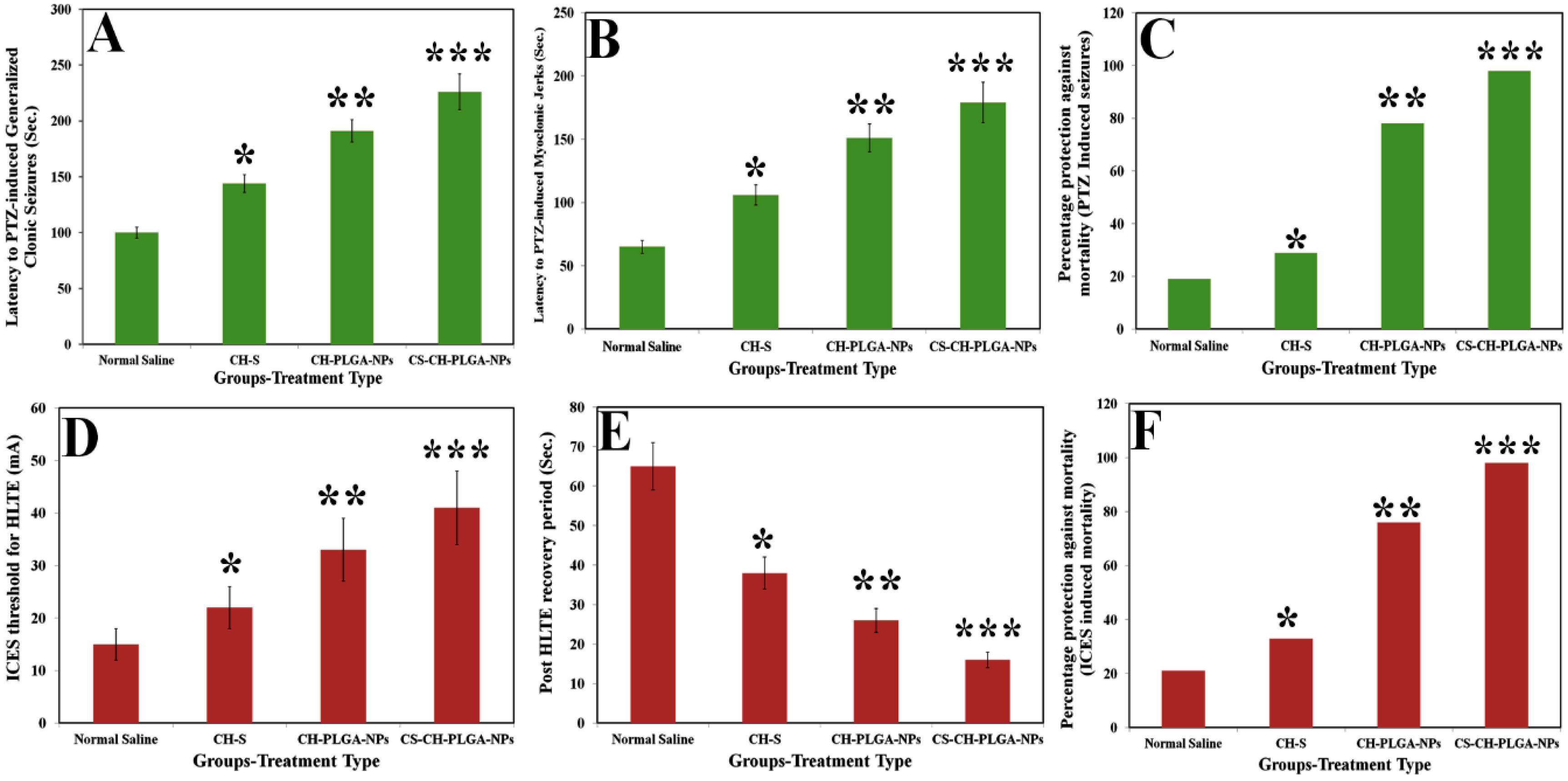
Figure 13.
Effect of different nanoformulations of catechin hydrate on pentylenetetrazole (PTZ)-induced latency to generalized seizures (A), myoclonic jerks (B), % against mortality owing to PTZ-induced seizures (C), increasing current electroshock (ICES)-induced threshold for HLTE (D), ICES-induced post-HLTE recovery time (E), and % protective effect of different nanoformulations of catechin hydrate against ICES-induced mortality in rats (F). *** (p < 0.001), ** (p < 0.01), and * (p < 0.05).
