Berberine-Loaded Liposomes for the Treatment of Leishmania infantum-Infected BALB/c Mice

 , ,
, ,  ,
,  and
and 
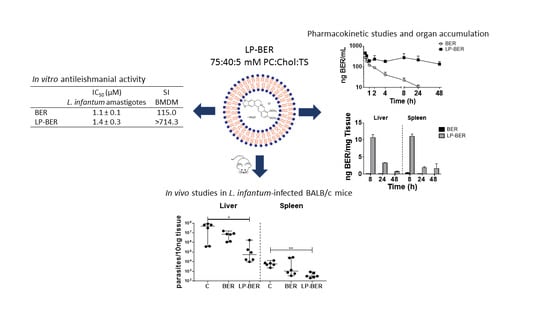
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Material
2.2. Parasites
2.3. Obtainment and Culture of Bone Marrow-Derived Macrophages (BMDM)
2.4. Animals
2.5. Preparation of BER-Containing Liposomes
2.6. Characterization of BER Liposomes
2.7. In Vitro Cytotoxicity of BER and BER Liposomes
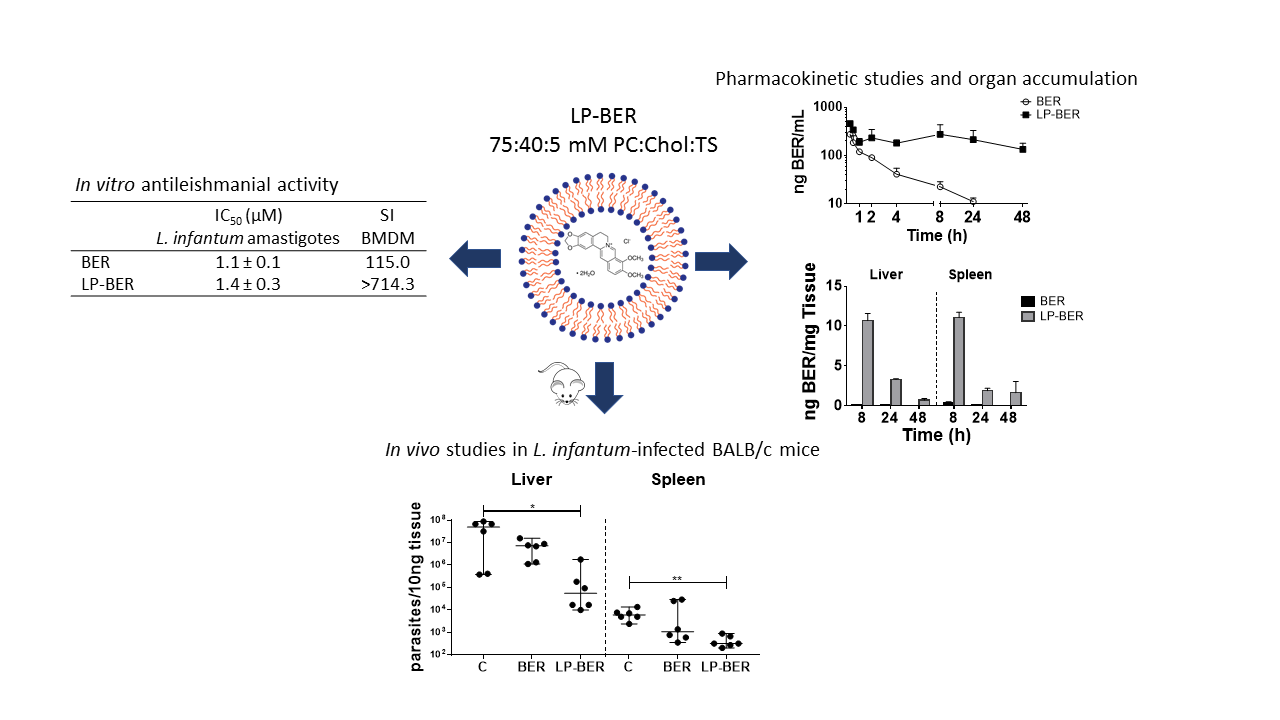
2.8. Nitric Oxide Production
2.9. In Vitro Antileishmanial Activity of BER and LP-BER Against L. infantum Promastigotes
2.10. In Vitro Antileishmanial Activity of BER and LP-BER Against L. infantum Amastigotes: Back Transformation Assay (BTA)
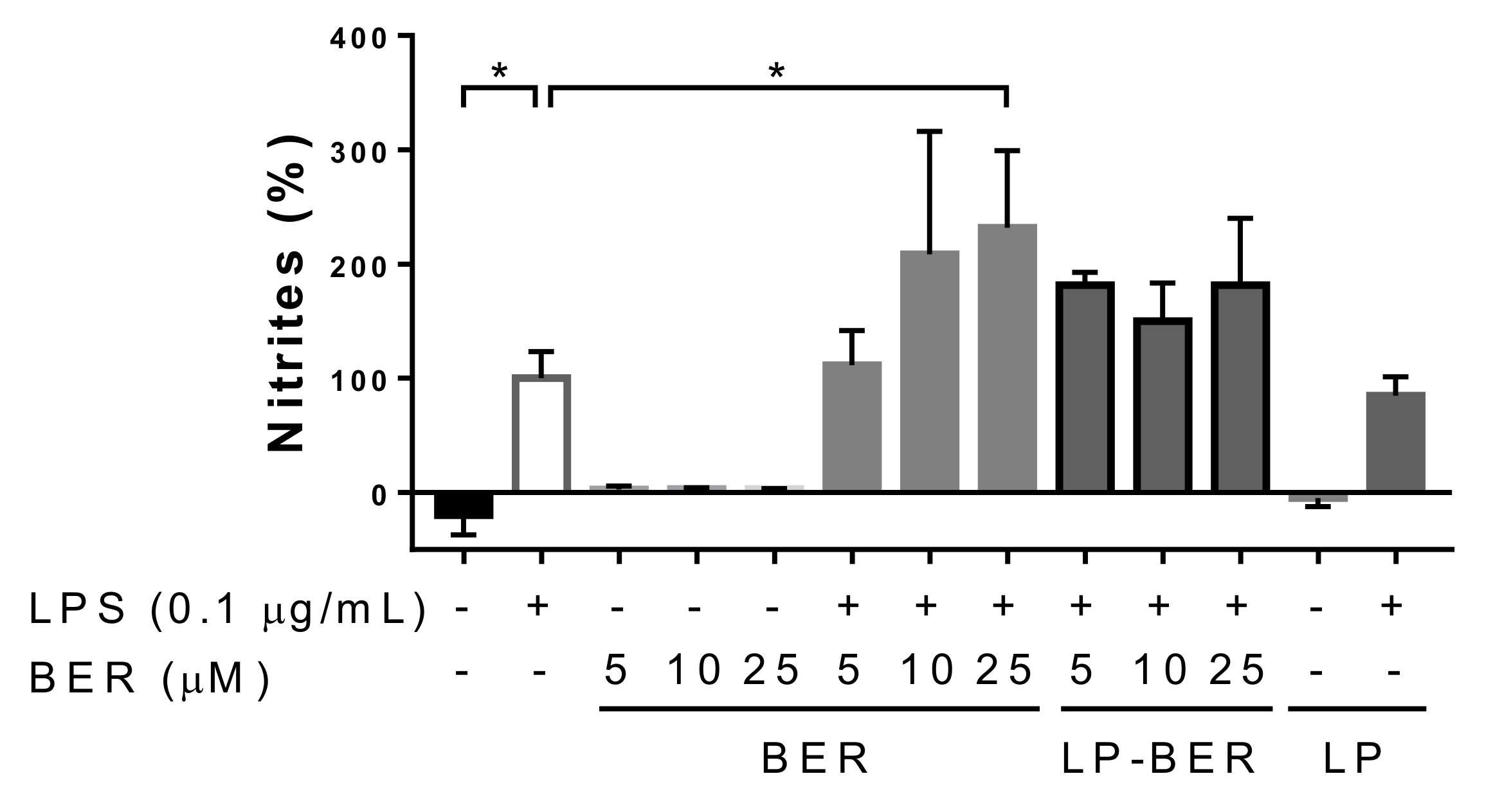
2.11. In Vitro BER Release Study in Serum
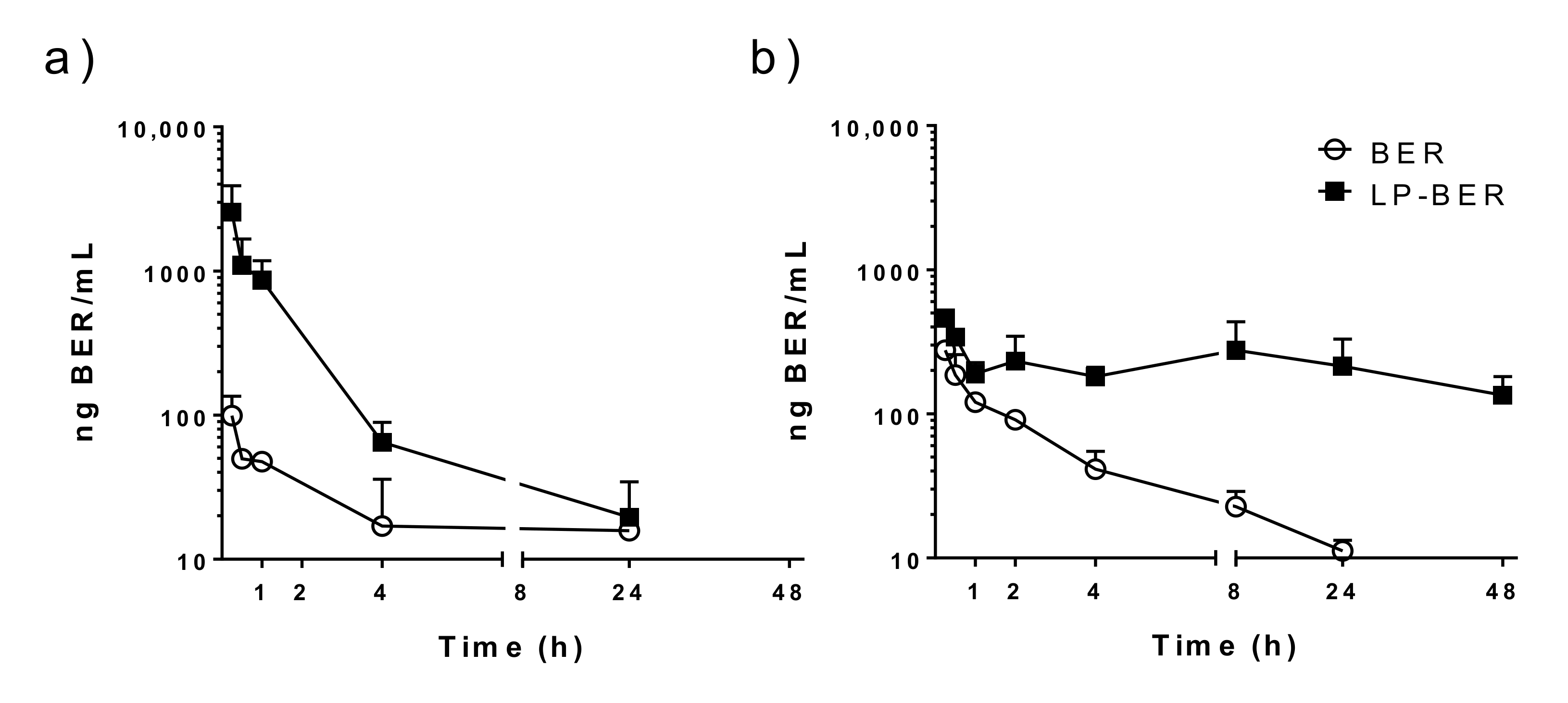
2.12. Pharmacokinetic Studies of BER and LP-BER
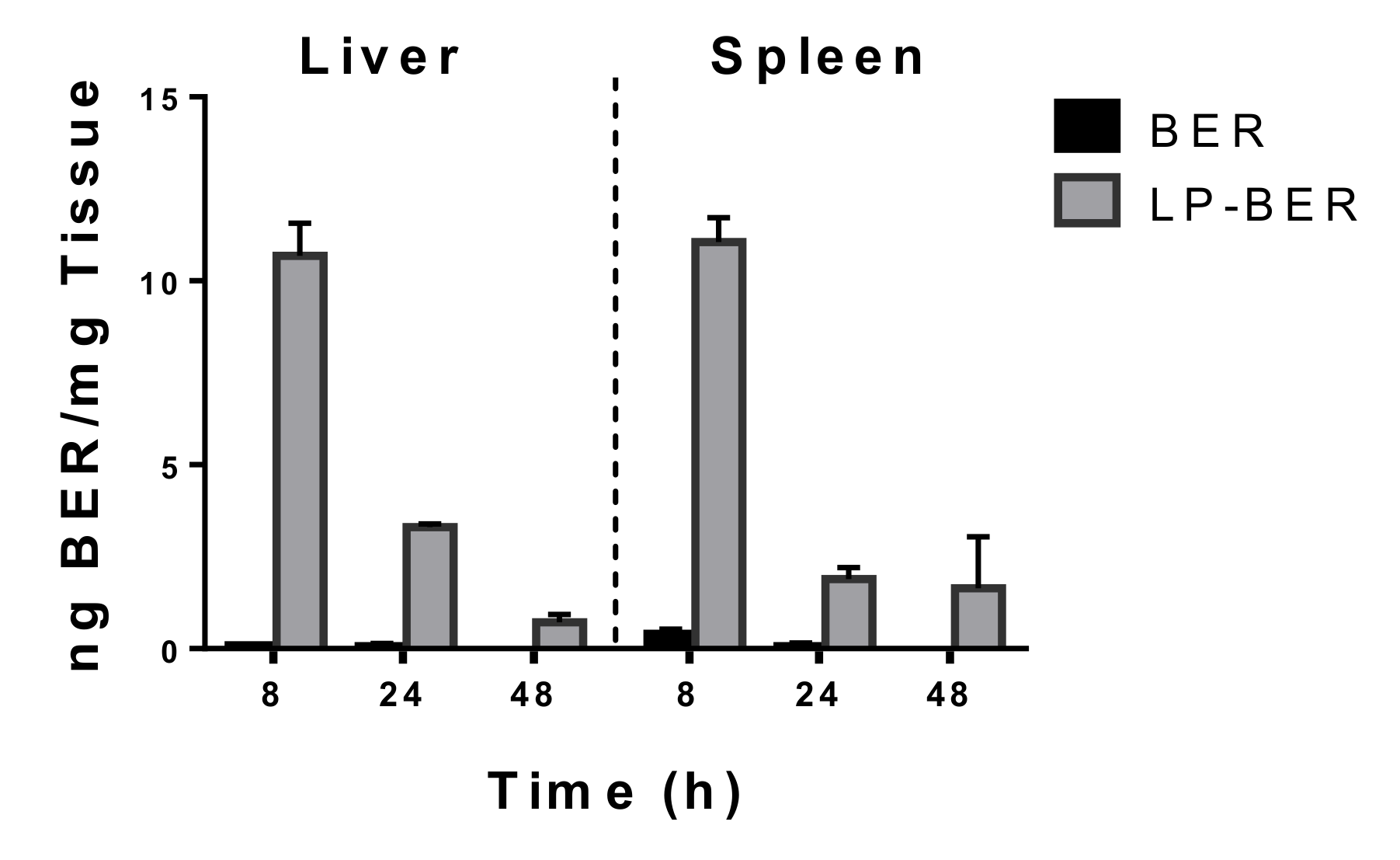
2.13. Tissue Distribution of BER and LP-BER
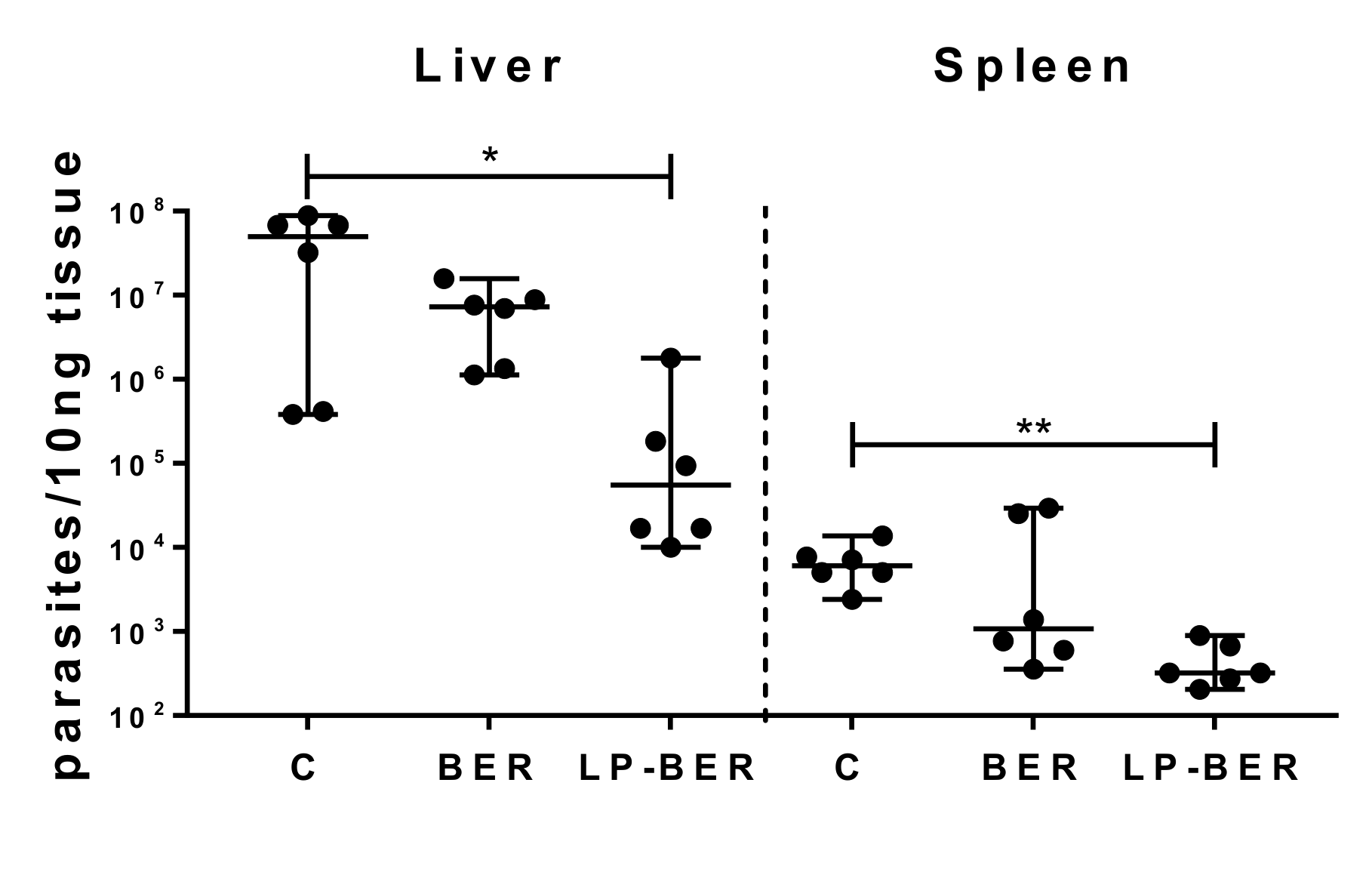
2.14. In Vivo Efficacy Studies of BER and LP-BER
2.15. DNA Extraction and Parasite Quantification
2.16. Biochemical Analysis after BER and LP-BER Administration: In Vivo Toxicity Studies
2.17. Statistical Analyses
3. Results
3.1. Characterization of BER Liposomes
3.2. In Vitro Cytotoxicity of BER and BER Liposomes and NO Production
3.3. In Vitro Drug Release Study in Serum
3.4. In Vitro Antileishmanial Activity of BER and LP-BER
3.5. Pharmacokinetic Studies
3.6. In Vivo Efficacy Studies of BER and LP-BER
3.7. In Vivo Toxicity Studies
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alvar, J.; Velez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; Boer, M.D. Leishmaniasis Worldwide and Global Estimates of Its Incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.-C.; Barrett, M.P.; Lopez-Vélez, R.; García-Hernández, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. Plos Negl. Trop. Dis. 2017, 11, e0006052. [Google Scholar] [CrossRef]
