
Calorimetric Investigation of the Relaxation Phenomena in Amorphous Lyophilized Solids


Abstract
:1. Introduction
2. Theoretical Background
2.1. Nomenclature of Relaxation Modi
2.2. DSC
- At temperatures higher than 60 °C, residual moisture will evaporate out of the measured matrix, which lowers the significantly and finally delivers data for the thermal behavior of a dry sample. However, this is not the property of the samples that is stored in the product vials after freeze-drying.
- In the context of the thermal history of an amorphous solid, the value of at is relevant. It is the comparison of the (“baseline”) between pre- and post- and can be determined as shown in Figure 2a. It must not be confused with the value itself, which is a substance constant and declares the needed heat to raise 1 g of sample by 1 K. With a pierced lid, the values are determined isobaric in contrast to a hermetically sealed pan. Moreover, it has to be kept in mind that at depends on the absolute sample weight. Thus, in multiple measurements (e.g., triplicate), the values of will differ more strongly than the temperature due to additional weighing errors. The results of are quite mass-independent.
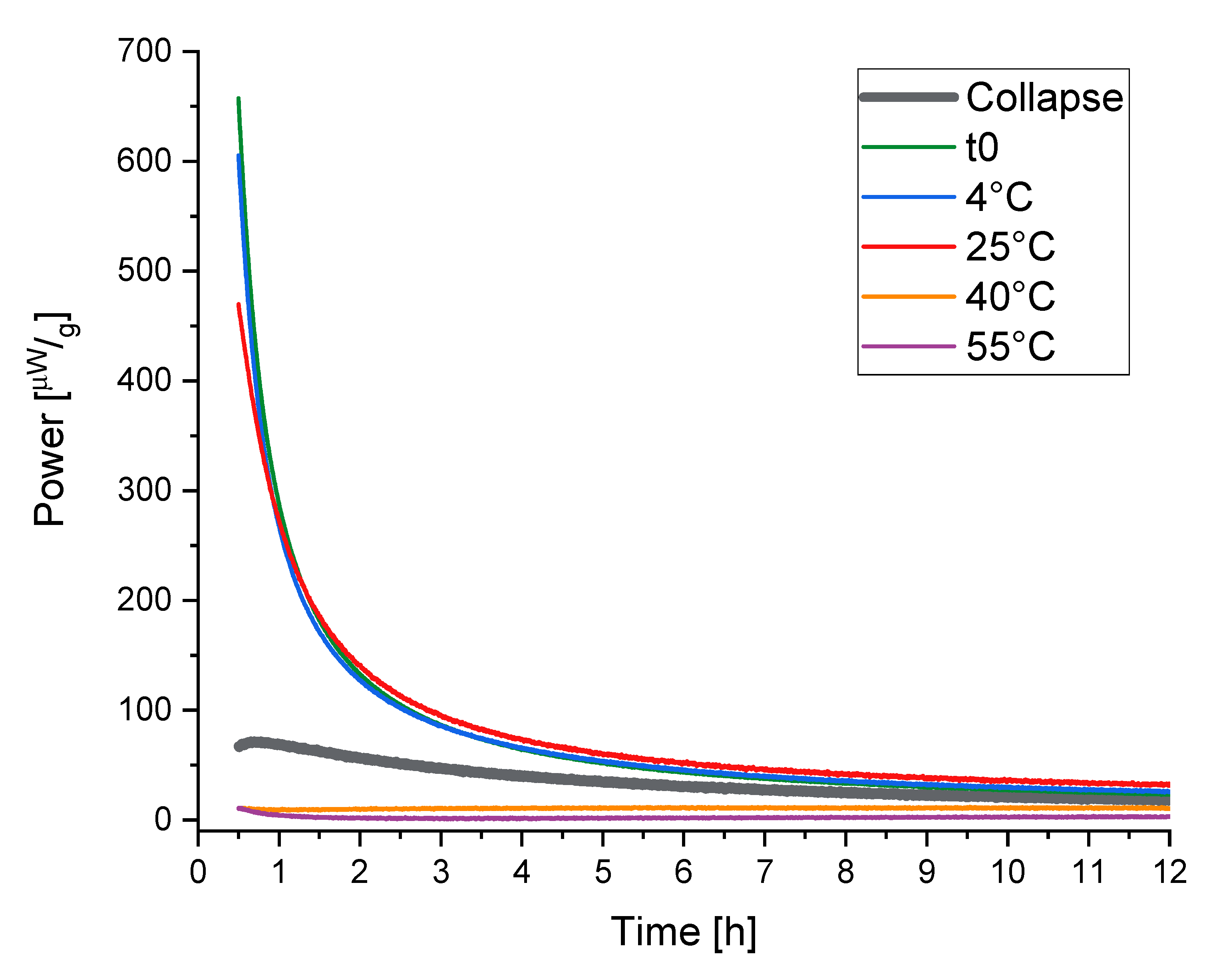
2.3. Isothermal Microcalorimetry (IMC), Direct Measurement of Relaxation
2.4. Comparison of DSC and IMC for -Relaxation Analysis
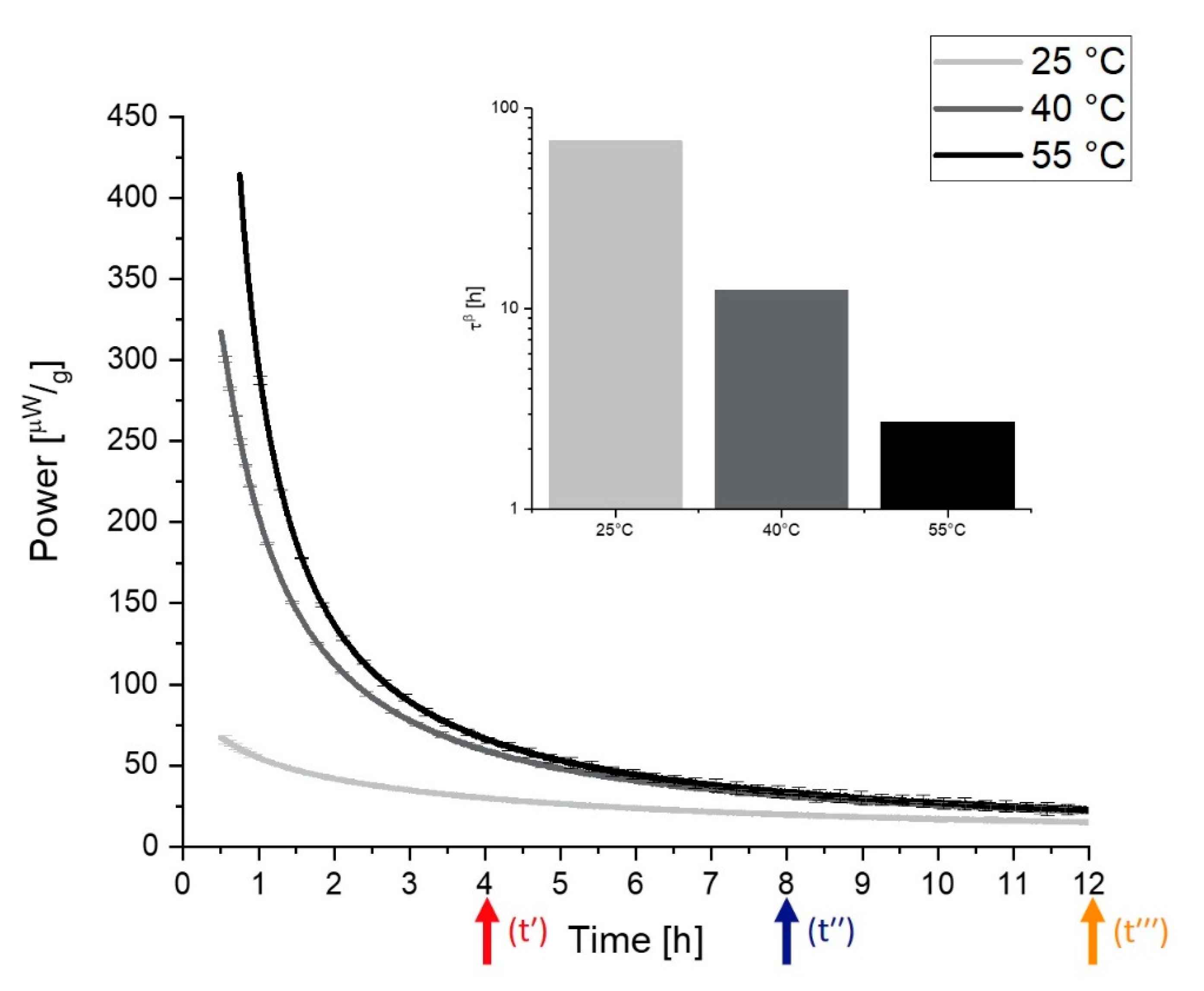
2.4.1. The Behavior of τ and β
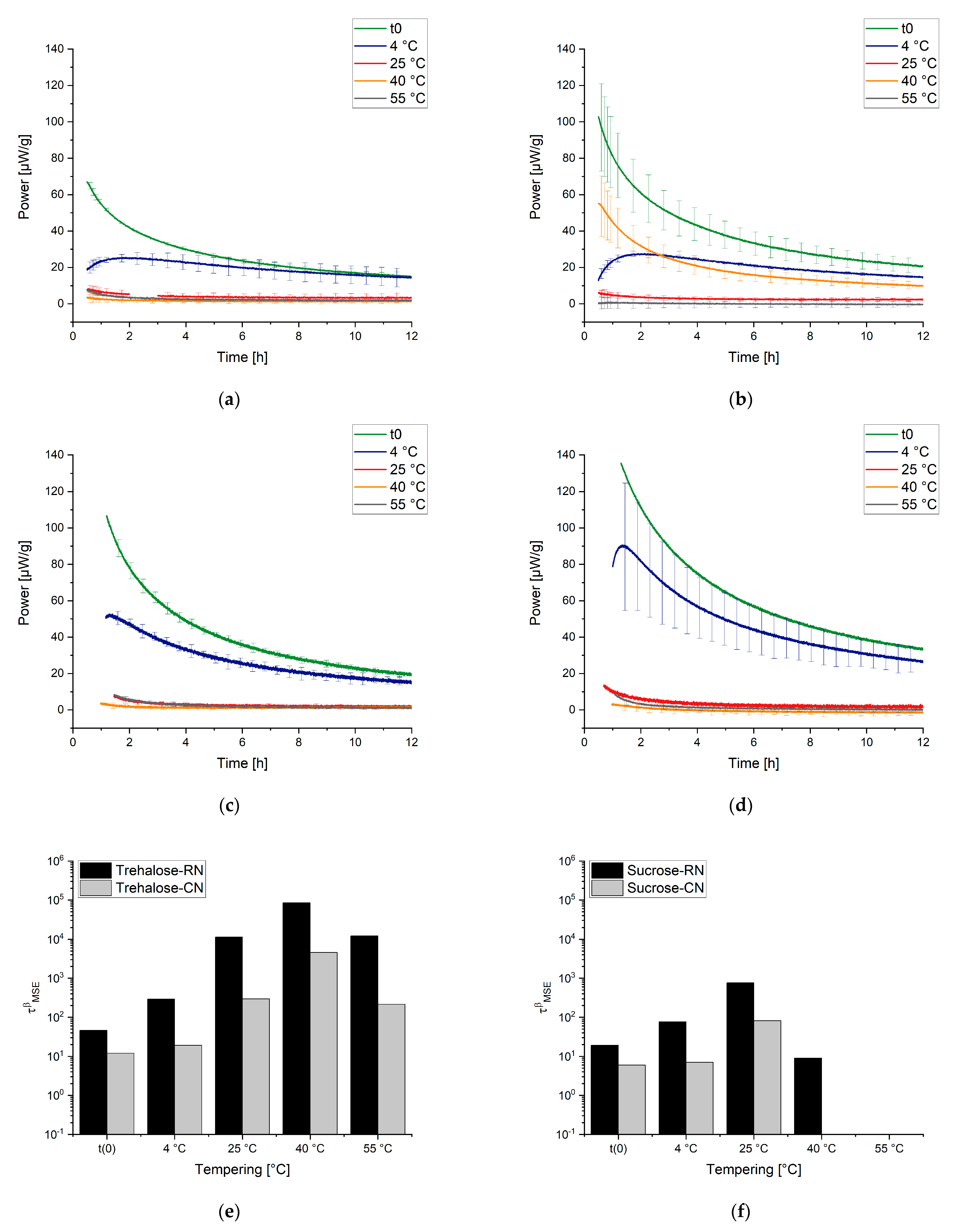
2.4.2. General Observations for Samples with Different Thermal History
3. Application of α-Relaxation Analysis
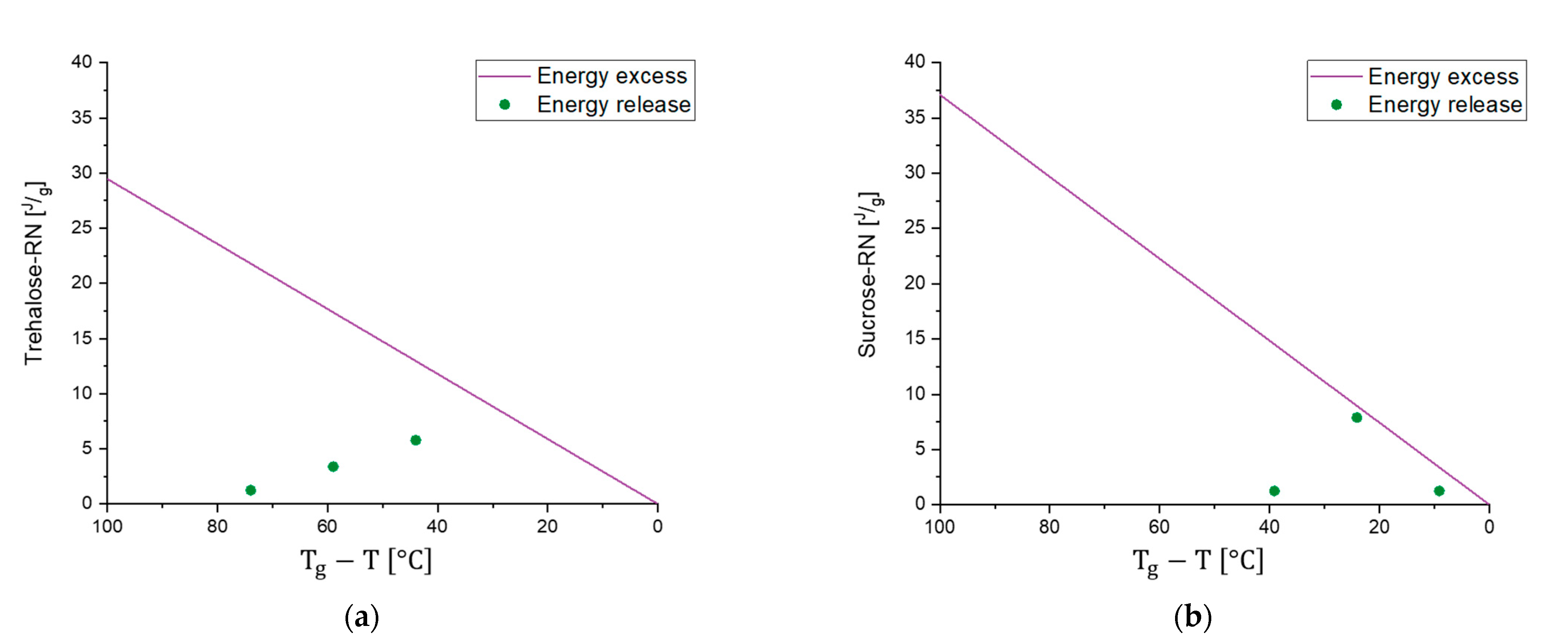
3.1. Correlation of with Storage Stability of Active Pharmaceutical Ingredients
3.2. Further Applications of -Relaxation Studies
3.2.1. Crystallization
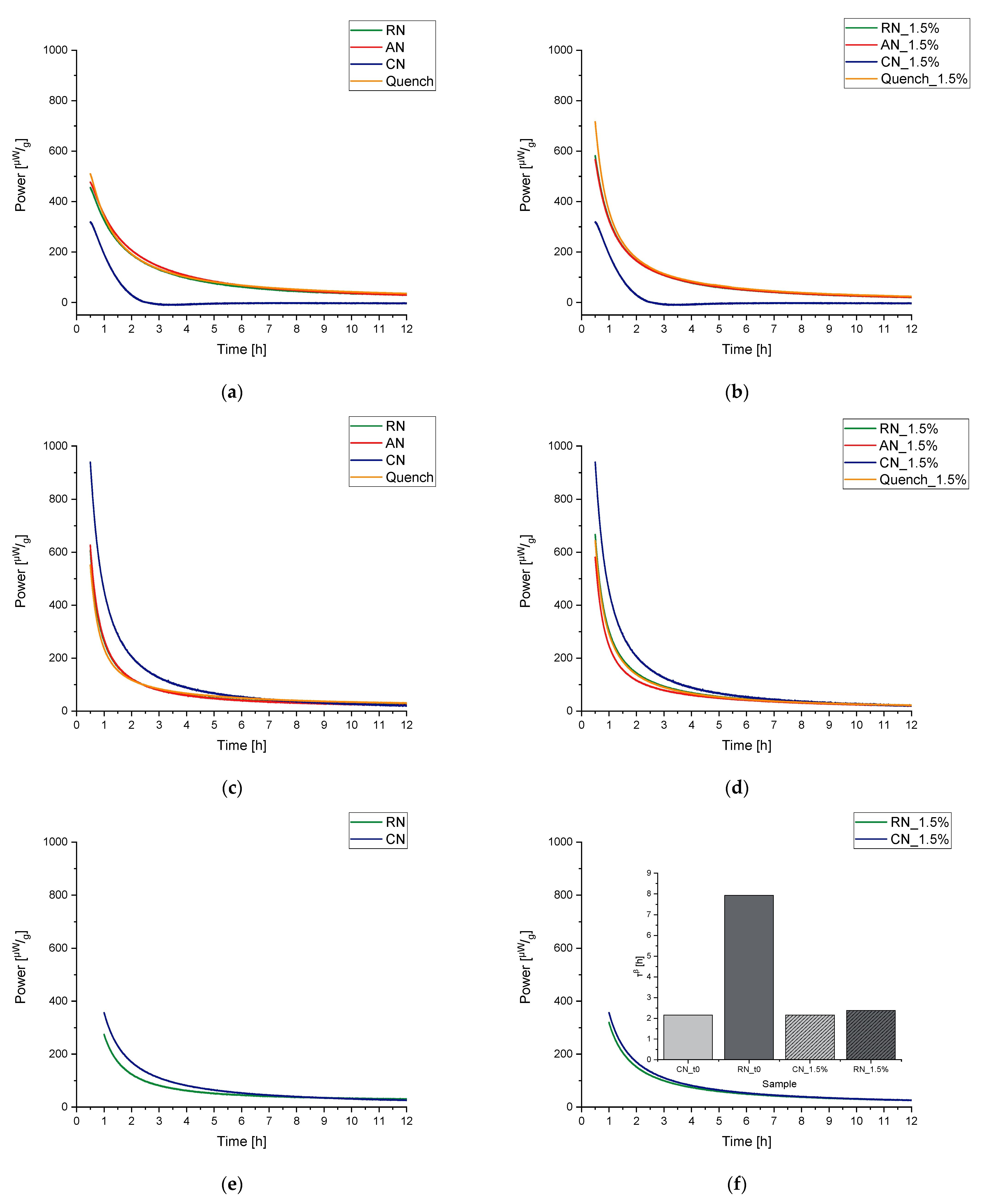
3.2.2. Influence of the Freezing Step on ɑ-Relaxation
3.3. Collapse as a Tempering Process at Relatively Low Temperatures
4. Conclusions
5. Outlook
6. Materials and Methods
6.1. Materials
6.2. Preparation of Formulations
6.3. Freeze-Drying
6.4. Sample Tempering
6.5. Differential Scanning Calorimetry
6.6. Isothermal Microcalorimetry
6.7. Curve Fitting
6.8. Karl Fischer Titration
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Purpose | Sample Weight [mg] | Lid | Heating Rates [K/min] | Hold Temperatures above Tg [K] | Tempering Temperature below Tg [K] |
|---|---|---|---|---|---|---|
| Hancock et al. 1995. [48] | Investigate relaxation times of pure, melt-quenched substances with DSC | 2–10 | Pierced | 20 | 10–25 | 16–47 |
| Van den Mooter et al. [90] | Investigate relaxation times of pure, melt-quenched APIs with DSC | 2–6 | Closed | 10 | 20 | 16–66 |
| Shamblin et al. [16] | Temperature + enthalpy of fusion of lyophilized substances | 5 | Closed | 10 | n.a. | n.a. |
| values of lyophilized substances | 5 | n.a. | 2 * (60, 0.5) | n.a. | n.a. | |
| Relaxation times of pure lyophilized substances with mDSC | 5 | n.a. | n.a. | 15 | 15 | |
| Aso et al. [111] | Correlation of mobile mobility and crystallization of quench-cooled samples | 5 | Pierced | 20 | 45 | 25 |
| Liu et al. [17] | Investigation and comparison of relaxation times of quench-cooled and freeze-dried samples. Additionally to DSC, also IMC was used for determination | 5–10 | Closed | n.a. | n.a. | n.a. |
| Zhou et al. [98] | values of lyophilized and quench-cooled substances | 10 | Closed | 1 * (100, 0.5) | 50 | 20–50 |
| Surana et al. [99,100] | Investigate relaxation times and crystallinity onset of freeze-dried trehalose | 4–5 | Pierced | 10 | n.a. | 20–60 |
| Chang et al. [21] | determination for IMC | 5–10 | Closed | 1 * (100, 0.5) | n.a. | n.a. |
| Luthra et al. [41] | Enthalpy recovery and Tg mDSC were used, where enthalpy recovery might be superimposed by Tg | 5–10 | Closed | 10 | 25 | 50-15 |
| 1 * (80, 0.85) | ||||||
| Wang et al. [70] | determination for IMC | n.a. | n.a. | 2 * (120, 1) | 30 | n.a. |
Appendix B
| Author | Method | Formulation | Outcome |
|---|---|---|---|
| Abdul-Fattah et al. [31] | MSE | Freeze-dried moxalactam mannitol system | Tempering (called annealing in the original literature) increased the structural relaxation time of the samples, leading to improved long-term stability. |
