1. Introduction
Dual centrifugation (DC) was first described by Massing et al. in 2007 [
1] as a powerful method for the preparation of vesicular phospholipid gels (VPGs) and, after simple dilution of a VPG, of a liposomal dispersion. DC homogenization of viscous lipid/buffer mixtures along with ceramic beads in small disposable vials is achieved by the additional, fast turning of the sample vial around its own axis (axis: 90° with respect to the longitudinal direction of the vial) during normal centrifugation. This results in a frequent and very powerful movement of the viscous sample material from top to bottom of the vial and vice versa. Strong bead–bead interactions take place and the sample material in between the beads becomes homogenized, particularly when the lipid mixture and the cloud of beads reach the distal regions of the vials (cloud homogenization). This very powerful movement of the potentially highly viscous samples is based on the high centrifugal acceleration of up to 1000×
g caused by the rotation around the primary axis, and is more than one magnitude higher than the sample acceleration that can be reached by the most powerful lab shakers [
2].
A more traditional method to prepare VPGs has been high-pressure homogenization (HPH) [
3]. DC produces VPGs and liposomes that are very similar to those made by high-pressure homogenization but does so in a gentler way. This results from the quasi-continuous action of comparatively moderate shear forces in the DC compared to a limited number of extreme-shear cycles in HPH. This way, DC reduces the risk of lipid breakdown products such as lysophosphatidylcholine and allows the entrapment of sensitive drugs. Additionally, the rather long duration of the DC process (typically 15–30 min for liposomes) makes it possible to efficiently cool the samples during DC [
2], which allows the entrapment of sensitive proteins [
4,
5].
DC allows the simultaneous preparation of up to 40 samples in one run. The liposomes can be prepared sterile and in very small batch sizes in closed vials [
1], which is especially advantageous when small batches of liposomes are needed (e.g., for biological tests) or when the lipids or the drug compound are rare and/or expensive. Preparations of extremely small amounts of sterile siRNA-containing liposomes by DC (1 mg) with high entrapping efficiency were reported [
6,
7]. While both DC and HPH yield equal VPGs and liposomes, small-scale development favors the smaller individual batch sizes possible with DC whereas HPH may facilitate the scale-up to big batches for production.
Shelf life is a major issue in the development of liposomal formulations. DC provides two possible solutions to this problem. First, VPGs prepared by DC can be used as a more stable storage form and can be diluted to form liposomes right before administration [
8,
9]. When VPGs are stored, there is no loss of encapsulation because there is no concentration gradient between the internal and external aqueous phase of the vesicles [
10]. Second, liposomes can be produced by DC in the hospital pharmacy immediately before the administration. To this end, the aqueous medium can be added to sterile containers, which can be stored including the active pharmaceutical ingredient (API), ceramic beads, and the lipid mixtures to be subjected to DC [
1,
2]. These special advantages of liposome preparation by DC also had direct impact on the design of our study. Whereas the ability to retain their cargo over significant storage times is a key parameter for most liposomal formulations, it is not of primary importance for DC liposomes and has not been addressed here.
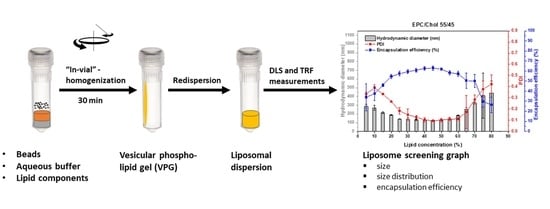
Determination of
EE after liposome production using a fluorescence dye like calcein typically requires the removal of the non-trapped fluorescence dye, e.g., by centrifugation, dialysis, or column separation [
11]. Due to the mechanical stress caused by the separation, liposome sizes and
EE might be influenced and it is often not possible to remove the non-entrapped fluorescence dye completely [
11]. Thus, methods have been described to determine
EE without removing the non-entrapped cargo. Oku et al. described a method for the determination of the trapped volume of liposomes using cobalt (Ⅱ) ions, which quench the fluorescence of accessible calcein by chelation [
12]. Zhang et al. developed a non-fluorescent method for a similar purpose by using homocarnosine as a marker. It shows a pH-sensitive
1H chemical shift in proton NMR in the presence of a pH gradient across the vesicle membrane or after addition of the chemical shift reagent TmDOTO
5− [
13]. However, both methods are very time consuming and subject to errors that render them unfavorable for a screening method where hundreds of samples have to be tested. Moreover, the samples might be affected by the addition of reagents or the formation of a pH gradient.