- Singh, I.P.; Mahajan, S. Berberine and its derivatives: A patent review (2009–2012). Expert Opin. Ther. Pat. 2012, 23, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Imenshahidi, M.; Hosseinzadeh, H. Berberis Vulgarisand Berberine: An Update Review. Phytother. Res. 2016, 30, 1745–1764. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, A.; Purohit, S.K.; Yadav, J.S.; Netra, P.R. Cutaneous leishmaniasis in domestic dogs. Indian J. Public Health 1993, 37, 29–31. [Google Scholar]
- Salehabadi, A.; Karamian, M.; Farzad, M.H.; Namaei, M.H. Effect of root bark extract of Berberis vulgaris L. on Leishmania major on BALB/c mice. Parasitol. Res. 2013, 113, 953–957. [Google Scholar] [CrossRef]
- Mahmoudvand, H.; Sharififar, F.; Sharifi, I.; Ezatpour, B.; Harandi, M.F.; Makki, M.S.; Zia-Ali, N.; Jahanbakhsh, S. In Vitro Inhibitory Effect of Berberis vulgaris (Berberidaceae) and Its Main Component, Berberine against Different Leishmania Species. Iran. J. Parasitol. 2014, 9, 28–36. [Google Scholar]
- Saha, P.; Bhattacharjee, S.; Sarkar, A.; Manna, A.; Majumder, S.; Chatterjee, M. Berberine Chloride Mediates Its Anti-Leishmanial Activity via Differential Regulation of the Mitogen Activated Protein Kinase Pathway in Macrophages. PLoS ONE 2011, 6, e18467. [Google Scholar] [CrossRef]
- Saha, P.; Sen, R.; Hariharan, C.; Kumar, D.; Das, P.; Chatterjee, M. Berberine chloride causes a caspase-independent, apoptotic-like death in Leishmania donovani promastigotes. Free. Radic. Res. 2009, 43, 1101–1110. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Bhattacharyya, F.K.; Ghosh, D.K. Leishmania donovani: Amastigote inhibition and mode of actior of berberine. Exp. Parasitol. 1985, 60, 404–413. [Google Scholar] [CrossRef]