| Abdul-Fattah et al. [56] | MSE | Mixtures of monoclonal antibodies with increasing sucrose content prepared by freeze-drying, spray-drying, and vacuum drying | “Initial endothermic response” in sucrose-rich vacuum-dried formulations appeared with unclear origin. With increased sucrose fraction, stability of the antibodies increased. Correlation between relaxation times and protein depending of the process was better for fast dynamics (not the topic of this review) than for global α-relaxation times. |
| Bhugra et al. [53] | KWW and MSE | Pure quench-cooled systems of indomethacin, nifedipin, ketoconazol and flopropione | Relaxation time constants above and below Tg are highly correlated. |
| Duddu et al. [92] | KWW | Trehalose- or sucrose-based formulations of a monoclonal antibody | Trehalose-based formulations showed Arrhenius-like behavior while sucrose showed non-Arrhenius kinetics. Thus, although sucrose is in general the more fragile glass, at temperatures below 5 °C, trehalose seems to be more fragile. |
| Hancock et al. [48] | KWW | Pure quench-cooled systems of indomethacin, PVP, and sucrose | Relaxation time at is up to many years. Relaxations could predict stability of some pharmaceutical products. |
| Liu et al. [17] | KWW and MSE | Pure quench-cooled or freeze-dried systems of saccharides (sucrose, trehalose, raffinose, lactose, stachyose) | is obtained with less labor, more data points than is furthermore precise and enables the measurement of samples that relax very slowly with only a minor release of heat. Further, a rational development of peptide and protein formulations should be possible. |
| Kawakami and Pikal [23] | KWW and MSE | Review | The evaluation with is superior compared to . In general, is suggested as parameter of choice instead of comparing the single parameters τ and β, respectively. |
| Shamblin et al. [93] | MSE | Freeze-dried amorphous drug mixtures | The correlation of α-relaxation with protein stability is dependent on the rate controlling step of the degradation process. The latter should be coupled to molecular mobility as well as α-relaxation. |
| Luthra et al. [41,95] | KWW and MSE | Freeze dries sucrose/aspartam and trehalose/aspartame formulations | Tempering (called annealing in the original study) significantly decreases the degradation rate and an optimum temperature for the tempering process was set . |
| Van den Mooter et al. [90] | KWW | Pure quench-cooled systems of diazepam, temazepam, and triazolam | Molecular mobility might be unimportant for temper-atures less than . Relaxations could predict stability of some pharmaceutical products. |
| Wang et al. [94] | KWW and MSE | Sucrose:protein mixtures in different ratios | MSE correlates with protein stability up to a su-crose/protein mass ratio of 1:1 (increase in ) well as FTIR. With higher sucrose concentration be-yond 1:1, decreases although protein stability further increases; thus, did not correlate at this ratio anymore. |
References
- Advancing Health through Innovation: New Drug Therapy Approvals 2020. Available online: https://fda.report/media/144982/final+FINAL+NewDrugsApprovalReport_Final2020_210108_0948_FINAL.pdf (accessed on 15 September 2021).
- Peters, C.; Brown, S. Antibody–Drug Conjugates as Novel Anti-Cancer Chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef] [Green Version]
- Schirrmacher, V. Cancer Vaccines and Oncolytic Viruses Exert Profoundly Lower Side Effects in Cancer Patients than Other Systemic Therapies: A Comparative Analysis. Biomedicines 2020, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Fritzsching, B. Personalized Medicine in Allergic Asthma: At the Crossroads of Allergen Immunotherapy and “Biologicals”. Front. Pediatr. 2017, 5, 3–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kis, Z.; Kontoravdi, C.; Shattock, R.; Shah, N. Resources, Production Scales and Time Required for Producing RNA Vaccines for the Global Pandemic Demand. Vaccines 2020, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Butreddy, A.; Janga, K.Y.; Ajjarapu, S.; Sarabu, S.; Dudhipala, N. Instability of Therapeutic Proteins—An Overview of Stresses, Stabilization Mechanisms and Analytical Techniques Involved in Lyophilized Proteins. Int. J. Biol. Macromol. 2021, 167, 309–325. [Google Scholar] [CrossRef]
