Barriers to Implementing Clinical Pharmacogenetics Testing in Sub-Saharan Africa. A Critical Review




Abstract
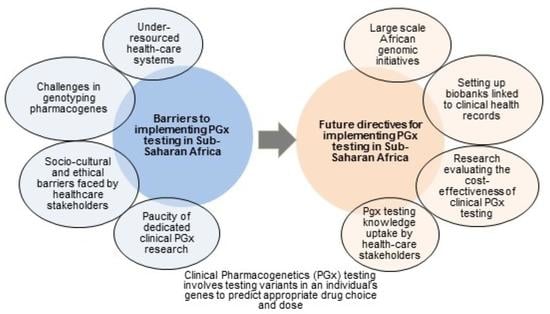
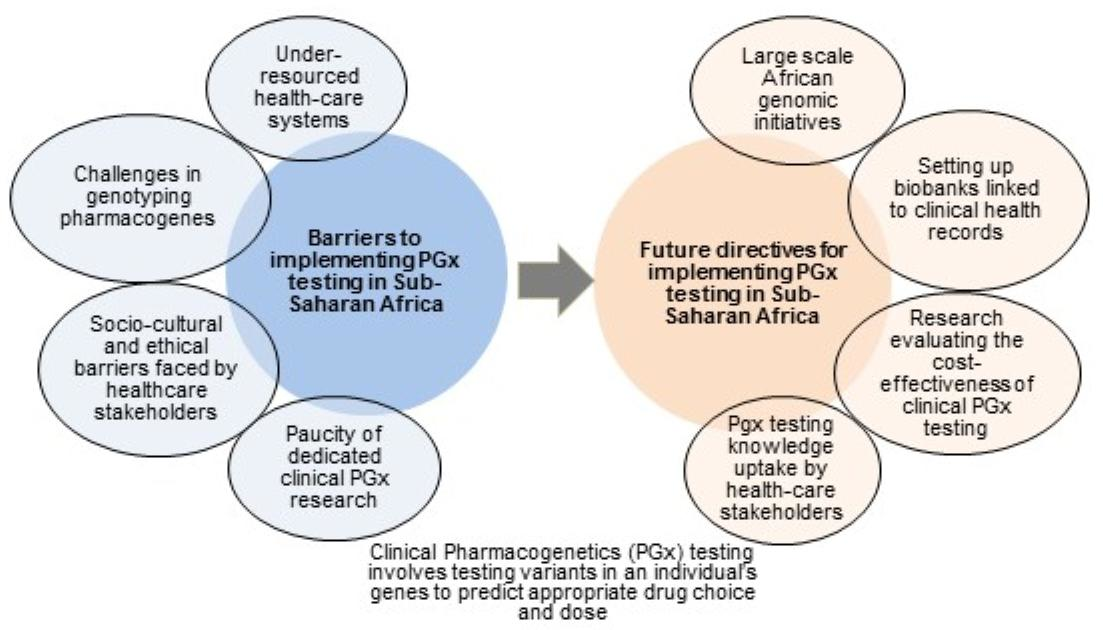
:
1. Introduction
2. Under-Resourced Clinical Health-Care Systems
3. Paucity of Clinical Pharmacogenetics Studies in SSA

{kind=link}
{kind=link}
{kind=link}
| Drugs | Clinical Study Outcome | References |
|---|---|---|
| Efavirenz | Pharmacogenetic determinants of response to Efavirenz in Black South African HIV/AIDS patients. | [41] |
| Gender, weight, and CYP2B6 genotype influence Efavirenz HIV/AIDS and TB treatment in Zimbabwe. | [26] | |
| CYP2B6 variants impact plasma Efavirenz concentrations in HIV/TB patients in Tanzania. | [27] | |
| CYP2B6 variants correlate with Efavirenz plasma concentrations in HIV patients in Zimbabwe. | [42] | |
| CYP2B6 variants and pregnancy impact on Efavirenz plasma concentrations in Nigerian patients. | [28] | |
| Novel variants in pharmacogenes are associated with Efavirenz metabolism in HIV patients in South Africa. | [30] | |
| Composite CYP2B6 alleles are significantly associated with Efavirenz-mediated central nervous system toxicity in HIV patients in Botswana. | [52] | |
| Nevirapine | CYP2B6 and CYP1A2 variants impact Nevirapine plasma concentrations and HIV progression respectively in an HIV patient cohort in Zimbabwe. | [29] |
| PEGylated Interferon-alpha/Ribavirin | IL28B SNPs correlate with treatment response in Hepatitis C patients from SSA. | [48] |
| ARV/TB | GWAS study identified SNPs linked to drug-induced hepatoxicity in HIV/TB patients in Ethiopia. | [47] |
| ARV/TB/Antimalarials | CYP2B6*6 variant and Efavirenz concentration impact on Lumefantrine plasma levels in HIV/Malaria patients in Tanzania. | [25] |
| High frequency of the CYP2B6*6 allele is associated with poor clinical response in HIV/TB/Malaria patient cohort in Congo. | [46] | |
| Lumefantrine | CYP3A4, CYP3A5 variants impact Lumefantrine response in a cohort of pregnant women with malaria in Tanzania. | [25] |
| Imatinib | CYP3A5*3 and ABCB1 C3435T variants influence clinical outcomes and plasma concentrations of Imatinib in Nigerian patients with chronic myeloid leukaemia. | [44] |
| Risperidone | CYP2D6 variants did not significantly impact the incidence of ADRs in a South African cohort. | [53] |
| Amitriptyline | CYP2D6 variants influence ADR incidence in patients with painful diabetic peripheral neuropathy in a South African cohort. | [54] |
| Rosuvastatin | Specific pharmacogene variants influencing rosuvastatin response in African populations. | [24] |
