PLGA Microspheres with Alginate-Coated Large Pores for the Formulation of an Injectable Depot of Donepezil Hydrochloride

 ,
, 
Abstract
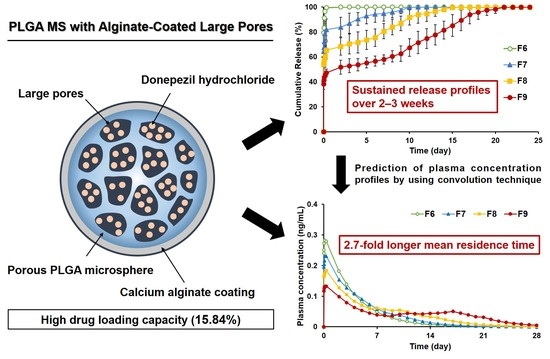
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Porous PLGA MS
2.3. Evaluation of Encapsulation Efficiency (EE) and Loading Capacity (LC) in Porous PLGA MS
2.4. HPLC Analysis of Donepezil
2.5. Preparation of PLGA MS with Closed Pores
2.6. Observation of Morphology of Porous or Pore-Closed PLGA MS
2.7. Assessment of Pore Size and Porosity of Porous PLGA MS
2.8. Evaluation of Particle Size Distribution of Porous or Pore-Closed PLGA MS
2.9. Viscosity Measurements of Spraying Suspensions Containing PLGA MS
2.10. Injectability
2.11. In Vitro Drug Release of Porous or Pore-Closed PLGA MS
2.12. Prediction of Plasma Drug Concentration Profiles by Using Convolution Technique
2.13. Statistical Analysis
3. Results and Discussion
3.1. Characterization of Porous PLGA MS
3.1.1. Surface Morphology and Pore Characteristics of Porous PLGA MS
3.1.2. EE and LC of Donepezil in Porous PLGA MS
3.2. Characterization of PLGA MS with Closed Pores
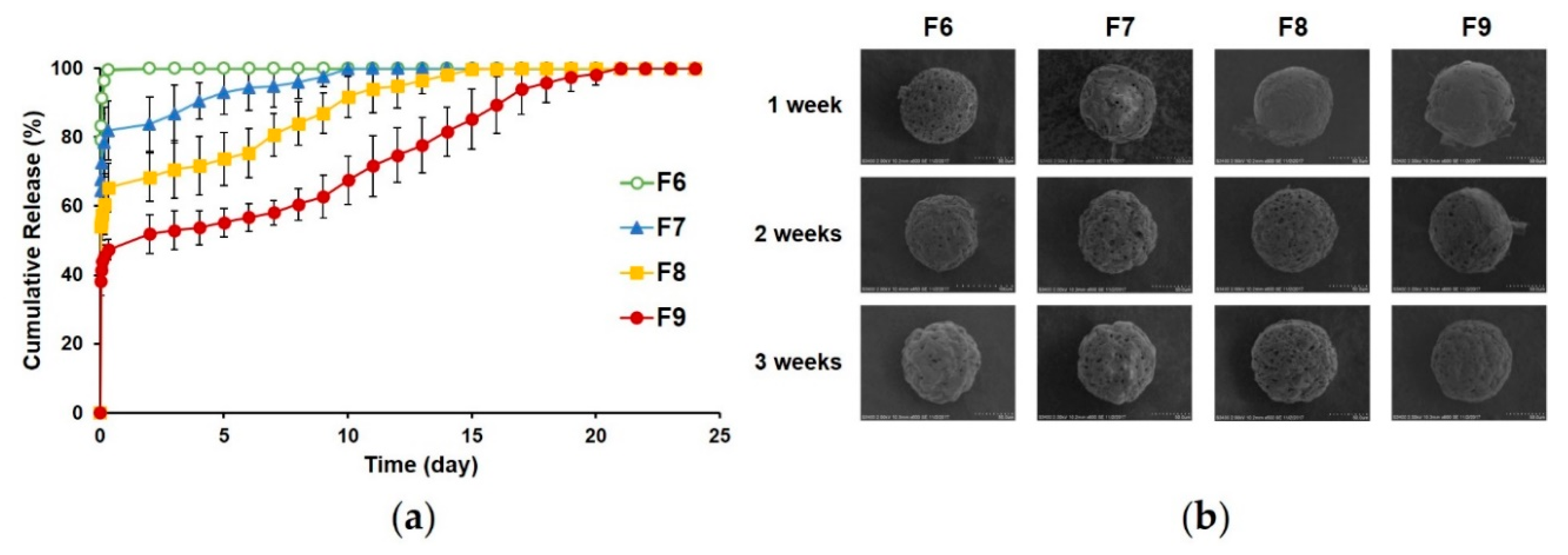
3.2.1. Surface Morphology of PLGA MS with Pores Closed by Calcium Alginate Coating
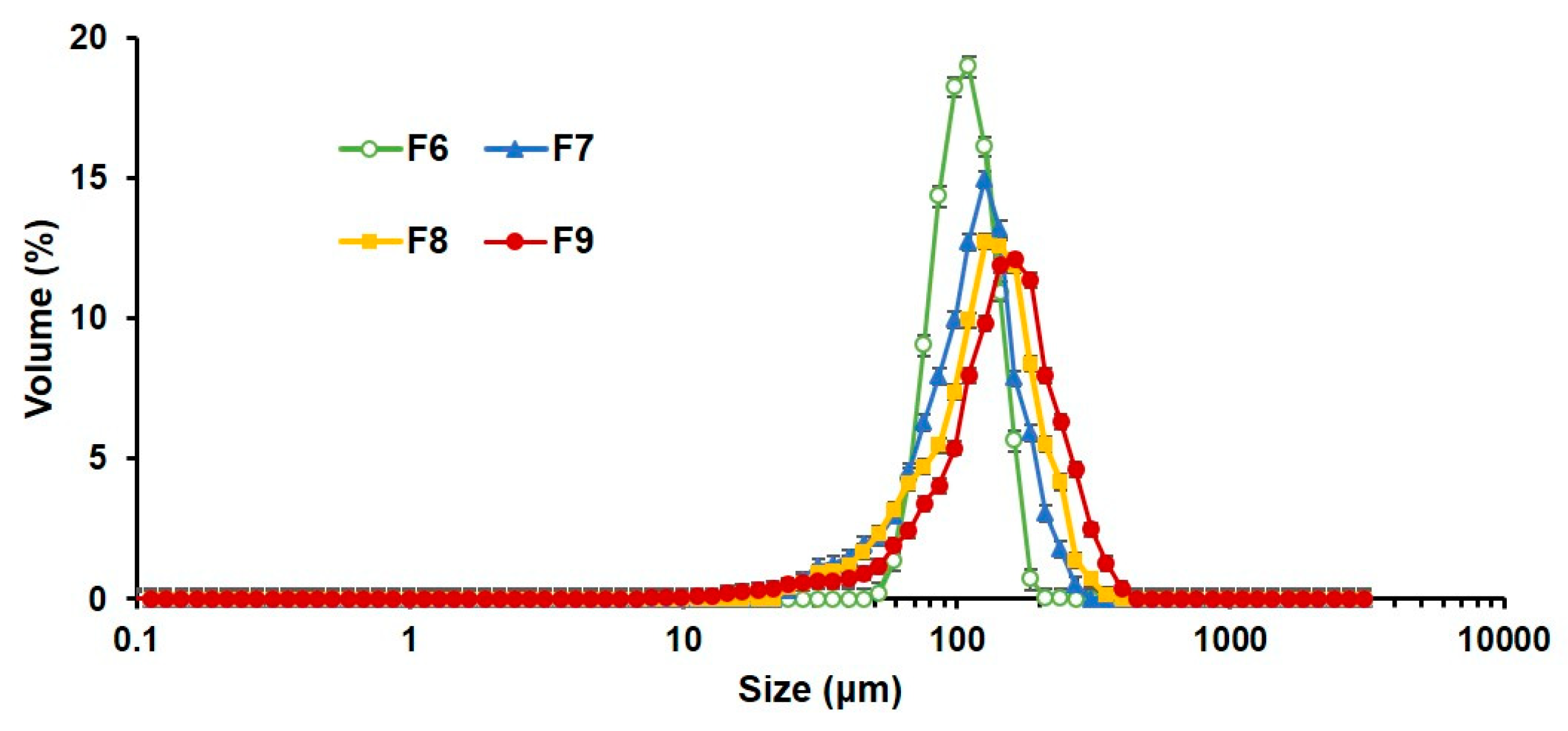
3.2.2. Particle Size of PLGA MS with Closed Pores and Viscosity of Spraying Suspensions
3.2.3. Injectability of PLGA MS with Closed Pores
3.2.4. In Vitro Release
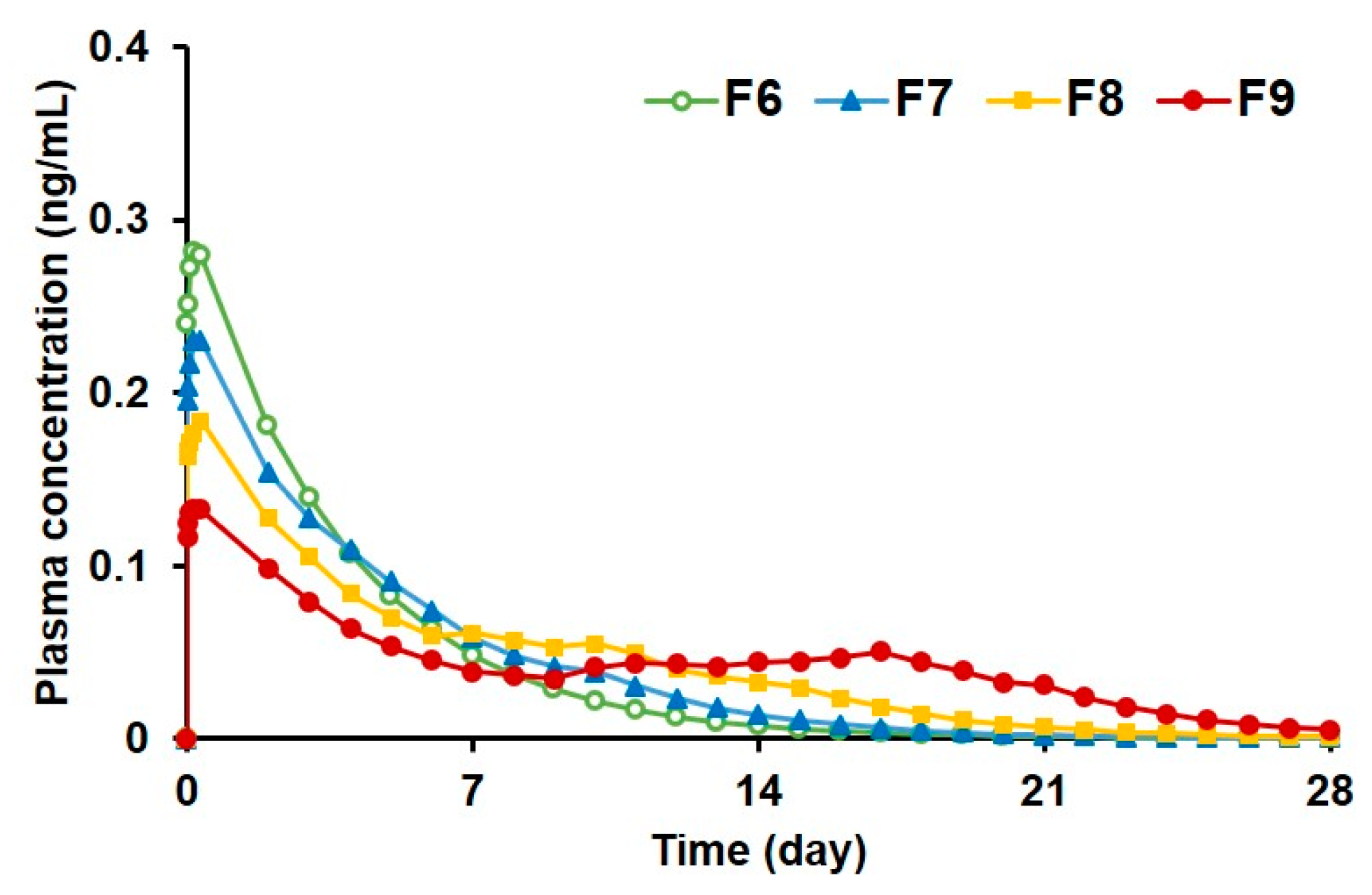
3.2.5. Prediction of Plasma Drug Concentrations for PLGA MS Depot Formulations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isik, A.T.; Yildiz, G.B.; Bozoglu, E.; Yay, A.; Aydemir, E. Cardiac safety of donepezil in elderly patients with Alzheimer disease. Intern. Med. 2012, 51, 575–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, P.T. The interplay of neurotransmitters in Alzheimer’s disease. CNS Spectr. 