Time-resolved fluorescence is used for measuring fluorescence intensity decays. The sample is exposed to a very short pulse of laser light that is shorter than the decay time of the sample. The intensity decay is recorded on a nanosecond timescale using a high-speed photon detection system. In contrast, the commonly used steady-state fluorescence measurements are performed with constant illumination and detection and represent an average over the intensity decays of the time-resolved method. Time-resolved fluorescence measurements can provide additional information about the chemical surroundings or the concentration of the fluorescence dye [
14]. Collisional self-quenching fluorescence dyes like calcein show concentration-dependent lifetimes [
15] because the likelihood of quenching an excited dye by a collision increases with the time after excitation and with concentration.
Hence, self-quenching reduces the fluorescence lifetime, τ, of calcein from 4 ns in a dilute solution to, e.g., 0.4 ns at 70 mM calcein [
15]. This effect can be used to differentiate between highly concentrated calcein trapped in liposomes and non-entrapped calcein which becomes diluted as VPGs are diluted to obtain a liposomal dispersion.
Patel et al. developed a leakage assay for calcein-loaded vesicles which allowed the parallel quantification of free and entrapped calcein using a biexponential fit of calcein fluorescence decays and thus the correlation of efflux with local dye concentration [
15]. They prepared the liposomes by extrusion [
16]. Since extrusion typically resulted in rather low liposome concentrations, the concentration gradient necessary for efflux studies cannot be established by a simple dilution of the liposome dispersion. Instead, the non-entrapped calcein had to be removed by using a desalting column [
15]. Based on the higher liposome concentration within VPGs obtained by DC homogenization, it appeared possible to determine the
EE after dilution of VPGs to liposomes without removing the non-entrapped calcein. This was achieved by using the idea of Patel et al. to use a parallel quantification of free and entrapped calcein [
15] to directly determine the
EE of a calcein containing liposome dispersion obtained by dilution of DC-prepared VPGs.
Considering that DC allows very intense and thorough homogenization of a mixed sample, we investigated whether it could eliminate the need for prior lipid film preparation. Virtually all other techniques to produce cholesterol-containing liposomes require a prior co-dissolution of all ingredients in an organic solvent. For extrusion or sonication, the solvent is removed to establish a molecularly mixed lipid film on the wall of the container prior to adding buffer and downstream processing. Reverse phase evaporation is based on injecting the organic solution into buffer and removing the solvent afterwards [
17]. Another recent strategy to produce drug loaded liposomes at reduced utilization of organic solvents is the supercritical fluid technology (SCF) with supercritical carbon dioxide (scCO
2) [
18]. Frederiksen et al. described the preparation of liposomes loaded with water-soluble compounds using the SCF technology with scCO
2 and could reduce the amount of ethanol needed as a co-solvent [
19]. However, phospholipids are known to have a rather poor solubility in scCO
2 so that the addition of some organic solvents may still be required [
20,
21]. For cholesterol-free liposomes of L-α-dioleoyl phosphatidylcholine (DOPC), Otake et al. achieved an encapsulation efficiency of ~36% in a co-solvent free SCF preparation utilizing an improved supercritical reverse phase evaporation [
22].