- Vennerstrom, J.L.; Lovelace, J.K.; Waits, V.B.; Hanson, W.L.; Klayman, D.L. Berberine derivatives as antileishmanial drugs. Antimicrob. Agents Chemother. 1990, 34, 918–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgohary, M.M.; Helmy, M.W.; Mortada, S.M.; Elzoghby, A.O. Dual-targeted nano-in-nano albumin carriers enhance the efficacy of combined chemo/herbal therapy of lung cancer. Nanomedicine 2018, 13, 2221–2224. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, P.; Kumari, M.; Pahuja, R.; Pant, A.B.; Shukla, Y.; Kumar, P.; Gupta, K.C. Hyaluronic acid-grafted PLGA nanoparticles for the sustained delivery of berberine chloride for an efficient suppression of Ehrlich ascites tumors. Drug Deliv. Transl. Res. 2018, 8, 565–579. [Google Scholar] [CrossRef]
- Zhu, J.-X.; Tang, D.; Feng, L.; Zheng, Z.-G.; Wang, R.-S.; Wu, A.-G.; Duan, T.-T.; He, B.; Zhu, Q. Development of self-microemulsifying drug delivery system for oral bioavailability enhancement of berberine hydrochloride. Drug Dev. Ind. Pharm. 2012, 39, 499–506. [Google Scholar] [CrossRef]
- Li, J.; Yang, L.; Shen, R.; Gong, L.; Tian, Z.; Qiu, H.; Shi, Z.; Gao, L.; Sun, H.-W.; Zhang, G. Self-nanoemulsifying system improves oral absorption and enhances anti-acute myeloid leukemia activity of berberine. J. Nanobiotechnol. 2018, 16, 76. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.-H.; Feng, C.-L.; Zhang, W.; Luo, Z.-G.; Zhang, H.-J.; Zhang, T.-T.; Ma, C.; Zhan, Y.; Li, R.; Wu, S.; et al. Liver-target nanotechnology facilitates berberine to ameliorate cardio-metabolic diseases. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Allijn, I.E.; Czarny, B.; Wang, X.; Chong, S.Y.; Weiler, M.; Da Silva, A.E.; Metselaar, J.M.; Lam, C.S.P.; Pastorin, G.; De Kleijn, D.P.V.; et al. Liposome encapsulated berberine treatment attenuates cardiac dysfunction after myocardial infarction. J. Control. Release 2017, 247, 127–133. [Google Scholar] [CrossRef]