- Stadtman, E.R. Protein Oxidation and Aging. Free Radic. Res. 2006, 40, 1250–1258. [Google Scholar] [CrossRef] [Green Version]
- Adem, Y.T.; Molina, P.; Liu, H.; Patapoff, T.W.; Sreedhara, A.; Esue, O. Hexyl Glucoside and Hexyl Maltoside Inhibit Light-Induced Oxidation of Tryptophan. J. Pharm. Sci. 2014, 103, 409–416. [Google Scholar] [CrossRef]
- Daugherty, A.L.; Mrsny, R.J. Formulation and Delivery Issues for Monoclonal Antibody Therapeutics. Adv. Drug Deliv. Rev. 2006, 58, 686–706. [Google Scholar] [CrossRef]
- Bis, R.L.; Mallela, K.M.G. Antimicrobial Preservatives Induce Aggregation of Interferon Alpha-2a: The Order in Which Preservatives Induce Protein Aggregation Is Independent of the Protein. Int. J. Pharm. 2014, 472, 356–361. [Google Scholar] [CrossRef] [Green Version]
- Chi, E.Y.; Krishnan, S.; Randolph, T.W.; Carpenter, J.F. Physical Stability of Proteins in Aqueous Solution: Mechanism and Driving Forces in Nonnative Protein Aggregation. Pharm. Res. 2003, 20, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Lyophilization and Development of Solid Protein Pharmaceuticals. Int. J. Pharm. 2000, 203, 1–60. [Google Scholar] [CrossRef]
- Constantino, H.R. Excipients for Use in Lyophilized Pharmaceutical Peptide, Protein, and other Bioproducts. In Lyophilization of Biopharmaceuticals; Constantino, H.R., Pikal, M.J., Eds.; AAPS Press: Arlington, VA, USA, 2004; pp. 139–228. ISBN 0-97117676-0. [Google Scholar]
- Liu, B.; Zhou, X. Freeze-Drying of Proteins. In Cryopreservation and Freeze-Drying Protocols; Wolkers, W.F., Ed.; Springer: New York, NY, USA, 2015; pp. 459–476. ISBN 978-1-4939-2193-5. [Google Scholar]
- Emami, F.; Vatanara, A.; Park, E.J.; Na, D.H. Drying Technologies for the Stability and Bioavailability of Biopharmaceuticals. Pharmaceutics 2018, 10, 131. [Google Scholar] [CrossRef] [Green Version]
- Shamblin, S.L.; Tang, X.; Chang, L.; Hancock, B.C.; Pikal, M.J. Characterization of the Time Scales of Molecular Motion in Pharmaceutically Important Glasses. J. Phys. Chem. B 1999, 103, 4113–4121. [Google Scholar] [CrossRef]
- Liu, J.; Rigsbee, D.R.; Stotz, C.; Pikal, M.J. Dynamics of Pharmaceutical Amorphous Solids: The Study of Enthalpy Relaxation by Isothermal Microcalorimetry. J. Pharm. Sci. 2002, 91, 1853–1862. [Google Scholar] [CrossRef]
- Pikal, M.J.; Rigsbee, D.R. The Stability of Insulin in Crystalline and Amorphous Solids: Observation of Greater Stability for the Amorphous Form. Pharm. Res. 1997, 14, 1379–1387. [Google Scholar] [CrossRef]
- Cicerone, M.T.; Douglas, J.F. β-Relaxation Governs Protein Stability in Sugar-Glass Matrices. Soft Matter 2012, 8, 2983–2991. [Google Scholar] [CrossRef]
- Allison, S.D.; Chang, B.; Randolph, T.W.; Carpenter, J.F. Hydrogen Bonding between Sugar and Protein Is Responsible for Inhibition of Dehydration-Induced Protein Unfolding. Arch. Biochem. Biophys. 1999, 365, 289–298. [Google Scholar] [CrossRef]
- Chang, L.; Shepherd, D.; Sun, J.; Ouellette, D.; Grant, K.L.; Tang, X.; Pikal, M.J. Mechanism of Protein Stabilization by Sugars during Freeze-Drying and Storage: Native Structure Preservation, Specific Interaction, and/or Immobilization in a Glassy Matrix? J. Pharm. Sci. 2005, 94, 1427–1444. [Google Scholar] [CrossRef] [PubMed]
- Chieng, N.; Teo, X.J.; Cheah, M.H.; Choo, M.L.; Chung, J.; Hew, T.K.; Keng, P.S. Molecular Dynamics and Physical Stability of Pharmaceutical Co-Amorphous Systems: Correlation Between Structural Relaxation Times Measured by Kohlrausch-Williams-Watts With the Width of the Glass Transition Temperature (ΔTg) and the Onset of Crystallization. J. Pharm. Sci. 2019, 108, 3848–3858. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Pikal, M.J. Calorimetric Investigation of the Structural Relaxation of Amorphous Materials: Evaluating Validity of the Methodologies. J. Pharm. Sci. 2005, 94, 948–965. [Google Scholar] [CrossRef]
- Chang, L.L.; Pikal, M.J. Mechanisms of Protein Stabilization in the Solid State. J. Pharm. Sci. 2009, 98, 2886–2908. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Beig, A.; Ioffe-Dahan, V.; Agbaria, R.; Miller, J.M. The Twofold Advantage of the Amorphous Form as an Oral Drug Delivery Practice for Lipophilic Compounds: Increased Apparent Solubility and Drug Flux through the Intestinal Membrane. AAPS J. 2013, 15, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Karagianni, A.; Kachrimanis, K.; Nikolakakis, I. Co-Amorphous Solid Dispersions for Solubility and Absorption Improvement of Drugs: Composition, Preparation, Characterization and Formulations for Oral Delivery. Pharmaceutics 2018, 10, 98. [Google Scholar] [CrossRef] [Green Version]