Figure 13.
Effect of different nanoformulations of catechin hydrate on pentylenetetrazole (PTZ)-induced latency to generalized seizures (A), myoclonic jerks (B), % against mortality owing to PTZ-induced seizures (C), increasing current electroshock (ICES)-induced threshold for HLTE (D), ICES-induced post-HLTE recovery time (E), and % protective effect of different nanoformulations of catechin hydrate against ICES-induced mortality in rats (F). *** (p < 0.001), ** (p < 0.01), and * (p < 0.05).
Table 1.
Variables in “Design Expert” software for the preparation and optimization of catechin hydrate (CH) PLGA–nanoparticles (NPs). PVA, polyvinyl alcohol; PDI, polydispersity index.
Table 1.
Variables in “Design Expert” software for the preparation and optimization of catechin hydrate (CH) PLGA–nanoparticles (NPs). PVA, polyvinyl alcohol; PDI, polydispersity index.
| Factors | Levels |
| Independent Variables | Low (−1) | Medium (0) | High (+1) |
| X1 = PLGA (mg) | 50 | 175 | 300 |
| X2 = PVA (%) | 0.5 | 2.0 | 4.0 |
| X3 = Sonication Time (s) | 30 | 150 | 300 |
| X4 = Temperature (°C) | 20 | 35 | 50 |
| | Constraints | Importance |
| Independent Variables |
| X1 = PLGA (mg) | Minimize | + + + + + + |
| X2 = PVA (%) | Minimize | + + + + |
| X3 = Sonication Time (s) | Minimize within the range | + + + + + + + |
| X4 = Temperature (°C) | In range | − − − − − − |
| Dependent variables |
| Y1 = Particle size (nm) | Minimize | + + + + + |
| Y2 = PDI | Minimize | + + + + |
| Y3 = Zeta Potential (mV) | Maximize | + + + + + |
Table 2.
Formulations recommended by “Design Expert” software, independent variables, and their responses.
Table 2.
Formulations recommended by “Design Expert” software, independent variables, and their responses.
| Formulation Code | Independent Variables | Dependent Variables |
|---|
| Coded Factors | Observed Responses | Predicted Responses |
|---|
| X1 | X2 | X3 | X4 | Y1 | Y2 | Y3 | Y1 | Y2 | Y3 |
|---|
| PLGA1 | 50 | 0.5 | 30 | 20 | 36.00 ± 2.01 | 0.063 ± 0.001 | −8.12 ± 0.09 | 38.56 | 0.051 | −38.53 |
| PLGA2 | 300 | 0.5 | 30 | 20 | 341.08 ± 11.01 | 0.394 ± 0.021 | −31.09 ± 3.11 | 340.16 | 0.395 | −31.13 |
| PLGA3 | 50 | 4 | 30 | 20 | 266.89 ± 7.35 | 0.412 ± 0.032 | −28.43 ± 2.91 | 266.60 | 0.413 | −28.36 |
| PLGA4 | 300 | 4 | 30 | 20 | 451.43 ± 18.54 | 0.506 ± 0.053 | −30.18 ± 2.98 | 453.41 | 0.505 | −31.08 |
| PLGA5 | 50 | 0.5 | 300 | 20 | 170.52 ± 4.27 | 0.316 ± 0.026 | −21.42 ± 2.18 | 171.64 | 0.318 | −21.50 |
| PLGA6 | 300 | 0.5 | 300 | 20 | 396.47 ± 13.54 | 0.539 ± 0.041 | −27.35 ± 3.11 | 397.10 | 0.540 | −27.13 |