- Xue, M.; Zhang, L.; Yang, M.-X.; Zhang, W.; Li, X.-M.; Ou, Z.-M.; Li, Z.-P.; Liu, S.-H.; Li, X.-J.; Yang, S.-Y. Berberine-loaded solid lipid nanoparticles are concentrated in the liver and ameliorate hepatosteatosis in db/db mice. Int. J. Nanomed. 2015, 10, 5049–5057. [Google Scholar] [CrossRef] [Green Version]
- Mirhadi, E.; Rezaee, M.; Malaekeh-Nikouei, B. Nano strategies for berberine delivery, a natural alkaloid of Berberis. Biomed. Pharmacother. 2018, 104, 465–473. [Google Scholar] [CrossRef]
- Saleem, K.; Khursheed, Z.; Hano, C.; Anjum, I.; Anjum, S. Applications of Nanomaterials in Leishmaniasis: A Focus on Recent Advances and Challenges. Nanomaterials 2019, 9, 1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borborema, S.E.T.; Junior, J.A.O.; Tempone, A.G.; Júnior, H.F.D.A.; Nascimento, N.D. Pharmacokinetic of meglumine antimoniate encapsulated in phosphatidylserine-liposomes in mice model: A candidate formulation for visceral leishmaniasis. Biomed. Pharmacother. 2018, 103, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Burza, S.; Sinha, P.K.; Mahajan, R.; Lima, M.A.; Mitra, G.; Verma, N.; Balasegarem, M.; Das, P. Five-Year Field Results and Long-Term Effectiveness of 20 mg/kg Liposomal Amphotericin B (Ambisome) for Visceral Leishmaniasis in Bihar, India. PLoS Negl. Trop. Dis. 2014, 8, e2603. [Google Scholar] [CrossRef] [PubMed]