2005, 10, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry Res. 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Tsuno, N. Donepezil in the treatment of patients with Alzheimer’s disease. Expert Rev. Neurother. 2009, 9, 591–598. [Google Scholar] [CrossRef]
- Ritchie, C.W.; Ames, D.; Clayton, T.; Lai, R. Metaanalysis of randomized trials of the efficacy and safety of donepezil, galantamine, and rivastigmine for the treatment of Alzheimer disease. Am. J. Geriatr. Psychiatry 2004, 12, 358–369. [Google Scholar] [CrossRef]
- Qizilbash, N.; Birks, J.; Lopez Arrieta, J.; Lewington, S.; Szeto, S. Withdrawn: Tacrine for Alzheimer’s disease. Cochrane Database Syst. Rev. 2007, 3, CD000202. [Google Scholar]
- Guo, W.J.; Quan, P.; Fang, L.; Cun, D.M.; Yang, M.S. Sustained release donepezil loaded PLGA microspheres for injection: Preparation, in vitro and in vivo study. Asian J. Pharm. Sci. 2015, 10, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Chen, R.; An, W.; Wang, C.; Liao, G.; Dong, X.; Bi, A.; Yin, Z.; Luo, L. A novel acetylcholinesterase inhibitor and calcium channel blocker SCR-1693 improves Abeta25-35-impaired mouse cognitive function. Psychopharmacology 2016, 233, 599–613. [Google Scholar] [CrossRef]
- Farlow, M.R.; Salloway, S.; Tariot, P.N.; Yardley, J.; Moline, M.L.; Wang, Q.; Brand-Schieber, E.; Zou, H.; Hsu, T.; Satlin, A. Effectiveness and tolerability of high-dose (23 mg/d) versus standard-dose (10 mg/d) donepezil in moderate to severe Alzheimer’s disease: A 24-week, randomized, double-blind study. Clin. Ther. 2010, 32, 1234–1251. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Chen, L.; Gu, W.; Xu, Z.; Gao, Y.; Li, Y. In vitro and in vivo evaluation of donepezil-sustained release microparticles for the treatment of Alzheimer’s disease. Biomaterials 2007, 28, 1882–1888. [Google Scholar] [CrossRef] [PubMed]