| PLGA7 | 50 | 4 | 300 | 20 | 241.67 ± 7.84 | 0.394 ± 0.019 | −24.51 ± 2.01 | 241.07 | 0.395 | −24.87 |
| PLGA8 | 300 | 4 | 300 | 20 | 496.37 ± 19.64 | 0.643 ± 0.073 | −32.64 ± 4.06 | 497.64 | 0.644 | −32.62 |
| PLGA9 | 50 | 1.1 | 90 | 25 | 93.46 ± 3.94 | 0.106 ± 0.010 | –12.63 ± 0.08 | 94.14 | 0.108 | –12.61 |
| PLGA10 | 300 | 0.5 | 30 | 50 | 371.46 ± 12.64 | 0.367 ± 0.013 | −28.45 ± 2.93 | 370.75 | 0.368 | −28.43 |
| PLGA11 | 50 | 4 | 30 | 50 | 291.58 ± 8.64 | 0.423 ± 0.034 | −26.47 ± 2.16 | 289.29 | 0.424 | −26.45 |
| PLGA12 | 300 | 4 | 30 | 50 | 446.48 ± 19.31 | 0.583 ± 0.050 | −27.94 ± 3.18 | 445.10 | 0.584 | −27.91 |
| PLGA13 | 50 | 0.5 | 300 | 50 | 121.34 ± 3.59 | 0.416 ± 0.031 | −19.34 ± 1.97 | 120.98 | 0.417 | −19.31 |
| PLGA14 | 300 | 0.5 | 300 | 50 | 366.49 ± 12.26 | 0.411 ± 0.016 | −16.49 ± 1.64 | 368.09 | 0.412 | −16.46 |
| PLGA15 | 50 | 4 | 300 | 50 | 221.64 ± 6.78 | 0.389 ± 0.026 | −18.64 ± 1.77 | 222.34 | 0.388 | −18.61 |
| PLGA16 | 300 | 4 | 300 | 50 | 549.67 ± 21.97 | 0.678 ± 0.040 | −32.14 ± 3.09 | 548.15 | 0.676 | −32.13 |
| Axial Points |
| PLGA17 | 75 | 2.25 | 165 | 35 | 348.64 ± 11.19 | 0.496 ± 0.033 | −21.45 ± 2.05 | 349.06 | 0.495 | −21.44 |
| PLGA18 | 425 | 2.25 | 165 | 35 | 414.68 ± 18.67 | 0.506 ± 0.042 | −23.64 ± 2.37 | 415.64 | 0.507 | −23.61 |
| PLGA19 | 175 | 1.25 | 165 | 35 | 313.54 ± 9.91 | 0.436 ± 0.029 | −14.56 ± 2.69 | 312.47 | 0.438 | −14.50 |
| PLGA20 | 175 | 5.75 | 165 | 35 | 496.89 ± 20.54 | 0.694 ± 0.043 | −33.54 ± 4.68 | 495.05 | 0.696 | −33.55 |
| PLGA21 | 175 | 2.25 | 105 | 35 | 233.65 ± 6.92 | 0.405 ± 0.031 | −16.64 ± 1.94 | 234.24 | 0.404 | −16.65 |
| PLGA22 | 175 | 2.25 | 435 | 35 | 311.69 ± 9.68 | 0.589 ± 0.053 | −21.64 ± 2.11 | 313.74 | 0.590 | −21.62 |
| PLGA23 | 175 | 2.25 | 165 | 5 | 468.64 ± 19.16 | 0.711 ± 0.064 | −31.64 ± 3.64 | 469.56 | 0.712 | −31.61 |
| PLGA24 | 175 | 2.25 | 165 | 65 | 331.54 ± 11.09 | 0.642 ± 0.039 | −19.34 ± 1.91 | 330.05 | 0.641 | −19.32 |
| Centre Points |
| PLGA25 | 175 | 2.25 | 165 | 35 | 289.64 ± 12.08 | 0.569 ± 0.059 | –20.56 ± 2.09 | 291.01 | 0.570 | –21.06 |
| PLGA26 | 175 | 2.25 | 165 | 35 | 289.64 ± 12.08 | 0.569 ± 0.059 | –20.56 ± 2.09 | 291.01 | 0.570 | –21.06 |
| PLGA27 | 175 | 2.25 | 165 | 35 | 289.64 ± 12.08 | 0.569 ± 0.059 | –20.56 ± 2.09 | 291.01 | 0.570 | –21.06 |
| PLGA28 | 175 | 2.25 | 165 | 35 | 289.64 ± 12.08 | 0.569 ± 0.059 | –20.56 ± 2.09 | 291.01 | 0.570 | –21.06 |
| PLGA29 | 175 | 2.25 | 165 | 35 | 289.64 ± 12.08 | 0.569 ± 0.059 | –20.56 ± 2.09 | 291.01 | 0.570 | –21.06 |
| PLGA30 | 175 | 2.25 | 165 | 35 | 289.64 ± 12.08 | 0.569 ± 0.059 | –20.56 ± 2.09 | 291.01 | 0.570 | –21.06 |
Table 3.