- Reis, L.E.S.; De Brito, R.C.F.; Cardoso, J.M.D.O.; Mathias, F.A.S.; Aguiar-Soares, R.D.D.O.; Carneiro, C.M.; Vieira, P.M.D.A.; Ramos, G.S.; Frézard, F.J.G.; Roatt, B.M.; et al. Mixed Formulation of Conventional and Pegylated Meglumine Antimoniate-Containing Liposomes Reduces Inflammatory Process and Parasite Burden in Leishmania infantum-Infected BALB/c Mice. Antimicrob. Agents Chemother. 2017, 61, e00962-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaspar, M.M.; Calado, S.; Pereira, J.; Ferronha, H.; Correia, I.; Castro, H.; Tomás, A.; Cruz, M.E.M. Targeted delivery of paromomycin in murine infectious diseases through association to nano lipid systems. Nanomedicine 2015, 11, 1851–1860. [Google Scholar] [CrossRef]
- Osada, Y.; Omachi, S.; Sanjoba, C.; Matsumoto, Y.; Noiri, E.; Jha, T. Animal Models of Visceral Leishmaniasis and Applicability to Disease Control. In Kala Azar in South Asia; Springer Science and Business Media LLC: Berlin, Germany, 2016; pp. 287–296. [Google Scholar]
- Zhang, H. Thin-Film Hydration Followed by Extrusion Method for Liposome Preparation. Methods Mol. Biol. 2017, 1522, 17–22. [Google Scholar]
- Rouser, G.; Fkeischer, S.; Yamamoto, A.; Fleischer, S. Two dimensional thin layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids 1970, 5, 494–496. [Google Scholar] [CrossRef]
- Hendrickx, S.; Eberhardt, E.; Mondelaers, A.; Rijal, S.; Bhattarai, N.R.; Dujardin, J.C.; Delputte, P.L.; Cos, P.; Maes, L. Lack of correlation between the promastigote back-transformation assay and miltefosine treatment outcome. J. Antimicrob. Chemother. 2015, 70, 3023–3026. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Q.; Liu, Z.; Zheng, X. Preparation, pharmacokinetics and tumour-suppressive activity of berberine liposomes. J. Pharm. Pharmacol. 2017, 69, 625–632. [Google Scholar] [CrossRef]
- Charles River. Available online: https://www.criver.com/sites/default/files/Technical%20Resources/Clinical%20Pathology%20Data%20for%20BALB_c%20Mouse%20Colonies%20in%20North%20America%20for%20January%202008%20-%20December%202012.pdf (accessed on 20 May 2020).