- Schwalbe, O.; Scheerans, C.; Freiberg, I.; Schmidt-Pokrzywniak, A.; Stang, A.; Kloft, C. Compliance assessment of ambulatory Alzheimer patients to aid therapeutic decisions by healthcare professionals. BMC Health Serv. Res. 2010, 10, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sozio, P.; Cerasa, L.S.; Marinelli, L.; Di Stefano, A. Transdermal donepezil on the treatment of Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2012, 8, 361–368. [Google Scholar]
- Nicoll, L.H.; Hesby, A. Intramuscular injection: An integrative research review and guideline for evidence-based practice. Appl. Nurs. Res. 2002, 15, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Cocoman, A.; Murray, J. Intramuscular injections: A review of best practice for mental health nurses. J. Psychiatr. Ment. Health Nurs. 2008, 15, 424–434. [Google Scholar] [CrossRef]
- Patel, R.M. Parenteral suspension: An overview. Int. J. Curr. Pharm. Res. 2010, 2, 3–13. [Google Scholar]
- Hippalgaonkar, K.; Majumdar, S.; Kansara, V. Injectable lipid emulsions-advancements, opportunities and challenges. AAPS PharmSciTech 2010, 11, 1526–1540. [Google Scholar] [CrossRef] [Green Version]
- Barcia, E.; Herrero-Vanrell, R.; Diez, A.; Alvarez-Santiago, C.; Lopez, I.; Calonge, M. Downregulation of endotoxin-induced uveitis by intravitreal injection of polylactic-glycolic acid (PLGA) microspheres loaded with dexamethasone. Exp. Eye Res. 2009, 89, 238–245. [Google Scholar] [CrossRef]
- Schwendeman, S.P.; Shah, R.B.; Bailey, B.A.; Schwendeman, A.S. Injectable controlled release depots for large molecules. J. Control. Release 2014, 190, 240–253. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Wang, J.; Ke, H.; Wang, S.; Dai, Z. Graphene oxide modified PLA microcapsules containing gold nanoparticles for ultrasonic/CT bimodal imaging guided photothermal tumor therapy. Biomaterials 2013, 34, 4794–4802. [Google Scholar] [CrossRef]