| Warfarin | CYP2C9 and VK0RC1 variants are associated with dose–response in Warfarin-treated Sudanese patients. | [55] |
| Novel CYP2C9/VK0RC1 variants influence Warfarin response in a black South African cohort. | [56] | |
| CYP2C9/VKORC1 variants did not correlate with Warfarin dose–response in a Ghanaian cohort. | [57] |
4. Challenges in Genotyping Pharmacogene Variants
5. Socio-Cultural and Ethical Challenges vis-à-vis Clinicians
6. Socio-Cultural and Ethical Challenges vis-à-vis Patients
7. Socio-Cultural and Ethical Challenges vis-à-vis Health-Care Authorities and Insurers
8. Conclusions and Future Directives
Author Contributions
Funding
Conflicts of Interest
References
- Cavallari, L.H.; Beitelshees, A.L.; Blake, K.V.; Dressler, L.G.; Duarte, J.D.; Elsey, A.; Eichmeyer, J.N.; Empey, P.E.; Franciosi, J.P.; Hicks, J.K.; et al. The IGNITE Pharmacogenetics Working Group: An Opportunity for Building Evidence with Pharmacogenetic Implementation in a Real-World Setting. Clin. Transl. Sci. 2017, 10, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Swanson, K.M.; Rojas, R.L.; Wang, Z.; St Sauver, J.L.; Visscher, S.L.; Prokop, L.J.; Bielinski, S.J.; Wang, L.; Weinshilboum, R.; et al. Systematic review of the evidence on the cost-effectiveness of pharmacogenomics-guided treatment for cardiovascular diseases. Genet. Med. 2020, 22, 475–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Relling, M.V.; Klein, T.E. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin. Pharmacol. Ther. 2011, 89, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Swen, J.J.; Wilting, I.; de Goede, A.L.; Grandia, L.; Mulder, H.; Touw, D.J.; de Boer, A.; Conemans, J.M.; Egberts, T.C.; Klungel, O.H.; et al. Pharmacogenetics: From bench to byte. Clin. Pharmacol. Ther. 2008, 83, 781–787. [Google Scholar] [CrossRef]
- PharmGKB. Available online: https://www.pharmgkb.org/ (accessed on 10 November 2019).
- EU-PIC. Available online: https://www.eu-pic.net/ (accessed on 9 August 2020).
- U-PGx. Available online: http://upgx.eu/ (accessed on 9 August 2020).
- CYTED. Available online: http://www.cyted.org/es/relivaf (accessed on 9 August 2020).
- Chumnumwat, S.; Lu, Z.H.; Sukasem, C.; Winther, M.D.; Capule, F.R.; Abdul Hamid, A.; Bhandari, B.; Chaikledkaew, U.; Chanhom, N.; Chantarangsu, S.; et al. Southeast Asian Pharmacogenomics Research Network (SEAPharm): Current Status and Perspectives. Public Health Genom. 2019, 22, 132–139. [Google Scholar] [CrossRef]
- FDA. Available online: https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling (accessed on 10 December 2019).