- Imanshahidi, M.; Hosseinzadeh, H. Pharmacological and therapeutic effects of Berberis vulgaris and its active constituent, berberine. Phytother. Res. 2008, 22, 999–1012. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Khadka, D.B.; Cho, W.-J. Pharmacological effects of berberine and its derivatives: A patent update. Expert Opin. Ther. Pat. 2016, 26, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Feng, X.; Chai, L.; Cao, S.; Qiu, F. The metabolism of berberine and its contribution to the pharmacological effects. Drug Metab. Rev. 2017, 49, 139–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-S.; Zheng, Y.-R.; Zhang, Y.-F.; Long, X.-Y. Research progress on berberine with a special focus on its oral bioavailability. Fitoterapia 2016, 109, 274–282. [Google Scholar] [CrossRef]
- Malebo, H.M.; Wenzler, T.; Cal, M.; Swaleh, S.M.; Omolo, M.O.; Hassanali, A.; Séquin, U.; Häussinger, D.; Dalsgaard, P.W.; Hamburger, M.; et al. Anti-protozoal activity of aporphine and protoberberine alkaloids from Annickia kummeriae (Engl. & Diels) Setten & Maas (Annonaceae). BMC Complement. Altern. Med. 2013, 13, 48. [Google Scholar]
- De Souza, A.; Marins, D.S.S.; Mathias, S.; Monteiro, L.M.; Yukuyama, M.N.; Scarim, C.B.; Löbenberg, R.; Bou-Chacra, N.A. Promising nanotherapy in treating leishmaniasis. Int. J. Pharm. 2018, 547, 421–431. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, N.; Leroux, J.-C. The journey of a drug-carrier in the body: An anatomo-physiological perspective. J. Control. Release 2012, 161, 152–163. [Google Scholar] [CrossRef]
- Swami, A.; Reagan, M.R.; Basto, P.; Mishima, Y.; Kamaly, N.; Glavey, S.; Zhang, S.; Moschetta, M.; Seevaratnam, D.; Zhang, Y.; et al. Engineered nanomedicine for myeloma and bone microenvironment targeting. Proc. Natl. Acad. Sci. USA 2014, 111, 10287–10292. [Google Scholar] [CrossRef] [Green Version]
- Mandl, H.K.; Quijano, E.; Suh, H.W.; Sparago, E.; Oeck, S.; Grun, M.; Glazer, P.M.; Saltzman, W.M. Optimizing biodegradable nanoparticle size for tissue-specific delivery. J. Control. Release 2019, 314, 92–101. [Google Scholar] [CrossRef]
- Banerjee, A.; De, M.; Ali, N. Complete cure of experimental visceral leishmaniasis with amphotericin B in stearylamine-bearing cationic liposomes involves down-regulation of IL-10 and favorable T cell responses. J. Immunol. 2008, 181, 1386–1398. [Google Scholar] [CrossRef]
- Banerjee, A.; De, M.; Ali, N. Combination Therapy with Paromomycin-Associated Stearylamine-Bearing Liposomes Cures Experimental Visceral Leishmaniasis through Th1-Biased Immunomodulation. Antimicrob. Agents Chemother. 2011, 55, 1661–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, R.; Roychoudhury, J.; Palit, P.; Ali, N. Cationic Liposomal Sodium Stibogluconate (SSG), a Potent Therapeutic Tool for Treatment of Infection by SSG-Sensitive and -Resistant Leishmania donovani. Antimicrob. Agents Chemother. 2015, 59, 344–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, M. Lipids in Nanotechnology, 1st ed.; AOCS Press: Urbana, IL, USA, 2012; pp. 221–268. [Google Scholar]