- Schreier, H.; Levy, M.; Mihalko, P. Sustained release of liposome-encapsulated gentamicin and fate of phospholipid following intramuscular injection in mice. J. Control. Release 1987, 5, 187–192. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.Z.; Chen, W.P.; Lee, C.P.; Wu, S.L.; Wang, Y.C.; Chung, T.W. Effects of alginate coated on PLGA microspheres for delivery tetracycline hydrochloride to periodontal pockets. J. Microencapsul. 2004, 21, 643–652. [Google Scholar] [CrossRef]
- Kim, H.K.; Chung, H.J.; Park, T.G. Biodegradable polymeric microspheres with “open/closed” pores for sustained release of human growth hormone. J. Control. Release 2006, 112, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, Y.X.; Zhang, H.X.; Zhou, W.Q.; Ma, G.H. A novel strategy for the preparation of porous microspheres and its application in peptide drug loading. J. Colloid Interface Sci. 2016, 478, 46–53. [Google Scholar] [CrossRef] [PubMed]
- de Lima, I.A.; Khalil, N.M.; Tominaga, T.T.; Lechanteur, A.; Sarmento, B.; Mainardes, R.M. Mucoadhesive chitosan-coated PLGA nanoparticles for oral delivery of ferulic acid. Artif. Cells Nanomed. Biotechnol. 2018, 46, 993–1002. [Google Scholar] [CrossRef] [Green Version]
- Rafiee, A.; Alimohammadian, M.H.; Gazori, T.; Riazi-rad, F.; Fatemi, S.M.R.; Parizadeh, A.; Haririan, I.; Havaskary, M. Comparison of chitosan, alginate and chitosan/alginate nanoparticles with respect to their size, stability, toxicity and transfection. Asian Pac. J. Trop. Dis. 2014, 4, 372–377. [Google Scholar] [CrossRef]