Summary of regression analysis for responses Y1 (particle size in nm), Y2 (PDI), and Y3 (Zeta potential).
Table 3.
Summary of regression analysis for responses Y1 (particle size in nm), Y2 (PDI), and Y3 (Zeta potential).
| Quadratic Model | R2 | Adjusted R2 | Predicted R2 | SD | % Coefficient of Variation (CV) |
|---|
| Response (Y1) | 0.9938 | 0.9879 | 0.9833 | 2.52 | 4.02 |
| Response (Y2) | 0.9927 | 0.9858 | 0.9805 | 0.0177 | 4.13 |
| Response (Y3) | 0.9993 | 0.9986 | 0.9980 | 0.2578 | 1.20 |
Table 4.
Optimal predicted and optimized batch of CH–PLGA–NPs with independent variables and dependent variables with other characterizations of optimized CH–PLGA–NPs.
Table 4.
Optimal predicted and optimized batch of CH–PLGA–NPs with independent variables and dependent variables with other characterizations of optimized CH–PLGA–NPs.
| Batch | Independent Variables | Dependent Variables |
| | X1 | X2 | X3 | X4 | Y1 | Y2 | Y3 |
| Predicted | 50.7 | 1.20 | 100 | 28 | 94.14 | 0.108 | −12.61 |
| Optimized | 50.0 | 1.10 | 90 | 25 | 93.46 ± 3.94 | 0.106 ± 0.01 | −12.63 ± 0.08 |
| Optimized Dependent Variables | Actual/Observed Value | p-Value | Entrapment Efficiency | Drug Loading | pH | Process Yield (%) |
| Particle Size | 93.46 nm | <0.0001 | 73.69 ± 3.16 | 4.73 ± 0.11 | 7.4 ± 0.18 | 76.69 ± 3.41% |
| PDI | 0.106 | <0.0001 |
| Zeta Potential (mV) | −12.63 | <0.0001 |
Table 5.
Chitosan coating above selected optimized CH-loaded–PLGA–NPs tried 1% and 2% CS concentration.
Table 5.
Chitosan coating above selected optimized CH-loaded–PLGA–NPs tried 1% and 2% CS concentration.
| Formulation Code | Chitosan (%) | pH | Mean Particle Size (nm) ± SD | Polydispersity Index (PDI) ± SD | Zeta Potential (mV) | Entrapment Efficiency (EE%) ± SD | Drug Loading (DL%) ± SD |
|---|
| CS-coated-optimized–CH-loaded–PLGA–NPs 1 | 2 | 7.4 | 106.31 ± 2.91 | 0.239 ± 0.008 | 21.64 ± 1.04 | 80.36 ± 4.01 | 5.98 ± 0.36 |
| CS-coated-optimized–CH-loaded–PLGA–NPs 2 | 4 | 7.4 | 142.43 ± 3.94 | 0.364 ± 0.023 | 24.34 ± 1.59 | 81.66 ± 4.61 | 6.87 ± 0.49 |
Table 6.
Validation: precision and accuracy data for catechin hydrate (CH) in different biomatrixes. LOQQC, lower limit of quantification for quality control; LQC, lower QC; MQC, medium QC; HQC, high QC.
Table 6.