- Mhalu, F.S. Burden of diseases in poor resource countries: Meeting the challenges of combating HIV/AIDS, tuberculosis and malaria. Tanzan. Health Res. Bull. 2005, 7, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Gouda, H.N.; Charlson, F.; Sorsdahl, K.; Ahmadzada, S.; Ferrari, A.J.; Erskine, H.; Leung, J.; Santamauro, D.; Lund, C.; Aminde, L.N.; et al. Burden of non-communicable diseases in sub-Saharan Africa, 1990-2017: Results from the Global Burden of Disease Study 2017. Lancet Glob. Health 2019, 7, e1375–e1387. [Google Scholar] [CrossRef] [Green Version]
- Ampadu, H.H.; Hoekman, J.; de Bruin, M.L.; Pal, S.N.; Olsson, S.; Sartori, D.; Leufkens, H.G.; Dodoo, A.N. Adverse Drug Reaction Reporting in Africa and a Comparison of Individual Case Safety Report Characteristics Between Africa and the Rest of the World: Analyses of Spontaneous Reports in VigiBase(R). Drug Saf. 2016, 39, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Asiimwe, I.G.; Zhang, E.J.; Osanlou, R.; Krause, A.; Dillon, C.; Suarez-Kurtz, G.; Zhang, H.; Perini, J.A.; Renta, J.Y.; Duconge, J.; et al. Genetic Factors Influencing Warfarin Dose in Black-African Patients: A Systematic Review and Meta-Analysis. Clin. Pharmacol. Ther. 2020, 107, 1420–1433. [Google Scholar] [CrossRef]
- CPIC. Available online: https://cpicpgx.org/genes-drugs/ (accessed on 30 July 2020).
- Maranville, J.C.; Cox, N.J. Pharmacogenomic variants have larger effect sizes than genetic variants associated with other dichotomous complex traits. Pharm. J. 2016, 16, 388–392. [Google Scholar] [CrossRef]
- Quinones, L.A.; Lavanderos, M.A.; Cayun, J.P.; Garcia-Martin, E.; Agundez, J.A.; Caceres, D.D.; Roco, A.M.; Morales, J.E.; Herrera, L.; Encina, G.; et al. Perception of the usefulness of drug/gene pairs and barriers for pharmacogenomics in Latin America. Curr. Drug Metab. 2014, 15, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Matimba, A.; Oluka, M.N.; Ebeshi, B.U.; Sayi, J.; Bolaji, O.O.; Guantai, A.N.; Masimirembwa, C.M. Establishment of a biobank and pharmacogenetics database of African populations. Eur. J. Hum. Genet. 2008, 16, 780–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tishkoff, S.A.; Reed, F.A.; Friedlaender, F.R.; Ehret, C.; Ranciaro, A.; Froment, A.; Hirbo, J.B.; Awomoyi, A.A.; Bodo, J.M.; Doumbo, O.; et al. The genetic structure and history of Africans and African Americans. Science 2009, 324, 1035–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajman, I.; Knapp, L.; Morgan, T.; Masimirembwa, C. African Genetic Diversity: Implications for Cytochrome P450-mediated Drug Metabolism and Drug Development. EBioMedicine 2017, 17, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Dandara, C.; Masimirembwa, C.; Haffani, Y.Z.; Ogutu, R.; Mabuka, G.; Aklillu, E.; Bolaji, R. African Pharmacogenomics Consortium: Consolidating pharmacogenomics knowledge, capacity development and translation in Africa. AAS Open Res. 2019, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- H3Africa. Available online: https://h3africa.org/ (accessed on 10 November 2019).
- Soko, N.D.; Chimusa, E.; Masimirembwa, C.; Dandara, C. An African-specific profile of pharmacogene variants for rosuvastatin plasma variability: Limited role for SLCO1B1 c.521T>C and ABCG2 c.421A>C. Pharm. J. 2019, 19, 240–248. [Google Scholar] [CrossRef]
- Mutagonda, R.F.; Kamuhabwa, A.A.R.; Minzi, O.M.S.; Massawe, S.N.; Asghar, M.; Homann, M.V.; Farnert, A.; Aklillu, E. Effect of pharmacogenetics on plasma lumefantrine pharmacokinetics and malaria treatment outcome in pregnant women. Malar. J. 2017, 16, 267. [Google Scholar] [CrossRef]
- Nemaura, T.; Nhachi, C.; Masimirembwa, C. Impact of gender, weight and CYP2B6 genotype on efavirenz exposure in patients on HIV/AIDS and TB treatment: Implications for individualising therapy. Afr. J. Pharm. Pharmacol. 2012, 6, 2188–2193. [Google Scholar]
- Ngaimisi, E.; Mugusi, S.; Minzi, O.; Sasi, P.; Riedel, K.D.; Suda, A.; Ueda, N.; Janabi, M.; Mugusi, F.; Haefeli, W.E.; et al. Effect of rifampicin and CYP2B6 genotype on long-term efavirenz autoinduction and plasma exposure in HIV patients with or without tuberculosis. Clin. Pharmacol. Ther. 2011, 90, 406–413. [Google Scholar] [CrossRef]
- Olagunju, A.; Bolaji, O.; Amara, A.; Else, L.; Okafor, O.; Adejuyigbe, E.; Oyigboja, J.; Back, D.; Khoo, S.; Owen, A. Pharmacogenetics of pregnancy-induced changes in efavirenz pharmacokinetics. Clin. Pharmacol. Ther. 2015, 97, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Mhandire, D.; Lacerda, M.; Castel, S.; Mhandire, K.; Zhou, D.; Swart, M.; Shamu, T.; Smith, P.; Musingwini, T.; Wiesner, L.; et al. Effects of CYP2B6 and CYP1A2 Genetic Variation on Nevirapine Plasma Concentration and Pharmacodynamics as Measured by CD4 Cell Count in Zimbabwean HIV-Infected Patients. Omics 2015, 19, 553–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decloedt, E.H.; Sinxadi, P.Z.; van Zyl, G.U.; Wiesner, L.; Khoo, S.; Joska, J.A.; Haas, D.W.; Maartens, G. Pharmacogenetics and pharmacokinetics of CNS penetration of efavirenz and its metabolites. J. Antimicrob. Chemother. 2019, 74, 699–709. [Google Scholar] [CrossRef] [PubMed]
- WHO. Available online: https://www.who.int/bulletin/africanhealth/en/ (accessed on 25 July 2020).