- Löbmann, K.; Laitinen, R.; Strachan, C.; Rades, T.; Grohganz, H. Amino Acids as Co-Amorphous Stabilizers for Poorly Water-Soluble Drugs—Part 2: Molecular Interactions. Eur. J. Pharm. Biopharm. 2013, 85, 882–888. [Google Scholar] [CrossRef]
- Babu, N.J.; Nangia, A. Solubility Advantage of Amorphous Drugs and Pharmaceutical Cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- Sophocleous, A.M.; Zhang, J.; Topp, E.M. Localized Hydration in Lyophilized Myoglobin by Hydrogen–Deuterium Exchange Mass Spectrometry. 1. Exchange Mapping. Mol. Pharm. 2012, 9, 718–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schersch, K. Effect of Collapse on the Stability of Protein Lyophilisates. Ph.D. Thesis, Ludwig Maximilian University of Munich, Munich, Germany, 2009. [Google Scholar]
- Abdul-Fattah, A.M.; Dellerman, K.M.; Bogner, R.H.; Pikal, M.J. The Effect of Annealing on the Stability of Amorphous Solids: Chemical Stability of Freeze-Dried Moxalactam. J. Pharm. Sci. 2007, 96, 1237–1250. [Google Scholar] [CrossRef]
- Andronis, V.; Zografi, G. Molecular Mobility of Supercooled Amorphous Indomethacin, Determined by Dynamic Mechanical Analysis. Pharm. Res. 1997, 14, 410–414. [Google Scholar] [CrossRef]
- Oksanen, C.A.; Zografi, G. Molecular Mobility in Mixtures of Absorbed Water and Solid Poly(Vinylpyrrolidone). Pharm. Res. 1993, 10, 791–799. [Google Scholar] [CrossRef]
- Andronis, V.; Zografi, G. The Molecular Mobility of Supercooled Amorphous Indomethacin as a Function of Temperature and Relative Humidity. Pharm. Res. 1998, 15, 835–842. [Google Scholar] [CrossRef]
- Sibik, J.; Zeitler, J.A. Direct Measurement of Molecular Mobility and Crystallisation of Amorphous Pharmaceuticals Using Terahertz Spectroscopy. Adv. Drug Deliv. Rev. 2016, 100, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Correia, N.T.; Ramos, J.J.M.; Descamps, M.; Collins, G. Molecular Mobility and Fragility in Indomethacin: A Thermally Stimulated Depolarization Current Study. Pharm. Res. 2001, 18, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Bhugra, C.; Shmeis, R.; Krill, S.L.; Pikal, M.J. Different Measures of Molecular Mobility: Comparison between Calorimetric and Thermally Stimulated Current Relaxation Times Below Tg and Correlation with Dielectric Relaxation Times Above Tg. J. Pharm. Sci. 2008, 97, 4498–4515. [Google Scholar] [CrossRef] [PubMed]
- Roozen, M.J.G.W.; Hemminga, M.A.; Walstra, P. Molecular Motion in Glassy Water-Malto-Oligosaccharide (Maltodextrin) Mixtures as Studied by Conventional and Saturation-Transfer Spin-Probe e.s.r. Spectroscopy. Carbohydr. Res. 1991, 215, 229–237. [Google Scholar] [CrossRef]
- Cicerone, M.T.; Soles, C.L. Fast Dynamics and Stabilization of Proteins: Binary Glasses of Trehalose and Glycerol. Biophys. J. 2004, 86, 3836–3845. [Google Scholar] [CrossRef] [Green Version]
- Cicerone, M.T.; Soles, C.L.; Chowdhuri, Z.; Pikal, M.J.; Chang, L. Fast Dynamics as a Diagnostic for Excipients in Preservation of Dried Proteins. Am. Pharm. Rev. 2005, 8, 22–27. [Google Scholar]
- Luthra, S.A.; Hodge, I.M.; Pikal, M.J. Investigation of the Impact of Annealing on Global Molecular Mobility in Glasses: Optimization for Stabilization of Amorphous Pharmaceuticals. J. Pharm. Sci. 2008, 97, 3865–3882. [Google Scholar] [CrossRef]
- Chieng, N.; Mizuno, M.; Pikal, M. Characterization of Dynamics in Complex Lyophilized Formulations: I. Comparison of Relaxation Times Measured by Isothermal Calorimetry with Data Estimated from the Width of the Glass Transition Temperature Region. Eur. J. Pharm. Biopharm. 2013, 85, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luthra, S. Impact of Optimum Annealing on Chemical Stabilization of Model Amorphous Pharmaceuticals. Ph.D. Thesis, University of Connecticut, Storrs, CT, USA, 2007. [Google Scholar]
- Ediger, M.D.; Angell, C.A.; Nagel, S.R. Supercooled Liquids and Glasses. J. Phys. Chem. 1996, 100, 13200–13212. [Google Scholar] [CrossRef]