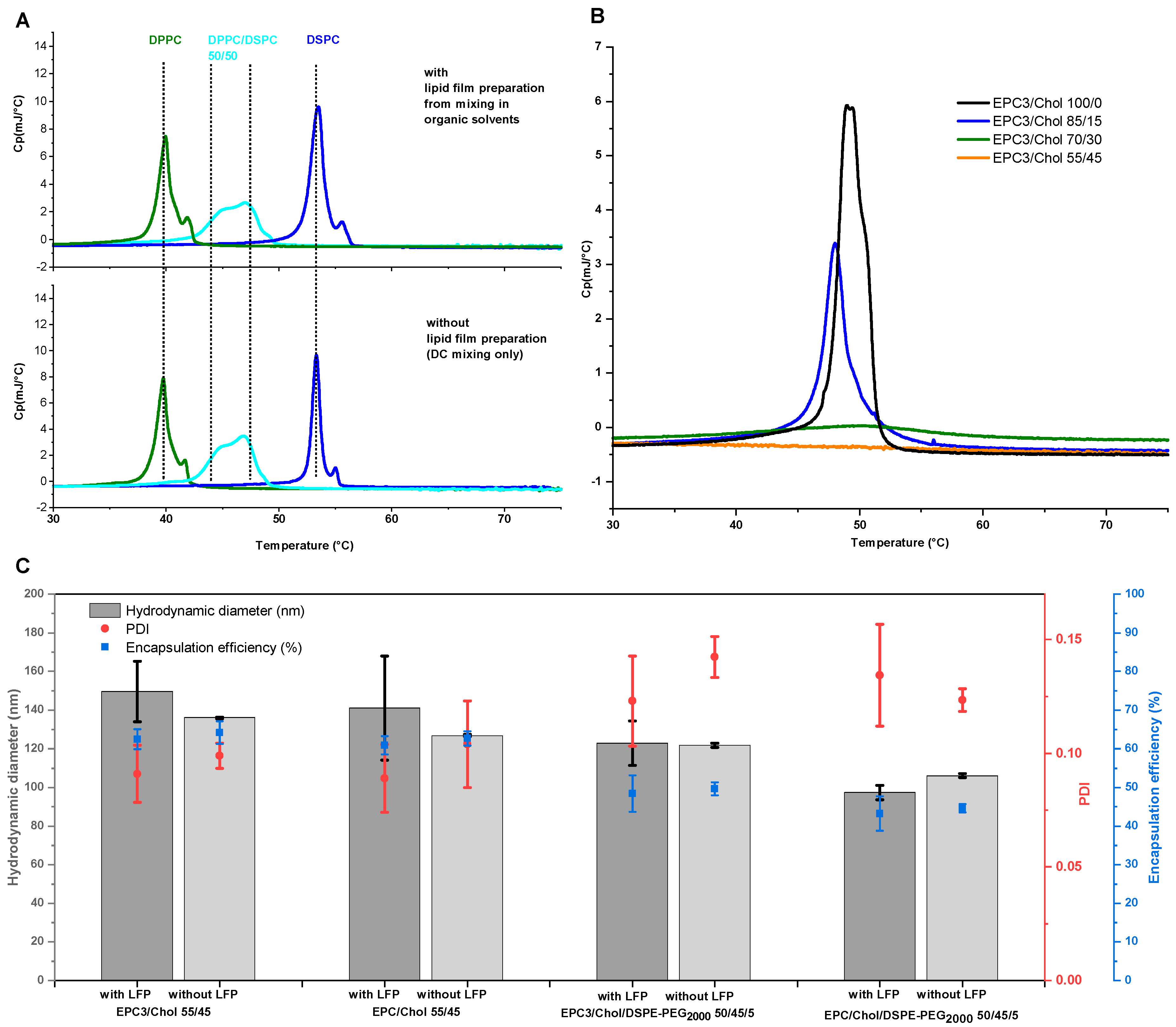
The hypothesis that DC is capable of generating molecularly homogeneous mixed liposomes without the prior dissolution in any organic solvents is tested using differential scanning calorimetry (DSC). This exploits the fact that lipids with different phase transitions that mix in one phase share a common characteristic melting peak at an intermediate temperature. When these lipids coexist in a dispersion without equilibrating at a molecular level, the result is a superposition of peaks representing different, individual local compositions.
To summarize, in this study, we developed a method for EE determination using time-resolved fluorescence measurements of calcein-loaded liposomes without removing the non-entrapped calcein. A very rapid screening method to find optimal lipid concentrations for DC homogenization of various lipid compositions was developed which allows the preparation of VPGs and liposomes without the need to first prepare a molecularly dispersed lipid-mixture. Furthermore, economic aspects of liposome preparation were considered by introducing the new parameter encapsulation capacity (EC).
4. Discussion
A first step in the design of a liposomal drug delivery system is the choice of its lipid constituents. The usage of saturated lipids and cholesterol renders liposomes particularly stable but may slow down drug release. PEG-grafted lipids may enhance circulation times by compromising the recognition of the liposomes by the immune system but may also hinder drug release by membrane fusion and cause adverse side effects. Consequently, there is no universal liposome formulation nor liposome composition and the production technology has to be adapted to a certain API, application route, and desired targeting and release characteristics.