- Singh, N.; Sharma, B. Toxicological Effects of Berberine and Sanguinarine. Front. Mol. Biosci. 2018, 5, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, P.J. The effect of tocopherol on the structure and permeability of phosphatidylcholine liposomes. J. Control. Release 2012, 160, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Spinozzi, S.; Colliva, C.; Camborata, C.; Roberti, M.; Ianni, C.; Neri, F.; Calvarese, C.; Lisotti, A.; Mazzella, G.; Roda, A. Berberine and Its Metabolites: Relationship between Physicochemical Properties and Plasma Levels after Administration to Human Subjects. J. Nat. Prod. 2014, 77, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Battu, S.K.; Repka, M.A.; Maddineni, S.; Chittiboyina, A.G.; Avery, M.A.; Majumdar, S. Physicochemical Characterization of Berberine Chloride: A Perspective in the Development of a Solution Dosage Form for Oral Delivery. AAPS PharmSciTech 2010, 11, 1466–1475. [Google Scholar] [CrossRef] [Green Version]
- Sarfarazi, A.; Lee, G.; Mirjalili, S.A.; Phillips, A.R.; Windsor, J.A.; Trevaskis, N.L. Therapeutic delivery to the peritoneal lymphatics: Current understanding, potential treatment benefits and future prospects. Int. J. Pharm. 2019, 567, 118456. [Google Scholar] [CrossRef]
- Lee, G.; Han, S.; Inocencio, I.; Cao, E.; Hong, J.; Phillips, A.R.J.; Windsor, J.A.; Porter, C.J.; Trevaskis, N.L. Lymphatic Uptake of Liposomes after Intraperitoneal Administration Primarily Occurs via the Diaphragmatic Lymphatics and is Dependent on Liposome Surface Properties. Mol. Pharm. 2019, 16, 4987–4999. [Google Scholar] [CrossRef]
- Shin, J.-S.; Choi, H.-E.; Seo, S.; Choi, J.-H.; Baek, N.-I.; Lee, K.-T. Berberine Decreased Inducible Nitric Oxide Synthase mRNA Stability through Negative Regulation of Human Antigen R in Lipopolysaccharide-Induced Macrophages. J. Pharmacol. Exp. Ther. 2016, 358, 3–13. [Google Scholar] [CrossRef]
- Kheir, M.M.; Wang, Y.; Hua, L.; Hu, J.; Li, L.; Lei, F.; Du, L. Acute toxicity of berberine and its correlation with the blood concentration in mice. Food Chem. Toxicol. 2010, 48, 1105–1110. [Google Scholar] [CrossRef]
- Tsai, P.I.-L.; Tsai, T.-H. Simultaneous determination of berberine in rat blood, liver and bile using microdialysis coupled to high-performance liquid chromatography. J. Chromatogr. A 2002, 961, 125–130. [Google Scholar] [CrossRef]
- Jeong, H.W.; Hsu, K.C.; Lee, J.-W.; Ham, M.; Huh, J.Y.; Shin, H.J.; Kim, W.S.; Kim, J.B. Berberine suppresses proinflammatory responses through AMPK activation in macrophages. Am. J. Physiol.Endocrinol. Metab. 2009, 296, E955–E964. [Google Scholar] [CrossRef] [PubMed]
- Hunter, P.M.; Hegele, R.A. Functional foods and dietary supplements for the management of dyslipidaemia. Nat. Rev. Endocrinol. 2017, 13, 278–288. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sense Primer (5′-3′) | Antisense Primer (5′-3′) |
|---|---|---|
| Leish kDNA | CCTATTTTACACCAACCCCCAGT | GGGTAGGGGCGTTCTGCGAAA |
| Liposomes (75:40:5 mM) | Mean Diameter (nm) | PDi | Z-Potential (mV) | BER EE (%) | nmol BER/ μmol lipid |
|---|---|---|---|---|---|
| PC:Chol:TS | 133.8 ± 1.4 | 0.19 | −22 ± 1 | – | – |
| PC:Chol:TS-BER | 121.6 ± 1.3 | 0.25 | −38 ± 1 | 14.1 ± 2.9 | 6.6 ± 1.8 |
| PC:Chol:DDAB | 159.6 ± 0.9 | 0.27 | 52 ± 1 | – | – |
| PC:Chol:DDAB-BER | 151.4 ± 0.8 | 0.27 | 58 ± 3 | 2.1 | 1.3 |
| PC:Chol:DP | 195.4 ± 4.8 | 0.28 | −47 ± 1 | – | – |
| PC:Chol:DP-BER | 245.2 ± 2.3 | 0.21 | −46 ± 1 | 28.7 ± 1.4 | 17.9 ± 3.8 |
| Drug | BMDM CC50 (μM) |
|---|---|
| BER | 125.3 ± 18.4 |
| PC:Chol:TS-BER | >1000 |
| PC:Chol:DDAB-BER | >1000 |
| PC:Chol:DP-BER | 140.2 ± 1.6 |
| AmB | 4.5 ± 1.2 |
| Drug | Promastigotes (μM) | Amastigotes (μM) | SI (CC50/IC50) |
|---|---|---|---|
| BER IC50 | 5.2 ± 0.1 | 1.1 ± 0.1 | 115.0 |
| BER IC90 | 46.6 ± 0.1 | 9.8 ± 0.1 | – |
| LP-BER IC50 | 6.8 ± 0.6 | 1.4 ± 0.2 | >714.3 |
| LP-BER IC90 | 60.8 ± 0.6 | 12.2 ± 0.2 | – |