- Hodge, I.M. Physical Aging in Polymer Glasses. Science 1995, 267, 1945–1947. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.P.; Shalaev, E.; Barnard, J. The Roles of Acid–Base Relationships, Interfaces, and Molecular Mobility in Stabilization During Drying and in the Solid State. In Drying Technologies for Biotechnology and Pharmaceutical Applications; Ohtake, S., Izutsu, K., Lechuga-Ballesteros, D., Eds.; Wiley: Weilheim, Germany, 2020; pp. 317–346. ISBN 978-3-527-34112-2. [Google Scholar]
- Cicerone, M.T.; Tellington, A.; Trost, L.; Sokolov, A. Substantially Improved Stability of Biological Agents in Dried Form: The Role of Glassy Dynamics in Preservation of Biopharmaceuticals. Bioprocess Int. 2003, 1, 36–47. [Google Scholar]
- Hancock, B.C.; Shamblin, S.L.; Zografi, G. Molecular Mobility of Amorphous Pharmaceutical Solids Below Their Glass-Transition Temperatures. Pharm. Res. 1995, 12, 799–806. [Google Scholar] [CrossRef]
- Johari, C.P.; Goldstein, M. Viscous Liquids and the Glass Transition. II. Secondary Relaxations in Glasses of Rigid Molecules. J. Chem. Phys. 1970, 53, 2372–2388. [Google Scholar] [CrossRef]
- Cicerone, M.T.; Pikal, M.J.; Qian, K.K. Stabilization of Proteins in Solid Form. Adv. Drug Deliv. Rev. 2015, 93, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Ngai, K.L. The Origin of the Faster Mechanism of Partial Enthalpy Recovery Deep in the Glassy State of Polymers. Phys. Chem. Chem. Phys. 2021, 23, 13468–13472. [Google Scholar] [CrossRef] [PubMed]
- Vyazovkin, S.; Dranca, I. Probing Beta Relaxation in Pharmaceutically Relevant Glasses by Using DSC. Pharm. Res. 2006, 23, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Bhugra, C.; Shmeis, R.; Krill, S.L.; Pikal, M.J. Predictions of Onset of Crystallization from Experimental Relaxation Times I-Correlation of Molecular Mobility from Temperatures Above the Glass Transition to Temperatures Below the Glass Transition. Pharm. Res. 2006, 23, 2277–2290. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Fattah, A.M.; Lechuga-Ballesteros, D.; Kalonia, D.S.; Pikal, M.J. The Impact of Drying Method and Formulation on the Physical Properties and Stability of Methionyl Human Growth Hormone in the Amorphous Solid State. J. Pharm. Sci. 2007, 97, 163–184. [Google Scholar] [CrossRef]
- Duddu, S.D.; Dal Monte, P.R. Effect of Glass Transition Temperature on the Stability of Lyophilized Formulations Containing a Chimeric Therapeutic Monoclonal Antibody. Pharm. Res. 1997, 14, 591–595. [Google Scholar] [CrossRef]
- Abdul-Fattah, A.M.; Truong-Le, V.; Yee, L.; Nguyen, L.; Kalonia, D.S.; Cicerone, M.T.; Pikal, M.J. Drying-Induced Variations in Physico-Chemical Properties of Amorphous Pharmaceuticals and Their Impact on Stability (I): Stability of a Monoclonal Antibody. J. Pharm. Sci. 2007, 96, 1983–2008. [Google Scholar] [CrossRef]
- Zografi, G.; Newman, A. Interrelationships Between Structure and the Properties of Amorphous Solids of Pharmaceutical Interest. J. Pharm. Sci. 2017, 106, 5–27. [Google Scholar] [CrossRef] [Green Version]
- Pikal, M.J.; Rigsbee, D.R.; Roy, M.L. Solid State Chemistry of Proteins: I. Glass Transition Behavior in Freeze Dried Disaccharide Formulations of Human Growth Hormone (HGH). J. Pharm. Sci. 2007, 96, 2765–2776. [Google Scholar] [CrossRef]
- Haeuser, C.; Goldbach, P.; Huwyler, J.; Friess, W.; Allmendinger, A. Impact of Dextran on Thermal Properties, Product Quality Attributes, and Monoclonal Antibody Stability in Freeze-Dried Formulations. Eur. J. Pharm. Biopharm. 2020, 147, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Pyne, A.; Surana, R.; Suryanarayanan, R. Crystallization of Mannitol below Tg′ during Freeze-Drying in Binary and Ternary Aqueous Systems. Pharm. Res. 2002, 19, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.; Zografi, G. Commentary: Considerations in the Measurement of Glass Transition Temperatures of Pharmaceutical Amorphous Solids. AAPS PharmSciTech 2020, 21, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sundaramurthi, P.; Suryanarayanan, R. Calorimetry and Complementary Techniques to Characterize Frozen and Freeze-Dried Systems. Adv. Drug Deliv. Rev. 2012, 64, 384–395. [Google Scholar] [CrossRef]
- Höhne, G.; Hemminger, W.F.; Flammersheim, H.-J. Differential Scanning Calorimetry, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2003; ISBN 978-3-662-06710-9. [Google Scholar]