4.1. DC Produces Liposomes from a Wide Variety of Lipid Compositions, Even without Lipid-Premixing in Organic Solvent
Formation of liposomes consisting of more than one lipid usually requires a molecularly dispersed lipid blend, especially when the poorly water-soluble cholesterol is part of the mixture. To get a so called “lipid film”, the lipids have to be dissolved in organic solvents (typically including the water-polluting chloroform) followed by a time consuming and careful removal of the solvents under vacuum, which involves the risk that lipid is lost by foaming over. Freeze drying from alcohol is also possible [
1,
6,
31] but requires expensive equipment. SCF technologies reduce or eliminate the need for organic solvents for certain lipid formulations but require very expensive equipment [
20], a complex optimization of the preparation conditions and are not suitable for bedside preparation. The efficient solubilization of the lipid mixture in scCO
2 depends on various parameters, including pressure, temperature, time and the amount of a co-solvent [
21].
Therefore, the finding that DC permits the formation of liposomes without the prior formation of a lipid film is a significant addition to the tool box of pharmaceutical liposome preparation. While the molecular mixing of different PC species could be expected during homogenization, the transfer of poorly water-soluble cholesterol from its crystalline form into the PC-bilayer was surprising. One explanation for that is the previous finding by Hagedorn et al. that DC is also a powerful tool for efficient nano-milling of water-insoluble compounds in an aqueous environment which contains detergents [
32]. The formation of nano-sized cholesterol crystals greatly enhances their surface area, which results in a strong increase of the water-solubility and the fast integration of cholesterol molecules within the PC-bilayers. This process was likely supported by PC, which might interact in a detergent-like manner and help to release cholesterol molecules from the nanocrystals.
4.2. Novel Screening Procedure Allows Tailoring the DC Protocol to the Specific Needs of a Given Lipid Formulation
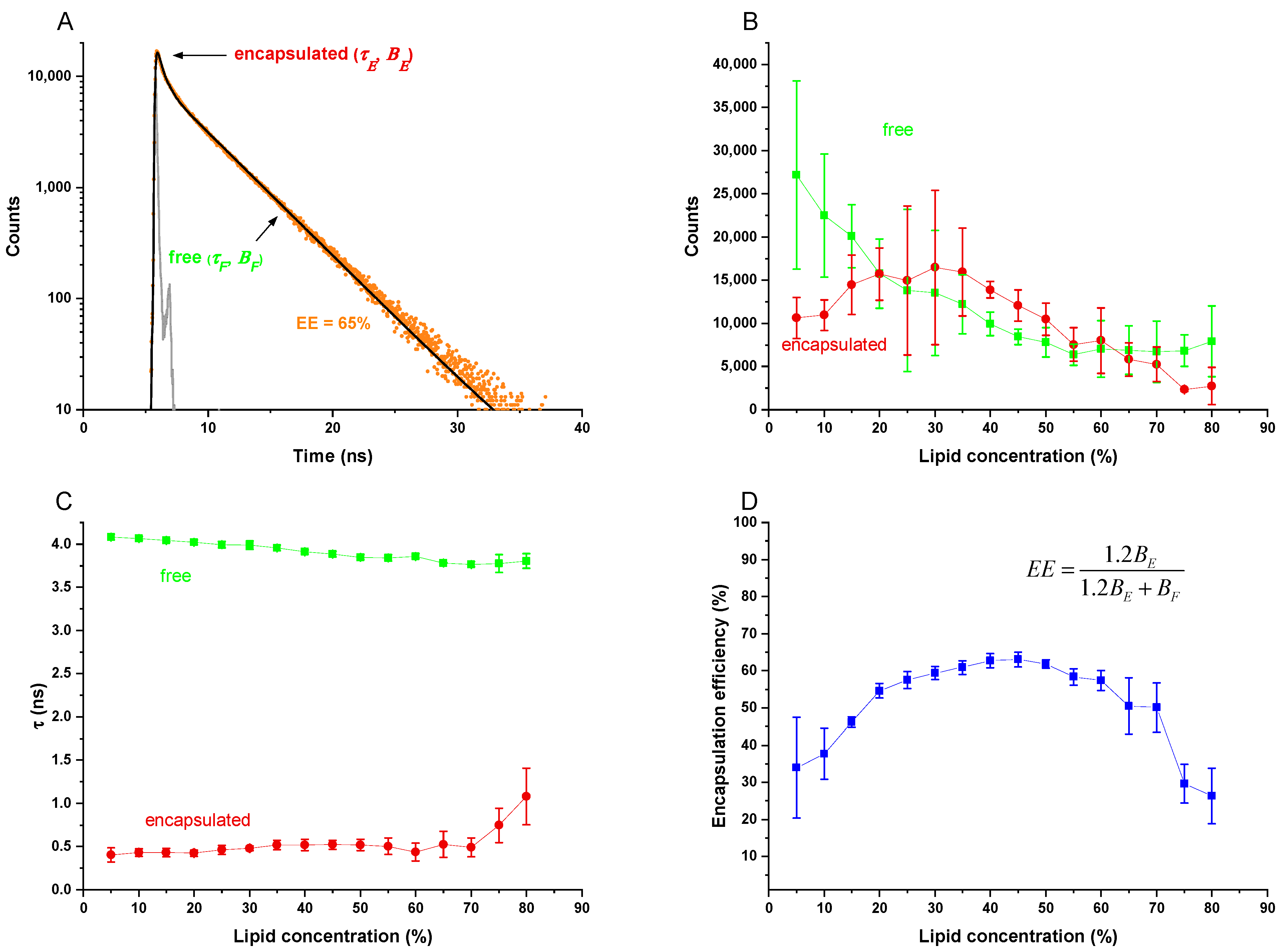