- Nakamoto, T. Essentials of Machine Olfaction and Taste; Wiley Press: Singapore, 2016. [Google Scholar]
- Katuwavila, N.P.; Perera, A.; Samarakoon, S.R.; Soysa, P.; Karunaratne, V.; Amaratunga, G.A.J.; Karunaratne, D.N. Chitosan-alginate nanoparticle system efficiently delivers doxorubicin to MCF-7 Cells. J. Nanomater. 2016, 2016, 3178904. [Google Scholar] [CrossRef] [Green Version]
- Gombotz, W.R.; Wee, S.F. Protein release from alginate matrices. Adv. Drug Deliv. Rev. 2012, 64, 194–205. [Google Scholar] [CrossRef]
- Qutachi, O.; Vetsch, J.R.; Gill, D.; Cox, H.; Scurr, D.J.; Hofmann, S.; Muller, R.; Quirk, R.A.; ShakeSheff, K.M.; Rahman, C.V. Injectable and porous PLGA microspheres that form highly porous scaffolds at body temperature. Acta Biomater. 2014, 10, 5090–5098. [Google Scholar] [CrossRef] [Green Version]
- Bahcecioglu, G.; Hasirci, N.; Hasirci, V. Cell behavior on the alginate-coated PLLA/PLGA scaffolds. Int. J. Biol. Macromol. 2019, 124, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Park, H.; Lee, J.; Kim, T.H.; Lee, E.S.; Oh, K.T.; Lee, K.C.; Youn, Y.S. Highly porous large poly(lactic-co-glycolic acid) microspheres adsorbed with palmityl-acylated exendin-4 as a long-acting inhalation system for treating diabetes. Biomaterials 2011, 32, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, B.R.; Shin, Y.J.; Cho, S.; Lee, J. Controlled formation of polylysinized inner pores in injectable microspheres of low molecular weight poly(lactide-co-glycolide) designed for efficient loading of therapeutic cells. Artif. Cells Nanomed. Biotechnol. 2018, 46, S233–S246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pistone, S.; Qoragllu, D.; Smistad, G.; Hiorth, M. Formulation and preparation of stable cross-linked alginate-zinc nanoparticles in the presence of a monovalent salt. Soft Matter 2015, 11, 5765–5774. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, S.; Oda, T.; Izumida, Y.; Aoyagi, Y.; Satake, M.; Ochiai, A.; Ohkohchi, N.; Nakajima, M. Size control of calcium alginate beads containing living cells using micro-nozzle array. Biomaterials 2005, 26, 3327–3331. [Google Scholar] [CrossRef]
- Park, C.W.; Lee, H.J.; Oh, D.W.; Kang, J.H.; Han, C.S.; Kim, D.W. Preparation and in vitro/in vivo evaluation of PLGA microspheres containing norquetiapine for long-acting injection. Drug Des. Dev. Ther. 2018, 12, 711–719. [Google Scholar] [CrossRef] [Green Version]
- Pervaiz, F.; Ahmad, M.; Li, L.H.; Murtaza, G. Development and characterization of olanzapine loaded poly(lactide-co-glycolide) microspheres for depot injection: In vitro and in vivo release profiles. Curr. Drug Deliv. 2019, 16, 375–383. [Google Scholar] [CrossRef]
- Cilurzo, F.; Selmin, F.; Minghetti, P.; Adami, M.; Bertoni, E.; Lauria, S.; Montanari, L. Injectability evaluation: An open issue. AAPS PharmSciTech 2011, 12, 604–609. [Google Scholar] [CrossRef]
- Overcashier, D.E.; Chan, E.K.; Hsu, C.C. Technical considerations in the development of prefilled syringes for protein products. Am. Pharm. Rev. 2006, 9, 77–83. [Google Scholar]
- Rastogi, V.; Yadav, P.; Lal, N.; Rastogi, P.; Singh, B.K.; Verma, N.; Verma, A. Mathematical prediction of pharmacokinetic parameters-an in-vitro approach for investigating pharmaceutical products for IVIVC. Future J. Pharm. Sci. 2018, 4, 175–184. [Google Scholar] [CrossRef]
- Qureshi, S.A. In vitro-in vivo correlation (IVIVC) and determining drug concentrations in blood from dissolution testing—A simple and practical approach. Open Drug Deliv. J. 2010, 4, 38–47. [Google Scholar] [CrossRef]
- CPhI Online. Available online: https://www.cphi-online.com/donepezil-depot-injection-prod965780.html (accessed on 9 March 2020).