Validation: precision and accuracy data for catechin hydrate (CH) in different biomatrixes. LOQQC, lower limit of quantification for quality control; LQC, lower QC; MQC, medium QC; HQC, high QC.
| Biomatrix | Quality Controls Samples | Theoretical Concentration (ng mL−1) or (ng g−1) | Intra-Batch Precision | Inter-Batch Precision | Recovery (%) |
|---|
| Observed Concentration (ng mL−1) or (ng g−1) ± S.D. | Accuracy (%) | Precision (%C.V.) | Observed Concentration (ng mL−1) or (ng g−1) ± S.D. | Accuracy (%) | Precision (%C.V.) |
|---|
| Brain Homogenate | LOQQC | 1.01 | 0.99 ± 0.03 | 98.02 | 3.03 | 0.98 ± 0.04 | 97.03 | 2.90 | 81.47 |
| LQC | 2.90 | 2.85 ± 0.09 | 98.28 | 3.16 | 2.84 ± 0.11 | 97.93 | 2.44 | 83.48 |
| MQC | 410.00 | 407.61 ± 11.36 | 99.41 | 2.79 | 403.17 ± 12.54 | 98.33 | 1.42 | 80.17 |
| HQC | 800.00 | 789.67 ± 18.64 | 98.47 | 3.81 | 784.69 ± 19.21 | 98.09 | 0.72 | 81.38 |
| Lungs Homogenate | LOQQC | 1.01 | 0.98 ± 0.04 | 97.03 | 4.08 | 0.97 ± 0.06 | 96.04 | 2.34 | 80.25 |
| LQC | 2.90 | 2.83 ± 0.11 | 97.59 | 3.89 | 2.81 ± 0.16 | 96.90 | 1.04 | 81.67 |
| MQC | 410.00 | 404.71 ± 13.02 | 98.71 | 3.22 | 402.37 ± 14.17 | 98.14 | 0.59 | 82.49 |
| HQC | 800.00 | 779.25 ± 19.61 | 97.41 | 2.52 | 775.43 ± 20.18 | 96.93 | 0.39 | 81.52 |
| Plasma | LOQQC | 1.01 | 0.96 ± 0.03 | 95.05 | 3.13 | 0.94 ± 0.04 | 93.07 | 2.32 | 80.57 |
| LQC | 2.90 | 2.86 ± 0.14 | 98.62 | 4.90 | 2.85 ± 0.15 | 98.28 | 1.94 | 82.61 |
| MQC | 410.00 | 402.33 ± 11.42 | 98.13 | 2.84 | 401.34 ± 12.69 | 97.89 | 0.66 | 83.46 |
| HQC | 800.00 | 778.67 ± 21.64 | 97.33 | 2.78 | 774.18 ± 22.37 | 96.77 | 0.39 | 84.51 |
Table 7.
Validation: stability data for catechin hydrate (CH) in different biomatrixes.
Table 7.
Validation: stability data for catechin hydrate (CH) in different biomatrixes.
| Exposure Condition | LQC (2.90 ng/mL or ng g−1) | MQC (410.00 ng/mL or ng g−1) | HQC (800.00 ng/mL or ng g−1) |
|---|
| Brain Homogenate | Lungs Homogenate | Plasma | Brain Homogenate | Lungs Homogenate | Plasma | Brain Homogenate | Lungs Homogenate | Plasma |
|---|
| Long-term stability; recovery (ng) after storage (−80 °C) |
| Previous day | 2.84 ± 0.11 | 2.86 ± 0.10 | 2.85 ± 0.10 | 406.35 ± 11.36 | 407.11 ± 10.08 | 406.93 ± 9.36 | 794.36 ± 13.38 | 792.48 ± 12.93 | 789.45 ± 13.64 |
| 30th Day | 2.81 ± 0.10 (98.94%) | 2.83 ± 0.14 (98.95%) | 2.82 ± 0.11 (98.95%) | 400.24 ± 9.21 (98.50%) | 399.25 ± 8.96 (98.07%) | 400.21 ± 9.28 (98.35%) | 777.43 ± 12.97 (97.87%) | 781.64 ± 11.67 (98.63%) | 776.78 ± 12.32 (98.40%) |
| Freeze–thaw stress; recovery (ng) after freeze–thaw cycles (−80 °C to 25 °C) |