With the ability to process 40 samples at once, DC turned out to be a rapid screening tool for lipid compositions and homogenization conditions to finally identify a suitable liposomal drug delivery system for a specific application. Detailed knowledge about optimal lipid blends in combination with optimal preparation conditions can later be used to produce liposomes in bigger batches either by DC (e.g., for animal experiments or for bedside preparation) or by related means such as HPH. Furthermore, it was found that entrapping efficiency of the marker molecule calcein can be determined by time resolved fluorescence spectroscopy of the diluted liposomes without removing of the non-entrapped marker. This is very helpful in establishing a rapid screening procedure.
The methods typically used to determine
EE require the time consuming and error prone separation of the non-trapped drug or marker molecules, e.g., by using desalting- or SEC-columns [
1]. The method developed here is based on the fluorescence dye calcein, which is widely in use as a model for water soluble APIs. The new method is based on different fluorescence lifetimes of concentrated calcein inside the liposomes (0.4 ns) and of the non-entrapped calcein (4 ns) after dilution of the resulting VPGs (1:5000), which can individually be determined by time-resolved fluorescence spectroscopy. This technique is perfectly applicable here since liposomes made by DC or HPH typically have high
EE values for water-soluble molecules. Thus, the ratio of trapped to free calcein is high enough to allow simultaneous counting of photons belonging to the fluorescence decay curves characterized by the different lifetimes (
τF and
τE) with high accuracy (compare
Figure 1A,C).
Patel et al. showed that the longer fluorescence lifetimes of diluted calcein molecules of 4 ns can be measured up to 1 mM of calcein [
15]. Since the calcein concentrations after dilution of the VPGs were much lower (below 10 µM), the new method is also expected to be applicable to liposomal formulations with about 100-times lower
EE-values, which includes most liposomal formulations. In addition to this new method’s speed and capability of rapid screening, it is of particular advantage that the samples only have to be diluted prior to
EE determination, which avoids the risk of liposome destruction during removing of non-entrapped molecules.
Another useful finding is that the same samples can also be used for DLS measurements (size and size distribution), using the same cuvette by dilution with additional buffer, which saves additional materials and time. Thus, the preparation as well as the determination of EE values, size, and size distribution of 40 liposomal formulations with different lipid compositions is now possible within one working day. The drawback of this method is that the equipment for time-resolved fluorescence spectroscopy is not available in many laboratories.