- Mpye, K.L.; Matimba, A.; Dzobo, K.; Chirikure, S.; Wonkam, A.; Dandara, C. Disease burden and the role of pharmacogenomics in African populations. Glob. Health Epidemiol. Genom. 2017, 2, e1. [Google Scholar] [CrossRef] [Green Version]
- Cavallari, L.H.; Van Driest, S.L.; Prows, C.A.; Bishop, J.R.; Limdi, N.A.; Pratt, V.M.; Ramsey, L.B.; Smith, D.M.; Tuteja, S.; Duong, B.Q.; et al. Multi-site investigation of strategies for the clinical implementation of CYP2D6 genotyping to guide drug prescribing. Genet. Med. 2019, 21, 2255–2263. [Google Scholar] [CrossRef]
- Odekunle, F.F.; Odekunle, R.O.; Shankar, S. Why sub-Saharan Africa lags in electronic health record adoption and possible strategies to increase its adoption in this region. Int. J. Health Sci. 2017, 11, 59–64. [Google Scholar]
- Aquilante, C.L.; Kao, D.P.; Trinkley, K.E.; Lin, C.T.; Crooks, K.R.; Hearst, E.C.; Hess, S.J.; Kudron, E.L.; Lee, Y.M.; Liko, I.; et al. Clinical implementation of pharmacogenomics via a health system-wide research biobank: The University of Colorado experience. Pharmacogenomics 2020, 21, 375–386. [Google Scholar] [CrossRef] [Green Version]
- Christoffels, A.; Abayomi, A. Careful governance of African biobanks. Lancet 2020, 395, 29–30. [Google Scholar] [CrossRef] [Green Version]
- Border, R.; Johnson, E.C.; Evans, L.M.; Smolen, A.; Berley, N.; Sullivan, P.F.; Keller, M.C. No Support for Historical Candidate Gene or Candidate Gene-by-Interaction Hypotheses for Major Depression Across Multiple Large Samples. Am. J. Psychiatry 2019, 176, 376–387. [Google Scholar] [CrossRef]
- Lewis, C.M.; Vassos, E. Polygenic risk scores: From research tools to clinical instruments. Genome Med. 2020, 12, 44. [Google Scholar] [CrossRef]
- Wilson, P.W.; Meigs, J.B.; Sullivan, L.; Fox, C.S.; Nathan, D.M.; D’Agostino, R.B., Sr. Prediction of incident diabetes mellitus in middle-aged adults: The Framingham Offspring Study. Arch. Intern. Med. 2007, 167, 1068–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, P.; Young, R.; Stitziel, N.O.; Padmanabhan, S.; Baber, U.; Mehran, R.; Sartori, S.; Fuster, V.; Reilly, D.F.; Butterworth, A.; et al. Polygenic Risk Score Identifies Subgroup With Higher Burden of Atherosclerosis and Greater Relative Benefit From Statin Therapy in the Primary Prevention Setting. Circulation 2017, 135, 2091–2101. [Google Scholar] [CrossRef] [Green Version]
- Swart, M.; Evans, J.; Skelton, M.; Castel, S.; Wiesner, L.; Smith, P.J.; Dandara, C. An Expanded Analysis of Pharmacogenetics Determinants of Efavirenz Response that Includes 3’-UTR Single Nucleotide Polymorphisms among Black South African HIV/AIDS Patients. Front. Genet. 2015, 6, 356. [Google Scholar] [CrossRef] [PubMed]
- Nyakutira, C.; Roshammar, D.; Chigutsa, E.; Chonzi, P.; Ashton, M.; Nhachi, C.; Masimirembwa, C. High prevalence of the CYP2B6 516G-->T(*6) variant and effect on the population pharmacokinetics of efavirenz in HIV/AIDS outpatients in Zimbabwe. Eur. J. Clin. Pharmacol. 2008, 64, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Soko, N.D.; Masimirembwa, C.; Dandara, C. Pharmacogenomics of Rosuvastatin: A Glocal (Global plus Local) African Perspective and Expert Review on a Statin Drug. Omics 2016, 20, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Adeagbo, B.A.; Bolaji, O.O.; Olugbade, T.A.; Durosinmi, M.A.; Bolarinwa, R.A.; Masimirembwa, C. Influence of CYP3A5*3 and ABCB1 C3435T on clinical outcomes and trough plasma concentrations of imatinib in Nigerians with chronic myeloid leukaemia. J. Clin. Pharm. Ther. 2016, 41, 546–551. [Google Scholar] [CrossRef]