- Gomolin, I.H.; Smith, C.; Jeitner, T.M. Donepezil dosing strategies: Pharmacokinetic considerations. J. Am. Med. Dir. Assoc. 2011, 12, 606–608. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.L.; Cooper, N.M.; Sukovaty, R.; Pederson, J.E.; Lee, J.N.; Friedhoff, L.T. Pharmacokinetic and pharmacodynamic profile of donepezil HCl following multiple oral doses. Br. J. Clin. Pharmacol. 1998, 46, 7–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, D.L.; Cao, X.D.; Gao, H.C.; Wang, Y.J. Engineering poly(lactic-co-glycolic acid)/calcium carbonate microspheres with controllable topography and their cell response. J. Mater. Chem. B 2013, 1, 3322–3329. [Google Scholar] [CrossRef]
- Cheng, D.L.; Cao, X.D.; Gao, H.C.; Wang, Y.J. Superficially porous poly(lactic-co-glycolic acid)/calcium carbonate microsphere developed by spontaneous pore-forming method for bone repair. RSC Adv. 2013, 3, 6871–6878. [Google Scholar] [CrossRef]
- Nam, Y.S.; Yoon, J.J.; Park, T.G. A novel fabrication method of macroporous biodegradable polymer scaffolds using gas foaming salt as a porogen additive. J. Biomed. Mater. Res. 2000, 53, 1–7. [Google Scholar] [CrossRef]
- Yoon, J.J.; Park, T.G. Degradation behaviors of biodegradable macroporous scaffolds prepared by gas foaming of effervescent salts. J. Biomed. Mater. Res. 2001, 55, 401–408. [Google Scholar] [CrossRef]
- Kim, T.K.; Yoon, J.J.; Lee, D.S.; Park, T.G. Gas foamed open porous biodegradable polymeric microspheres. Biomaterials 2006, 27, 152–159. [Google Scholar] [CrossRef]
- Gharsallaoui, A.; Roudaut, G.; Chambin, O.; Voilley, A.; Saurel, R. Applications of spray-drying in microencapsulation of food ingredients: An overview. Food Res. Int. 2007, 40, 1107–1121. [Google Scholar] [CrossRef]
- Hewitt, A.J. Spray optimization through application and liquid physical property variables–I. Environmentalist 2008, 28, 25–30. [Google Scholar] [CrossRef]
- ISO 7886-1:2017(E). Part 1: Syringes for manual use. In Sterile Hypodermic Syringes for Single Use, 2nd ed.; ISO: Geneva, Switzerland, 2017. [Google Scholar]
- Street, D.; Bangsbo, J.; Juel, C. Interstitial pH in human skeletal muscle during and after dynamic graded exercise. J. Physiol. 2001, 537, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, C.H.; Gao, J.Q.; Zhang, Y.P.; Liang, W.Q. A protein delivery system: Biodegradable alginate-chitosan-poly(lactic-co-glycolic acid) composite microspheres. Biochem. Biophys. Res. Commun. 2004, 323, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Engineer, C.; Parikh, J.; Raval, A. Effect of copolymer ratio on hydrolytic degradation of poly(lactide-co-glycolide) from drug eluting coronary stents. Chem. Eng. Res. Des. 2011, 89, 328–334. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLGA MS | Formulation Code | PLGA (%, w/v) | Ammonium Bicarbonate (%, w/v) | Donepezil Hydrochloride (mg/mL) | Sodium Alginate (%, w/v) |