| Pre-Cycle | 2.87 ± 0.09 | 2.89 ± 0.09 | 2.88 ± 0.09 | 407.14 ± 10.22 | 406.15 ± 9.34 | 405.33 ± 9.37 | 792.47 ± 13.57 | 790.45 ± 12.31 | 789.49 ± 11.64 |
| First Cycle | 2.84 ± 0.11 (98.95%) | 2.87 ± 0.08 (99.31%) | 2.85 ± 0.07 (98.96%) | 403.25 ± 9.24 (99.04%) | 402.34 ± 10.04 (99.06%) | 401.24 ± 9.01 (98.99%) | 784.19 ± 11.38 (98.96%) | 781.44 ± 12.10 (98.86%) | 777.41 ± 12.48 (98.47%) |
| Second Cycle | 2.81 ± 0.10 (97.91%) | 2.85 ± 0.09 (98.62%) | 2.84 ± 0.08 (98.61%) | 400.14 ± 9.04 (98.28) | 399.28 ± 9.03 (98.31%) | 396.32 ± 8.94 (97.78%) | 771.58 ± 12.02 (97.36%) | 773.24 ± 11.64 (97.82%) | 761.58 ± 14.64 (96.46%) |
| Third Cycle | 2.79 ± 0.08 (97.21%) | 2.82 ± 0.07 (97.58%) | 2.82 ± 0.07 (97.92%) | 397.16 ± 10.04 (97.55%) | 392.14 ± 11.04 (96.55%) | 390.18 ± 9.01 (96.26%) | 764.28 ± 12.09 (96.44) | 763.24 ± 13.08 (96.56%) | 753.49 ± 10.39 (95.44%) |
| Bench top stability; recovery (ng) at room temperature (25 °C) |
| 0 h | 2.88 ± 0.07 | 2.89 ± 0.09 | 2.88 ± 0.07 | 408.01 ± 11.01 | 405.97 ± 10.13 | 406.48 ± 10.17 | 791.41 ± 11.92 | 793.46 ± 13.01 | 791.61 ± 12.07 |
| 24 h | 2.85 ± 0.08 (96.96%) | 2.87 ± 0.07 (99.31%) | 2.83 ± 0.04 (98.26%) | 399.97 ± 10.16 (98.03%) | 399.46 ± 9.63 (98.40%) | 399.94 ± 9.64 (99.39%) | 779.94 ± 12.06 (98.55%) | 778.92 ± 11.64 (98.17%) | 780.06 ± 13.08 (98.54%) |
| Post-processing stability; recovery (ng) after storage in auto sampler (4 °C) |
| 0 h | 2.85 ± 0.07 | 2.87 ± 0.06 | 2.88 ± 0.09 | 404.36 ± 8.94 | 406.14 ± 9.43 | 407.36 ± 10.00 | 790.34 ± 11.56 | 789.64 ± 11.64 | 791.25 ± 12.32 |
| 4 h | 2.82 ± 0.06 (98.95%) | 2.84 ± 0.07 (98.95%) | 2.83 ± 0.06 (98.26%) | 398.99 ± 9.02 (98.67%) | 399.04 ± 8.81 (98.25%) | 401.03 ± 9.34 (98.45%) | 779.86 ± 12.34 (98.67%) | 774.64 ± 12.08 (98.10%) | 776.34 ± 10.05 (98.12%) |
Table 8.
Definitions and formulas used in the calculations.
Table 8.
Definitions and formulas used in the calculations.
| Accuracy (%) = Mean value of [(mean observed concentration)/(theoretical concentration)] × 100; |
| Precision (%): Coefficient of variance (percentage) = standard deviation divided by mean concentration found × 100; |
| Recovery (%) = Mean value of (peak height (mV) obtained from extracted biological sample)/(peak height (mV) obtained from aqueous sample) × 100. |
Table 9.
Pharmacokinetics (PKs) of catechin hydrate after i.n. and i.v. administration to rats at the dose of 10 mg kg−1 in the brain, lungs, and plasma (n = 6, mean ± SD). AUC, area under curve.
Table 9.
Pharmacokinetics (PKs) of catechin hydrate after i.n. and i.v. administration to rats at the dose of 10 mg kg−1 in the brain, lungs, and plasma (n = 6, mean ± SD). AUC, area under curve.