- Maganda, B.A.; Minzi, O.M.; Ngaimisi, E.; Kamuhabwa, A.A.; Aklillu, E. CYP2B6*6 genotype and high efavirenz plasma concentration but not nevirapine are associated with low lumefantrine plasma exposure and poor treatment response in HIV-malaria-coinfected patients. Pharm. J. 2016, 16, 88–95. [Google Scholar] [CrossRef]
- Peko, S.M.; Gueye, N.S.G.; Vouvoungui, C.; Koukouikila-Koussounda, F.; Kobawila, S.C.; Nderu, D.; Velavan, T.P.; Ntoumi, F. Cytochrome P450 CYP2B6*6 distribution among Congolese individuals with HIV, Tuberculosis and Malaria infection. Int. J. Infect. Dis. 2019, 82, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Petros, Z.; Lee, M.M.; Takahashi, A.; Zhang, Y.; Yimer, G.; Habtewold, A.; Amogne, W.; Aderaye, G.; Schuppe-Koistinen, I.; Mushiroda, T.; et al. Genome-wide association and replication study of anti-tuberculosis drugs-induced liver toxicity. BMC Genom. 2016, 17, 755. [Google Scholar] [CrossRef] [Green Version]
- Asselah, T.; De Muynck, S.; Broet, P.; Masliah-Planchon, J.; Blanluet, M.; Bieche, I.; Lapalus, M.; Martinot-Peignoux, M.; Lada, O.; Estrabaud, E.; et al. IL28B polymorphism is associated with treatment response in patients with genotype 4 chronic hepatitis C. J. Hepatol. 2012, 56, 527–532. [Google Scholar] [CrossRef]
- Carpenter, D.; Gonzalez, D.; Retsch-Bogart, G.; Sleath, B.; Wilfond, B. Methodological and Ethical Issues in Pediatric Medication Safety Research. Pediatrics 2017, 140, e20170195. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Mkrtchian, S.; Kumondai, M.; Hiratsuka, M.; Lauschke, V.M. An optimized prediction framework to assess the functional impact of pharmacogenetic variants. Pharm. J. 2019, 19, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Bernstein, D.; Cole, F.S.; Khokha, M.K.; Lee, F.S.; Lin, S.; McDonald, T.V.; Moskowitz, I.P.; Quertermous, T.; Sankaran, V.G.; et al. Functional Assays to Screen and Dissect Genomic Hits: Doubling Down on the National Investment in Genomic Research. Circ. Genom. Precis. Med. 2018, 11, e002178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vujkovic, M.; Bellamy, S.L.; Zuppa, A.F.; Gastonguay, M.R.; Moorthy, G.S.; Ratshaa, B.; Han, X.; Steenhoff, A.P.; Mosepele, M.; Strom, B.L.; et al. Polymorphisms in cytochrome P450 are associated with extensive efavirenz pharmacokinetics and CNS toxicities in an HIV cohort in Botswana. Pharm. J. 2018, 18, 678–688. [Google Scholar]
- Dodgen, T.M.; Eloff, A.; Mataboge, C.; Roos, L.J.; van Staden, W.C.; Pepper, M.S. Risperidone-associated adverse drug reactions and CYP2D6 polymorphisms in a South African cohort. Appl. Transl. Genom. 2015, 5, 40–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, M.; Alessandrini, M.; Rademan, J.; Dodgen, T.M.; Steffens, F.E.; van Zyl, D.G.; Gaedigk, A.; Pepper, M.S. Impact of CYP2D6 genotype on amitriptyline efficacy for the treatment of diabetic peripheral neuropathy: A pilot study. Pharmacogenomics 2017, 18, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Shrif, N.E.; Won, H.H.; Lee, S.T.; Park, J.H.; Kim, K.K.; Kim, M.J.; Kim, S.; Lee, S.Y.; Ki, C.S.; Osman, I.M.; et al. Evaluation of the effects of VKORC1 polymorphisms and haplotypes, CYP2C9 genotypes, and clinical factors on warfarin response in Sudanese patients. Eur. J. Clin. Pharmacol. 2011, 67, 1119–1130. [Google Scholar] [CrossRef]
- Mitchell, C.; Gregersen, N.; Krause, A. Novel CYP2C9 and VKORC1 gene variants associated with warfarin dosage variability in the South African black population. Pharmacogenomics 2011, 12, 953–963. [Google Scholar] [CrossRef]
- Kudzi, W.A.S.; Dzudzor, B.; Olayemi, E.; Nartey, E.T.; Asmah, R.H. Genetic polymorphisms of patients on stable warfarin maintenance therapy in a Ghanaian population. BMC Res. Notes 2019, 9, 507. [Google Scholar] [CrossRef] [Green Version]