| AmB IC50 | 1.4 ± 0.3 | 1.3 ± 0.2 | 3.5 |
| AmB IC90 | 12.6 ± 0.3 | 11.4 ± 0.2 | – |
| i.v. 7.5 mg/kg | i.p. 15 mg/kg | ||||
|---|---|---|---|---|---|
| Parameter | Unit | BER | LP-BER | BER | LP-BER |
| t1/2 | h | 12.9 ± 3.5 | 4.1 ± 1.4 * | 11.7 ± 4.9 | 16.2 ± 8.2 |
| tmax | h | 0.25 | 0.25 | 0.25 | 0.25 |
| Cmax | ng/mL | 99.2 ± 36.0 | 2565.1 ± 1349.6 * | 276.8 ± 29.7 | 461.5 ± 47.7 ** |
| Cl † | mL/h·kg | 9399.8 ± 3148.9 | 1716.8 ± 882.4 * | 9334.5 ± 250.4 | 1978.8 ± 265.3 *** |
| AUC0–t | ng/mL·h | 504.6 ± 213.8 | 4253.8 ± 1246.1 * | 806.8 ± 83.2 | 11,756.7 ± 976.4 ** |
| MRT0–t | h | 9.2 ± 1.5 | 2.2 ± 0.6 ** | 6.1 ± 0.5 | 25.3 ± 4.1 ** |
| Fr | % | – | – | 62.0 | 160.0 |
| Liver | Spleen | BM (Bone Marrow) | ||||||
|---|---|---|---|---|---|---|---|---|
| Dose (mg/kg) | Median | 95% IC | Median | 95% IC | Median | 95% IC | ||
| 5 Days | Control | 3.4 × 107 | 4 × 106–8 × 107 | 2.8 × 103 | 1 × 103–5 × 103 | 5.6 × 105 | 3 × 105–1 × 106 | |
| 5 | BER | 4.4 × 106 | 9 × 105–1 × 107 | 2.5 × 103 | 6 × 102–2 × 104 | 8.7 × 105 | 2 × 105–3 × 106 | |
| LP-BER | 3.5 × 107 | 1 × 107–1 × 108 | 2.9 × 103 | 2 × 103–1 × 104 | 1.1 × 106 | 2 × 105–4 × 106 | ||
| 10 | BER | 4.9 × 107 | 2 × 107–1 × 108 | 2.3 × 103 | 7 × 102–9 × 103 | 2.0 × 106 | 8 × 105–3 × 106 | |
| LP-BER | 1.7 × 107 | 7 × 106–5 × 107 | 2.5 × 103 | 2 × 103–6 × 103 | 6.6 × 105 | 1 × 105–1 × 106 | ||
| 1 | FGZ | 8.0 × 105 | 3 × 105–1 × 107 | 6.0 × 102 | 2 × 102–2 × 103 | 2.3 × 105 | 2 × 104–5 × 105 | |
| 10 Days | Control | 5.0 × 107 | 4 × 105–9 × 107 | 6.1 × 103 | 2 × 103–1 × 104 | 5.1 × 105 | 8 × 104–2 × 106 | |
| 15 | BER | 7.3 × 106 | 1 × 106–2 × 107 | 1.1 × 103 | 4 × 102–3 × 104 | 7.9 × 105 | 1 × 105–3 × 106 | |
| LP-BER | 5.6 × 104 * | 1 × 104–2 × 106 | 3.2 × 102 ** | 2 × 102–9 × 102 | 1.1 × 106 | 8 × 104–5 × 106 | ||
| 1 | FGZ | 1.8 × 102 *** | 1–7 × 102 | 5.2 **** | 1–1 × 101 | 1.3 × 101 * | 6–2 × 101 | |
| Parameter | Control | BER | LP-BER |
|---|---|---|---|
| ALT (U/L) | 94.7 ± 23.7 | 61.0 ± 12.2 | 60.8 ± 42.9 |
| AST (U/L) | 120.6 ± 16.4 | 105.2 ± 14.1 | 88.3 ± 24.2 |
| BUN (mg/dL) | 41.8 ± 7.3 | 26.0 ± 4.0 ** | 29.8 ± 3.0 |
| LDH (U/L) | 366.8 ± 55.4 | 372.5 ± 108.6 | 457.8 ± 245.3 |
| TRIG (mg/dL) | 109.5 ± 15.4 | 89.1 ± 25.1 | 70.3 ± 21.6 * |
| CHO (mg/dL) | 114.3 ± 8.5 | 111.4 ± 4.8 | 116.8 ± 16.3 |
| HDL (mmol/L) | 2.7 ± 0.1 | 2.6 ± 0.1 | 2.2 ± 0.3 |
| LDL (mmol/L) | 0.4 ± 0.1 | 0.5 ± 0.1 | 1.3 ± 0.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calvo, A.; Moreno, E.; Larrea, E.; Sanmartín, C.; Irache, J.M.; Espuelas, S. Berberine-Loaded Liposomes for the Treatment of Leishmania infantum-Infected BALB/c Mice. Pharmaceutics 2020, 12, 858. https://doi.org/10.3390/pharmaceutics12090858
Calvo A, Moreno E, Larrea E, Sanmartín C, Irache JM, Espuelas S. Berberine-Loaded Liposomes for the Treatment of Leishmania infantum-Infected BALB/c Mice. Pharmaceutics. 2020; 12(9):858. https://doi.org/10.3390/pharmaceutics12090858
Chicago/Turabian StyleCalvo, Alba, Esther Moreno, Esther Larrea, Carmen Sanmartín, Juan Manuel Irache, and Socorro Espuelas. 2020. "Berberine-Loaded Liposomes for the Treatment of Leishmania infantum-Infected BALB/c Mice" Pharmaceutics 12, no. 9: 858. https://doi.org/10.3390/pharmaceutics12090858
APA StyleCalvo, A., Moreno, E., Larrea, E., Sanmartín, C., Irache, J. M., & Espuelas, S. (2020). Berberine-Loaded Liposomes for the Treatment of Leishmania infantum-Infected BALB/c Mice. Pharmaceutics, 12(9), 858. https://doi.org/10.3390/pharmaceutics12090858