| Formulation Administration | Samples | Cmax (ng/mL g) | Tmax | t1/2 (h) | Ke (h−1) | AUC0–t (ng min/mL g) |
| CH–S (i.n.) | Brain | 81.68 ± 10.13 | 2.00 | 15.25 ± 2.06 | 0.04544 ± 0.0006 | 977.32 ± 33.14 |
| Lungs | 28.64 ± 3.09 | 2.00 | 21.77 ± 2.19 | 0.03184 ± 0.00011 | 373.85 ± 16.47 |
| Plasma | 32.02 ± 2.63 | 0.50 | 8.23 ± 3.10 | 0.08420 ± 0.0009 | 170.48 ± 18.43 |
| CH–S (i.v.) | Brain | 44.23 ± 7.04 | 2.00 | 11.16 ± 0.39 | 0.06210 ± 0.0009 | 395.26 ± 19.47 |
| Lungs | 95.43 ± 18.14 | 2.00 | 9.19 ± 0.81 | 0.07540 ± 0.00051 | 958.79 ± 34.64 |
| Plasma | 1141.54 ± 83.45 | 0.50 | 4.69 ± 0.29 | 0.14767 ± 0.0018 | 3167.33 ± 55.67 |
| CH–PLGA–NPs (i.n.) | Brain | 623.36 ± 24.21 | 2.00 | 24.71 ± 4.59 | 0.02806 ± 0.00006 | 7785.90 ± 83.64 |
| Lungs | 106.97 ± 11.97 | 2.00 | 11.00 ± 1.01 | 0.06303 ± 0.00011 | 1033.42 ± 44.67 |
| Plasma | 56.78 ± 4.64 | 2.00 | 8.26 ± 0.97 | 0.08391 ± 0.00008 | 409.41 ± 25.94 |
| CS–CH–PLGA–NPs (i.n.) | Brain | 996.24 ± 44.24 | 2.00 | 21.81 ± 3.94 | 0.03178 ± 0.00008 | 12444.50 ± 139.25 |
| Lungs | 121.64 ± 18.45 | 2.00 | 11.21 ± 2.88 | 0.06184 ± 0.0008 | 1187.01 ± 31.04 |
| Plasma | 99.67 ± 10.18 | 2.00 | 9.33 ± 0.94 | 0.07427 ± 0.00013 | 936.04 ± 86.79 |
| CH–PLGA–NPs (i.v.) | Brain | 98.36 ± 19.55 | 2.00 | 15.91 ± 3.06 | 0.04355 ± 0.00010 | 1170.58 ± 28.57 |
| Lungs | 109.94 ± 7.98 | 2.00 | 11.52 ± 2.97 | 0.06016 ± 0.0005 | 1091.68 ± 34.67 |
| Plasma | 757.54 ± 33.16 | 1.00 | 16.65 ± 1.06 | 0.04162 ± 0.0007 | 9430.18 ± 189.67 |
| CS–CH–PLGA–NPs (i.v.) | Brain | 126.48 ± 17.61 | 2.00 | 67.83 ± 5.47 | 0.01022 ± 0.00006 | 1959.57 ± 79.68 |
| Lungs | 136.48 ± 15.97 | 2.00 | 62.03 ± 2.91 | 0.01117 ± 0.0010 | 1974.57 ± 44.89 |
| Plasma | 1064.43 ± 96.43 | 1.00 | 15.83 ± 1.06 | 0.04378 ± 0.0006 | 12948.59 ± 277.13 |
| CH–S (i.n.) | Brain/Plasma | 2.55 | 4.00 | 1.85 | 0.54 | 5.73 |
| CH–S (i.v.) | Brain/Plasma | 0.039 | 4.00 | 2.38 | 0.42 | 0.13 |
| CH–PLGA–NPs (i.n.) | Brain/Plasma | 10.98 | 1.00 | 2.99 | 0.34 | 19.02 |
| CH–PLGA–NPs (i.v.) | Brain/Plasma | 0.13 | 2.00 | 0.96 | 1.05 | 0.12 |
| CS–CH–PLGA–NPs (i.n.) | Brain/Plasma | 10.00 | 1.00 | 2.34 | 0.43 | 13.29 |
| CS–CH–PLGA–NPs (i.v.) | Brain/Plasma | 0.13 | 2.00 | 4.29 | 0.23 | 0.15 |
| Comparative Bioavailability * (AUCi.n./AUCi.v.); (%) |
| Formulations | CH–S | CH–PLGA–NPs | CS–CH–PLGA–NPs |
| Blood | 5.38 | 4.34 | 7.23 |
| Brain | 247.26 * | 665.13 ** | 1063.11 *** |

 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}