- Maigetter, K.; Pollock, A.M.; Kadam, A.; Ward, K.; Weiss, M.G. Pharmacovigilance in India, Uganda and South Africa with reference to WHO’s minimum requirements. Int. J. Health Policy Manag. 2015, 4, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Hershman, D.; McBride, R.; Jacobson, J.S.; Lamerato, L.; Roberts, K.; Grann, V.R.; Neugut, A.I. Racial disparities in treatment and survival among women with early-stage breast cancer. J. Clin. Oncol. 2005, 23, 6639–6646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramamoorthy, A.; Pacanowski, M.A.; Bull, J.; Zhang, L. Racial/ethnic differences in drug disposition and response: Review of recently approved drugs. Clin. Pharmacol. Ther. 2015, 97, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Gaedigk, A.; Sangkuhl, K.; Whirl-Carrillo, M.; Klein, T.; Leeder, J.S. Prediction of CYP2D6 phenotype from genotype across world populations. Genet. Med. 2017, 19, 69–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thummel, K.E.; Lin, Y.S. Sources of interindividual variability. Methods Mol. Biol. 2014, 1113, 363–415. [Google Scholar]
- Wang, S.; Pitt, J.J.; Zheng, Y.; Yoshimatsu, T.F.; Gao, G.; Sanni, A.; Oluwasola, O.; Ajani, M.; Fitzgerald, D.; Odetunde, A.; et al. Germline variants and somatic mutation signatures of breast cancer across populations of African and European ancestry in the US and Nigeria. Int. J. Cancer 2019, 145, 3321–3333. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.R.; Smith, R.L. Inflammation-induced phenoconversion of polymorphic drug metabolizing enzymes: Hypothesis with implications for personalized medicine. Drug Metab. Dispos. 2015, 43, 400–410. [Google Scholar] [CrossRef] [Green Version]
- Kiss, A.F.; Vasko, D.; Deri, M.T.; Toth, K.; Monostory, K. Combination of CYP2C19 genotype with non-genetic factors evoking phenoconversion improves phenotype prediction. Pharmacol. Rep. 2018, 70, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Bedada, W.; de Andres, F.; Engidawork, E.; Hussein, J.; LLerena, A.; Aklillu, E. Effects of Khat (Catha edulis) use on catalytic activities of major drug-metabolizing cytochrome P450 enzymes and implication of pharmacogenetic variations. Sci. Rep. 2018, 8, 12726. [Google Scholar] [CrossRef]
- Bousman, C.A.; Jaksa, P.; Pantelis, C. Systematic evaluation of commercial pharmacogenetic testing in psychiatry: A focus on CYP2D6 and CYP2C19 allele coverage and results reporting. Pharm. Genom. 2017, 27, 387–393. [Google Scholar] [CrossRef]
- Nofziger, C.; Paulmichl, M. Accurately genotyping CYP2D6: Not for the faint of heart. Pharmacogenomics 2018, 19, 999–1002. [Google Scholar] [CrossRef] [Green Version]
- Tshabalala, S.; Choudhury, A.; Beeton-Kempen, N.; Martinson, N.; Ramsay, M.; Mancama, D. Targeted ultra-deep sequencing of a South African Bantu-speaking cohort to comprehensively map and characterize common and novel variants in 65 pharmacologically-related genes. Pharm. Genom. 2019, 29, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Rusk, N. Pan-African genome. Nat. Methods 2019, 16, 143. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Ramsay, M.; Hazelhurst, S.; Aron, S.; Bardien, S.; Botha, G.; Chimusa, E.R.; Christoffels, A.; Gamieldien, J.; Sefid-Dashti, M.J.; et al. Whole-genome sequencing for an enhanced understanding of genetic variation among South Africans. Nat. Commun. 2017, 8, 2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurdasani, D.; Carstensen, T.; Tekola-Ayele, F.; Pagani, L.; Tachmazidou, I.; Hatzikotoulas, K.; Karthikeyan, S.; Iles, L.; Pollard, M.O.; Choudhury, A.; et al. The African Genome Variation Project shapes medical genetics in Africa. Nature 2015, 517, 327–332. [Google Scholar] [CrossRef] [Green Version]
- MalariaGEN. Available online: https://www.malariagen.net/ (accessed on 23 July 2019).