|---|---|---|---|---|---|
| Porous PLGA MS | F1 | 4 | - | 1 | - |
| F2 | 4 | 1 | 1 | - | |
| F3 | 4 | 2 | 1 | - | |
| F4 | 4 | 4 | 1 | - | |
| F5 | 4 | 4 | 2 | - | |
| F6 | 4 | 4 | 4 | - | |
| Pore-closed PLGA MS | F7 | 4 | 4 | 4 | 1 |
| F8 | 4 | 4 | 4 | 1.5 | |
| F9 | 4 | 4 | 4 | 2 |
| Parameter | F1 | F2 | F3 | F4 |
|---|---|---|---|---|
| Average pore size (µm) | 1.45 ± 0.38 | 2.13 ± 0.64 | 2.70 ± 0.37 | 2.86 ± 0.33 |
| Porosity (%) | 8.93 ± 0.91 | 29.9 ± 7.26 | 47.3 ± 8.11 | 63.4 ± 9.08 |
| Apparent density (g/mL) | 1.74 ± 0.66 | 1.13 ± 0.20 | 0.79 ± 0.26 | 0.43 ± 0.13 |
| Parameter | F6 | F7 | F8 | F9 |
|---|---|---|---|---|
| ŋ0 (mPa·s) | - | 48.2 ± 2.57 | 162.6 ± 3.52 | 393.8 ± 5.49 |
| d10 (μm) | 71.2 ± 0.40 | 56.1 ± 1.28 | 57.0 ± 1.81 | 64.4 ± 3.30 |
| d50 (μm) | 99.8 ± 1.73 | 108.6 ± 5.96 | 120.6 ± 4.92 | 138.9 ± 3.42 |
| d90 (μm) | 138.2 ± 3.95 | 160.2 ± 1.44 | 194.2 ± 6.92 | 235.5 ± 9.08 |
| Span | 0.67 ± 0.02 | 0.96 ± 0.07 | 1.14 ± 0.03 | 1.23 ± 0.09 |
| Formulation | PBF (N) | Fmax (N) | DGF (N) |
|---|---|---|---|
| F6 | 1.65 ± 0.09 | 1.53 ± 0.09 | 1.29 ± 0.03 |
| F7 | 1.90 ± 0.24 | 1.76 ± 0.12 | 1.47 ± 0.09 |
| F8 | 1.97 ± 0.17 | 1.74 ± 0.10 | 1.41 ± 0.13 |
| F9 | 1.89 ± 0.17 | 1.86 ± 0.07 | 1.44 ± 0.14 |
| Parameter | F6 | F7 | F8 | F9 |
|---|---|---|---|---|
| Cmax (ng/mL) | 0.28 | 0.23 | 0.18 | 0.13 |
| AUC0-∞ (ng·day/mL) | 1.16 | 1.19 | 1.21 | 1.24 |
| MRT (day) | 3.75 | 4.75 | 6.64 | 10.14 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Han, T.H.; Hong, S.-C.; Park, S.J.; Lee, Y.H.; Kim, H.; Park, M.; Lee, J. PLGA Microspheres with Alginate-Coated Large Pores for the Formulation of an Injectable Depot of Donepezil Hydrochloride. Pharmaceutics 2020, 12, 311. https://doi.org/10.3390/pharmaceutics12040311
Kim D, Han TH, Hong S-C, Park SJ, Lee YH, Kim H, Park M, Lee J. PLGA Microspheres with Alginate-Coated Large Pores for the Formulation of an Injectable Depot of Donepezil Hydrochloride. Pharmaceutics. 2020; 12(4):311. https://doi.org/10.3390/pharmaceutics12040311
Chicago/Turabian StyleKim, Dohyun, Tae Hee Han, Seong-Chul Hong, Sun Jae Park, Yong Hak Lee, Hyeongmin Kim, Minwoo Park, and Jaehwi Lee. 2020. "PLGA Microspheres with Alginate-Coated Large Pores for the Formulation of an Injectable Depot of Donepezil Hydrochloride" Pharmaceutics 12, no. 4: 311. https://doi.org/10.3390/pharmaceutics12040311
APA StyleKim, D., Han, T. H., Hong, S. -C., Park, S. J., Lee, Y. H., Kim, H., Park, M., & Lee, J. (2020). PLGA Microspheres with Alginate-Coated Large Pores for the Formulation of an Injectable Depot of Donepezil Hydrochloride. Pharmaceutics, 12(4), 311. https://doi.org/10.3390/pharmaceutics12040311