- Simperler, A.; Kornherr, A.; Chopra, R.; Bonnet, P.A.; Jones, W.; Motherwell, W.D.S.; Zifferer, G. Glass Transition Temperature of Glucose, Sucrose, and Trehalose: An Experimental and in Silico Study. J. Phys. Chem. B 2006, 110, 19678–19684. [Google Scholar] [CrossRef]
- Richardson, M.J.; Savill, N.G. Derivation of Accurate Glass Transition Temperatures By Differential Scanning Calorimetry. Rubber Chem. Technol. 1976, 49, 224–232. [Google Scholar] [CrossRef]
- Hancock, B.C.; Zografi, G. The Relationship Between the Glass Transition Temperature and the Water Content of Amorphous Pharmaceutical Solids. Pharm. Res. 1994, 11, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Luthra, S.A.; Hodge, I.M.; Pikal, M.J. Effects of Annealing on Enthalpy Relaxation in Lyophilized Disaccharide Formulations: Mathematical Modeling of DSC Curves. J. Pharm. Sci. 2008, 97, 3084–3099. [Google Scholar] [CrossRef]
- Mizuno, M.; Pikal, M.J. Is the Pre-Tg DSC Endotherm Observed with Solid State Proteins Associated with the Protein Internal Dynamics? Investigation of Bovine Serum Albumin by Solid State Hydrogen/Deuterium Exchange. Eur. J. Pharm. Biopharm. 2013, 85, 170–176. [Google Scholar] [CrossRef]
- Shultz, A.R.; Young, A.L. DSC on Freeze-Dried Poly(Methyl Methacrylate)-Polystyrene Blends. Macromolecules 1980, 13, 663–668. [Google Scholar] [CrossRef]
- Wang, B.; Cicerone, M.T.; Aso, Y.; Pikal, M.J. The Impact of Thermal Treatment on the Stability of Freeze-Dried Amorphous Pharmaceuticals: II. Aggregation in an IgG1 Fusion Protein. J. Pharm. Sci. 2010, 99, 683–700. [Google Scholar] [CrossRef] [PubMed]
- Angell, C. Relaxation in Liquids, Polymers and Plastic Crystals—Strong/Fragile Patterns and Problems. J. Non. Cryst. Solids 1991, 131–133, 13–31. [Google Scholar] [CrossRef]
- Reddy, R.; Chang, L.L.; Luthra, S.; Collins, G.; Lopez, C.; Shamblin, S.L.; Pikal, M.J.; Gatlin, L.A.; Shalaev, E. The Glass Transition and Sub-Tg-Relaxation in Pharmaceutical Powders and Dried Proteins by Thermally Stimulated Current. J. Pharm. Sci. 2009, 98, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Hodge, I.M.; Berens, A.R. Effects of Annealing and Prior History on Enthalpy Relaxation in Glassy Polymers. 2. Mathematical Modeling. Macromolecules 1982, 15, 762–770. [Google Scholar] [CrossRef]
- Thiewes, H.J.; Steeneken, P.A.M. The Glass Transition and the Sub-Tg Endotherm of Amorphous and Native Potato Starch at Low Moisture Content. Carbohydr. Polym. 1997, 32, 123–130. [Google Scholar] [CrossRef]
- Chang, L.; Milton, N.; Rigsbee, D.; Mishra, D.S.; Tang, X.; Thomas, L.C.; Pikal, M.J. Using Modulated DSC to Investigate the Origin of Multiple Thermal Transitions in Frozen 10% Sucrose Solutions. Thermochim. Acta 2006, 444, 141–147. [Google Scholar] [CrossRef]
- Artiaga, R.; López-Beceiro, J.; Tarrío-Saavedra, J.; Gracia-Fernández, C.; Naya, S.; Mier, J.L. Estimating the Reversing and Non-Reversing Heat Flow from Standard DSC Curves in the Glass Transition Region. J. Chemom. 2011, 25, 287–294. [Google Scholar] [CrossRef]
- Gill, P.S.; Sauerbrunn, S.R.; Reading, M. Modulated Differential Scanning Calorimetry. J. Therm. Anal. 1993, 40, 931–939. [Google Scholar] [CrossRef]
- MODULATED DSC® (MDSC®): How Does It Work? Available online: http://www.tainstruments.com/pdf/literature/MDSC.pdf (accessed on 15 September 2021).
- Wilkinson, C.J.; Doss, K.; Gulbiten, O.; Allan, D.C.; Mauro, J.C. Fragility and Temperature Dependence of Stretched Exponential Relaxation in Glass-Forming Systems. J. Am. Ceram. Soc. 2021, 104, 4559–4567. [Google Scholar] [CrossRef]
- Williams, G.; Watts, D.C. Non-Symmetrical Dielectric Relaxation Behaviour Arising from a Simple Empirical Decay Function. Trans. Faraday Soc. 1970, 66, 80–85. [Google Scholar] [CrossRef]
- Kohlrausch, R. Theorie Des Elektrischen Rückstandes in Der Leidener Flasche. Ann. Phys. 1854, 167, 179–214. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C. Microscopic Theory of the Kohlrausch Relaxation Constant ΒK. J. Non. Cryst. Solids 1994, 172–174, 98–103. [Google Scholar] [CrossRef]
- Drogoń, A.; Skotnicki, M.; Skotnicka, A.; Pyda, M. Physical Ageing of Amorphous Indapamide Characterised by Differential Scanning Calorimetry. Pharmaceutics 2020, 12, 800. [Google Scholar] [CrossRef]