- Zhong, A.; Darren, B.; Loiseau, B.; He, L.Q.B.; Chang, T.; Hill, J.; Dimaras, H. Ethical, social, and cultural issues related to clinical genetic testing and counseling in low- and middle-income countries: A systematic review. Genet. Med. 2018. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.C.; Rideout, A.L.; Wilson, B.J.; Allanson, J.M.; Blaine, S.M.; Esplen, M.J.; Farrell, S.A.; Graham, G.E.; MacKenzie, J.; Meschino, W.; et al. Genetic education for primary care providers: Improving attitudes, knowledge, and confidence. Can. Fam. Physician 2009, 55, e92–e99. [Google Scholar]
- Jegede, A.S. Culture and genetic screening in Africa. Dev. World Bioeth. 2009, 9, 128–137. [Google Scholar] [CrossRef]
- Nembaware, V.; Johnston, K.; Diallo, A.A.; Kotze, M.J.; Matimba, A.; Moodley, K.; Tangwa, G.B.; Torrorey-Sawe, R.; Tiffin, N. A framework for tiered informed consent for health genomic research in Africa. Nat. Genet. 2019, 51, 1566–1571. [Google Scholar] [CrossRef]
- De Vries, J.; Munung, S.N.; Matimba, A.; McCurdy, S.; Ouwe Missi Oukem-Boyer, O.; Staunton, C.; Yakubu, A.; Tindana, P.; Consortium, H.A. Regulation of genomic and biobanking research in Africa: A content analysis of ethics guidelines, policies and procedures from 22 African countries. BMC Med. Ethics 2017, 18, 8. [Google Scholar] [CrossRef]
- Academy of Science of South Africa (ASSAf). Available online: http://hdl.handle.net/20.500.11911/106 (accessed on 25 July 2020).


| Gene | Allele | Functional Effect | Sub-Saharan Africa | African American/Afro-Caribbean | Caucasian | Central/South Asian |
|---|---|---|---|---|---|---|
| CYP2B6 | *4 | Increased function | 0.0000 | 0.0103 | 0.0409 | 0.0990 |
| *5 | Normal function | 0.0200 | 0.0621 | 0.1155 | 0.0110 | |
| *6 | Decreased function | 0.3749 | 0.3170 | 0.2330 | 0.1850 | |
| *9 | Decreased function | - | 0.0465 | 0.0147 | 0.0590 | |
| *16 | Decreased function | 0.0054 | 0.0000 | 0.0000 | 0.0000 | |
| *18 | No function | 0.0577 | 0.0330 | 0.0000 | 0.0000 | |
| CYP2C9 | *2 | Decreased function | 0.0131 | 0.0224 | 0.1273 | 0.1138 |
| *3 | No function | 0.0112 | 0.0301 | 0.0763 | 0.1099 | |
| *5 | Decreased function | 0.0131 | 0.0116 | 0.0003 | 0.0000 | |
| *11 | Decreased function | 0.0257 | 0.0139 | 0.0016 | 0.0010 | |
| CYP2C19 | *2 | No function | 0.1568 | 0.1815 | 0.1466 | 0.2699 |
| *3 | No function | 0.0027 | 0.0028 | 0.0017 | 0.0157 | |
| *4A/B | No function | 0.0000 | 0.0000 | 0.0020 | 0.0000 | |
| *5 | No function | 0.0000 | 0.0000 | 0.0000 | 0.0032 | |
| *6 | No function | 0.0000 | 0.0000 | 0.0003 | 0.0006 | |
| *8 | No function | 0.0000 | 0.0011 | 0.0034 | 0.0000 | |
| *9 | Decreased function | 0.0270 | 0.0143 | 0.0007 | 0.0001 | |
| *10 | Decreased function | 0.0000 | 0.0033 | 0.0000 | 0.0001 | |
| *17 | Increased function | 0.1733 | 0.2072 | 0.2164 | 0.1708 | |
| CYP2D6 | 2XN | Increased function | 0.0173 | 0.0188 | 0.0084 | 0.095 |
| *3 | No function | 0.0015 | 0.0032 | 0.0159 | 0.0011 | |
| *4 | No function | 0.0338 | 0.0482 | 0.1854 | 0.0906 | |
| *5 | No function | 0.0338 | 0.0482 | 0.1854 | 0.0459 | |
| *6 | No function | 0.0000 | 0.0029 | 0.0111 | 0.0000 | |
| *8 | No function | 0.0000 | 0.0000 | 0.0002 | 0.0000 | |