- Chung, H.-J.; Lim, S.-T. Physical Aging of Glassy Normal and Waxy Rice Starches: Effect of Aging Temperature on Glass Transition and Enthalpy Relaxation. Carbohydr. Polym. 2003, 53, 205–211. [Google Scholar] [CrossRef]
- Pikal, M.J.; Chang, L.; Tang, X. Evaluation of Glassy-State Dynamics from the Width of the Glass Transition: Results from Theoretical Simulation of Differential Scanning Calorimetry and Comparisons with Experiment. J. Pharm. Sci. 2004, 93, 981–994. [Google Scholar] [CrossRef]
- Thermometric 2277 Thermal Activity Monitor Instruction Manual; TA Instruments: New Castle, DE, USA, 2008.
- TAM IV Isothermal Microcalorimeter. Available online: https://www.tainstruments.com/wp-content/uploads/BROCH-TAM-lV.pdf (accessed on 15 September 2021).
- Thermometric 2277 Thermal Activity Monitor Service Manual; TA Instruments: New Castle, DE, USA, 2006.
- Peyron, M.; Pierens, G.K.; Lucas, A.J.; Hall, L.D.; Stewart, R.C. The Modified Stretched-Exponential Model for Characterization of NMR Relaxation in Porous Media. J. Magn. Reson. Ser. A 1996, 118, 214–220. [Google Scholar] [CrossRef]
- Van den Mooter, G.; Augustijns, P.; Kinget, R. Stability Prediction of Amorphous Benzodiazepines by Calculation of the Mean Relaxation Time Constant Using the Williams-Watts Decay Function. Eur. J. Pharm. Biopharm. 1999, 48, 43–48. [Google Scholar] [CrossRef]
- Chieng, N.; Cicerone, M.T.; Zhong, Q.; Liu, M.; Pikal, M.J. Characterization of Dynamics in Complex Lyophilized Formulations: II. Analysis of Density Variations in Terms of Glass Dynamics and Comparisons with Global Mobility, Fast Dynamics, and Positron Annihilation Lifetime Spectroscopy (PALS). Eur. J. Pharm. Biopharm. 2013, 85, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duddu, S.P.; Zhang, G.; del Monte, P.R. The Relationship Between Protein Aggregation and Molecular Mobility Below the Glass Transition Temperature of Lyophilized Formulations Containing a Monoclonal Antibody. Pharm. Res. 1997, 14, 596–600. [Google Scholar] [CrossRef]
- Shamblin, S.L.; Hancock, B.C.; Pikal, M.J. Coupling between Chemical Reactivity and Structural Relaxation in Pharmaceutical Glasses. Pharm. Res. 2006, 23, 2254–2268. [Google Scholar] [CrossRef]
- Wang, B.; Tchessalov, S.; Cicerone, M.T.; Warne, N.W.; Pikal, M.J. Impact of Sucrose Level on Storage Stability of Proteins in Freeze-Dried Solids: II. Correlation of Aggregation Rate with Protein Structure and Molecular Mobility. J. Pharm. Sci. 2009, 98, 3145–3166. [Google Scholar] [CrossRef] [PubMed]
- Luthra, S.A.; Hodge, I.M.; Utz, M.; Pikal, M.J. Correlation of Annealing with Chemical Stability in Lyophilized Pharmaceutical Glasses. J. Pharm. Sci. 2008, 97, 5240–5251. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Fattah, A.M.; Truong-Le, V.; Yee, L.; Pan, E.; Ao, Y.; Kalonia, D.S.; Pikal, M.J. Drying-Induced Variations in Physico-Chemical Properties of Amorphous Pharmaceuticals and Their Impact on Stability II: Stability of a Vaccine. Pharm. Res. 2007, 24, 715–727. [Google Scholar] [CrossRef]
- Borde, B.; Bizot, H.; Vigier, G.; Buléon, A. Calorimetric Analysis of the Structural Relaxation in Partially Hydrated Amorphous Polysaccharides. II. Phenomenological Study of Physical Ageing. Carbohydr. Polym. 2002, 48, 111–123. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, G.G.Z.; Law, D.; Grant, D.J.W.; Schmitt, E.A. Physical Stability of Amorphous Pharmaceuticals: Importance of Configurational Thermodynamic Quantities and Molecular Mobility. J. Pharm. Sci. 2002, 91, 1863–1872. [Google Scholar] [CrossRef]
- Surana, R.; Pyne, A.; Suryanarayanan, R. Effect of Aging on the Physical Properties of Amorphous Trehalose. Pharm. Res. 2004, 21, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Surana, R.; Pyne, A.; Suryanarayanan, R. Effect of Preparation Method on Physical Properties of Amorphous Trehalose. Pharm. Res. 2004, 21, 1167–1176. [Google Scholar] [CrossRef]
- Bhugra, C.; Rambhatla, S.; Bakri, A.; Duddu, S.P.; Miller, D.P.; Pikal, M.J.; Lechuga-Ballesteros, D. Prediction of the Onset of Crystallization of Amorphous Sucrose Below the Calorimetric Glass Transition Temperature from Correlations with Mobility. J. Pharm. Sci. 2007, 96, 1258–1269. [Google Scholar] [CrossRef]
- Bhugra, C.; Pikal, M.J. Role of Thermodynamic, Molecular, and Kinetic Factors in Crystallization From the Amorphous State. J. Pharm. Sci. 2008, 97, 1329–1349. [Google Scholar] [CrossRef] [PubMed]
- Grzybowska, K.; Capaccioli, S.; Paluch, M. Recent Developments in the Experimental Investigations of Relaxations in Pharmaceuticals by Dielectric Techniques at Ambient and Elevated Pressure. Adv. Drug Deliv. Rev. 2016, 100, 158–182. [Google Scholar] [CrossRef] [PubMed]
- Vollrath, I. Controlled Nucleation and Heat Flux Measurements as Innovative Technologies for Freeze-Drying. Ph.D. Thesis, Ludwig Maximilian University of Munich, Munich, Germany, 2018. [Google Scholar]
- Gitter, J.H.; Geidobler, R.; Presser, I.; Winter, G. A Comparison of Controlled Ice Nucleation Techniques for Freeze-Drying of a Therapeutic Antibody. J. Pharm. Sci. 2018, 107, 2748–2754. [Google Scholar] [CrossRef] [PubMed]
- Lo Presti, K.; Friess, W. Adjustment of Specific Residual Moisture Levels in Completely Freeze-Dried Protein Formulations by Controlled Spiking of Small Water Volumes. Eur. J. Pharm. Biopharm. 2021. (under review). [Google Scholar]
- Schersch, K.; Betz, O.; Garidel, P.; Muehlau, S.; Bassarab, S.; Winter, G. Systematic Investigation of the Effect of Lyophilizate Collapse on Pharmaceutically Relevant Proteins, Part 2: Stability During Storage at Elevated Temperatures. J. Pharm. Sci. 2012, 101, 2288–2306. [Google Scholar] [CrossRef]
- Beezer, A.E.; Hills, A.K.; O’Neill, M.A.A.; Morris, A.C.; Kierstan, K.T.E.; Deal, R.M.; Waters, L.J.; Hadgraft, J.; Mitchell, J.C.; Connor, J.A.; et al. The Imidazole Catalysed Hydrolysis of Triacetin: An Inter- and Intra-Laboratory Development of a Test Reaction for Isothermal Heat Conduction Microcalorimeters Used for Determination of Both Thermodynamic and Kinetic Parameters. Thermochim. Acta 2001, 380, 13–17. [Google Scholar] [CrossRef]
- Hills, A.K.; Beezer, A.E.; Connor, J.A.; Mitchell, J.C.; Wolf, G.; Baitalow, F. Microcalorimetric Study of a Proposed Test Reaction—The Imidazole Catalysed Hydrolysis of Triacetin. Temperature and Imidazole Concentration Dependence. Thermochim. Acta 2002, 386, 139–142. [Google Scholar] [CrossRef]
- Angberg, M.; Nyström, C.; Castensson, S. Evaluation of Heat-Conduction Microcalorimetry in Pharmaceutical Stability Studies (1) Precission and Accuracy for Static Experiments in Glass Vials. Available online: http://www.tainstruments.com/pdf/M142.pdf. (accessed on 15 September 2021).
- Aso, Y.; Yoshioka, S.; Kojima, S. Relationship between the Crystallization Rates of Amorphous Nifedipine, Phenobarbital, and Flopropione, and Their Molecular Mobility as Measured by Their Enthalpy Relaxation and 1H NMR Relaxation Times. J. Pharm. Sci. 2000, 89, 408–416. [Google Scholar] [CrossRef]













| Sucrose-Placebo | Trehalose-Placebo | Su5.5-2-0PS | Su5.5-2-8PS | Su5.5-2-16PS | Su7.5-2-8PS | Tr.5.5-2-8PS | |
|---|---|---|---|---|---|---|---|
| IgG1 | - | - | 2 | 2 | 2 | 2 | 2 |
| Sucrose | 79.45 | - | 79.45 | 79.45 | 79.45 | 79.45 | - |
| Trehalose | - | 79.45 | - | - | - | - | 79.45 |
| Methionine | 1.5 | 1.5 | - | - | - | - | - |
| Histidine | 0.42 | 0.42 | 0.42 | 0.42 | 0.42 | 0.42 | 0.42 |
| Polysorbate 20 | 0.4 | 0.4 | - | 0.8 | 1.6 | 0.8 | 0.8 |
| Fit time Onset [h]—Endset [h] | ||
|---|---|---|
| 0.5–12 | 11 | 41 |
| 0.5–25 | 11 | 37 |
| 0.5–50 | 11 | 37 |
| 0.5–100 | 11 | 37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Groёl, S.; Menzen, T.; Winter, G. Calorimetric Investigation of the Relaxation Phenomena in Amorphous Lyophilized Solids. Pharmaceutics 2021, 13, 1735. https://doi.org/10.3390/pharmaceutics13101735
Groёl S, Menzen T, Winter G. Calorimetric Investigation of the Relaxation Phenomena in Amorphous Lyophilized Solids. Pharmaceutics. 2021; 13(10):1735. https://doi.org/10.3390/pharmaceutics13101735
Chicago/Turabian StyleGroёl, Sebastian, Tim Menzen, and Gerhard Winter. 2021. "Calorimetric Investigation of the Relaxation Phenomena in Amorphous Lyophilized Solids" Pharmaceutics 13, no. 10: 1735. https://doi.org/10.3390/pharmaceutics13101735
APA StyleGroёl, S., Menzen, T., & Winter, G. (2021). Calorimetric Investigation of the Relaxation Phenomena in Amorphous Lyophilized Solids. Pharmaceutics, 13(10), 1735. https://doi.org/10.3390/pharmaceutics13101735