| *9 | Decreased function | 0.0000 | 0.0044 | 0.0276 | 0.0300 | |
| *10 | Decreased function | 0.0557 | 0.0382 | 0.0157 | 0.0867 | |
| *14 | Decreased function | - | 0.0000 | 0.0000 | - | |
| *17 | Decreased function | 0.1929 | 0.1688 | 0.0039 | 0.0007 | |
| *41 | Decreased function | 0.1147 | 0.0372 | 0.0924 | 0.1230 | |
| CYP3A5 | *3 | No function | 0.2409 | 0.3160 | 0.9249 | 0.6733 |
| *6 | No function | 0.1932 | 0.1112 | 0.0015 | 0.0000 | |
| *7 | No function | 0.0864 | 0.1200 | 0.0000 | - | |
| DPYD | *2A | No function | 0.0000 | 0.0031 | 0.0079 | 0.0051 |
| *13 | No function | 0.0000 | 0.0000 | 0.0006 | 0.0000 | |
| 2846A > T | Decreased function | - | 0.0031 | 0.0037 | 0.0006 | |
| 1236G > A | Decreased function | 0.0000 | 0.0031 | 0.0237 | - | |
| TPMT | *2 | No function | 0.0000 | 0.0053 | 0.0021 | 0.0002 |
| *3A | No function | 0.0016 | 0.0080 | 0.0343 | 0.0042 | |
| *3B | No function | 0.0000 | 0.0000 | 0.0027 | 0.0017 | |
| *3C | No function | 0.0529 | 0.0240 | 0.0047 | 0.0112 | |
| NUDT15 | *2* | No function | - | - | 0.000 | 0.035 |
| *3 | No function | - | - | 0.002 | 0.061 | |
| *6 | Uncertain function | - | - | 0.003 | 0.013 | |
| *9 | No function | - | - | 0.002 | 0.000 | |
| SLCO1B1 | *5 | Decreased function | 0.0000 | 0.0000 | 0.0083 | 0.0224 |
| *15 | Decreased function | 0.0297 | - | 0.0439 | 0.1214 | |
| *17 | Decreased function | - | 0.1330 | 0.0519 | - | |
| UGT1A1 | *28 | Decreased function | 0.4000 | 0.3734 | 0.3165 | 0.4142 |
| *6 | Decreased function | 0.0000 | 0.0040 | 0.0079 | 0.0449 | |
| *37 | Decreased function | 0.0371 | 0.0570 | 0.0007 | 0.0000 | |
| HLA-A/HLA-B | HLA-A*31:01 | High risk allele | 0.52 | 0.98 | 2.84 | 2.20 |
| HLA-B*15:02 | High risk allele | 0.00 | 0.10 | 0.04 | 4.64 | |
| HLA-B*57:01 | High risk allele | 0.79 | 0.10 | 3.23 | 4.49 | |
| HLA-B*58:01 | High risk allele | 5.54 | 3.89 | 1.32 | 4.54 | |
| IFNL3 | IL28B:CC | Increased response | 26.8 | 15.2 | 36.5 | 1.9 |
| IL28B:CT | Increased response | 52.4 | 40.62 | 47.6 | 23 | |
| IL28B:TT | Increased response | 20.8 | 43.75 | 15.9 | 75.1 | |
| G6PD | 376A>G | Deficiency | 0.312 | - | 0.0595 | - |
| VKORC1 | 1639G>A | Decreased Warfarin dose | 12.900 | 10.274 | 41.2242 | 15.317 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
B. Tata, E.; A. Ambele, M.; S. Pepper, M. Barriers to Implementing Clinical Pharmacogenetics Testing in Sub-Saharan Africa. A Critical Review. Pharmaceutics 2020, 12, 809. https://doi.org/10.3390/pharmaceutics12090809
B. Tata E, A. Ambele M, S. Pepper M. Barriers to Implementing Clinical Pharmacogenetics Testing in Sub-Saharan Africa. A Critical Review. Pharmaceutics. 2020; 12(9):809. https://doi.org/10.3390/pharmaceutics12090809
Chicago/Turabian StyleB. Tata, Emiliene, Melvin A. Ambele, and Michael S. Pepper. 2020. "Barriers to Implementing Clinical Pharmacogenetics Testing in Sub-Saharan Africa. A Critical Review" Pharmaceutics 12, no. 9: 809. https://doi.org/10.3390/pharmaceutics12090809
APA StyleB. Tata, E., A. Ambele, M., & S. Pepper, M. (2020). Barriers to Implementing Clinical Pharmacogenetics Testing in Sub-Saharan Africa. A Critical Review. Pharmaceutics, 12(9), 809. https://doi.org/10.3390/pharmaceutics12090809