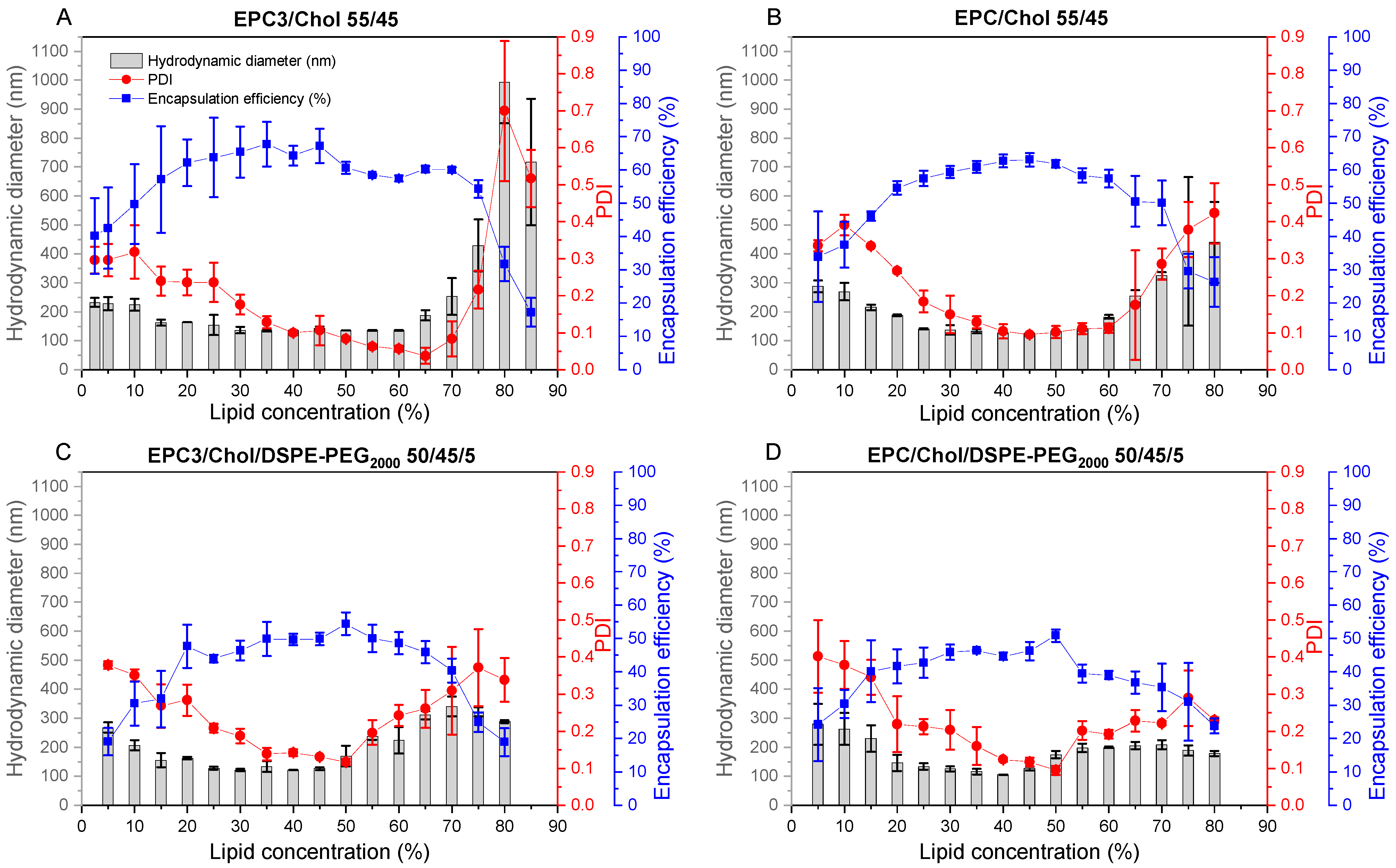
The results of screening a certain lipid composition were summarized in a liposome screening diagram, showing size, size distribution (PDI), and
EE values for a wide range of lipid concentrations (2.5–85%) at a glance. Liposome screening diagrams are not restricted to a certain lipid composition. Instead of varying the concentration of a defined lipid composition, one can also vary the API content or the composition itself (as done for investigating of the influence of DSPE-PEG2000 (
Figure 4)).
4.3. Lipid Concentration Determined Vesicle Size and Size Distribution
The liposome screening diagrams of the investigated phosphatidylcholine (PC)/Chol mixtures are similar and characterized by a decrease of size and narrower size distribution (lowered PDI) with increasing lipid concentration, followed by a concentration range in which the size and PDI are minimal. At higher lipid concentrations of about 65%, size and size distribution increase again.
The decrease of size and size distribution with increasing lipid concentrations can be explained by the increasing viscosity of the lipid mixtures, which allows the introduction of more energy during the DC process. The increase of size and PDI at very high lipid concentrations, for which a very high viscosity can also be assumed, can be explained by the presence of insufficient water to completely hydrate the membranes (discussed in detail below).
For each lipid mixture, a certain minimal liposome size seems to be a typical characteristic of a certain lipid composition which is reached between 25 and 60% lipid concentration. Even with longer homogenization times, this minimal size cannot further be reduced [
1]. This “homogenization limit” is related to the “grinding limit”observed during nano-milling of poorly soluble drugs by DC [
33]. The grinding limit represents an equilibrium between crystal downsizing and reformation of smaller crystals to larger ones. In analogy to that, the homogenization limit might be characterized by an equilibrium between the formation of small bilayer fragments from larger vesicles by disruption and correspondingly smaller vesicle formation as well as the reformation of larger vesicles from two or more of the smaller fragments.
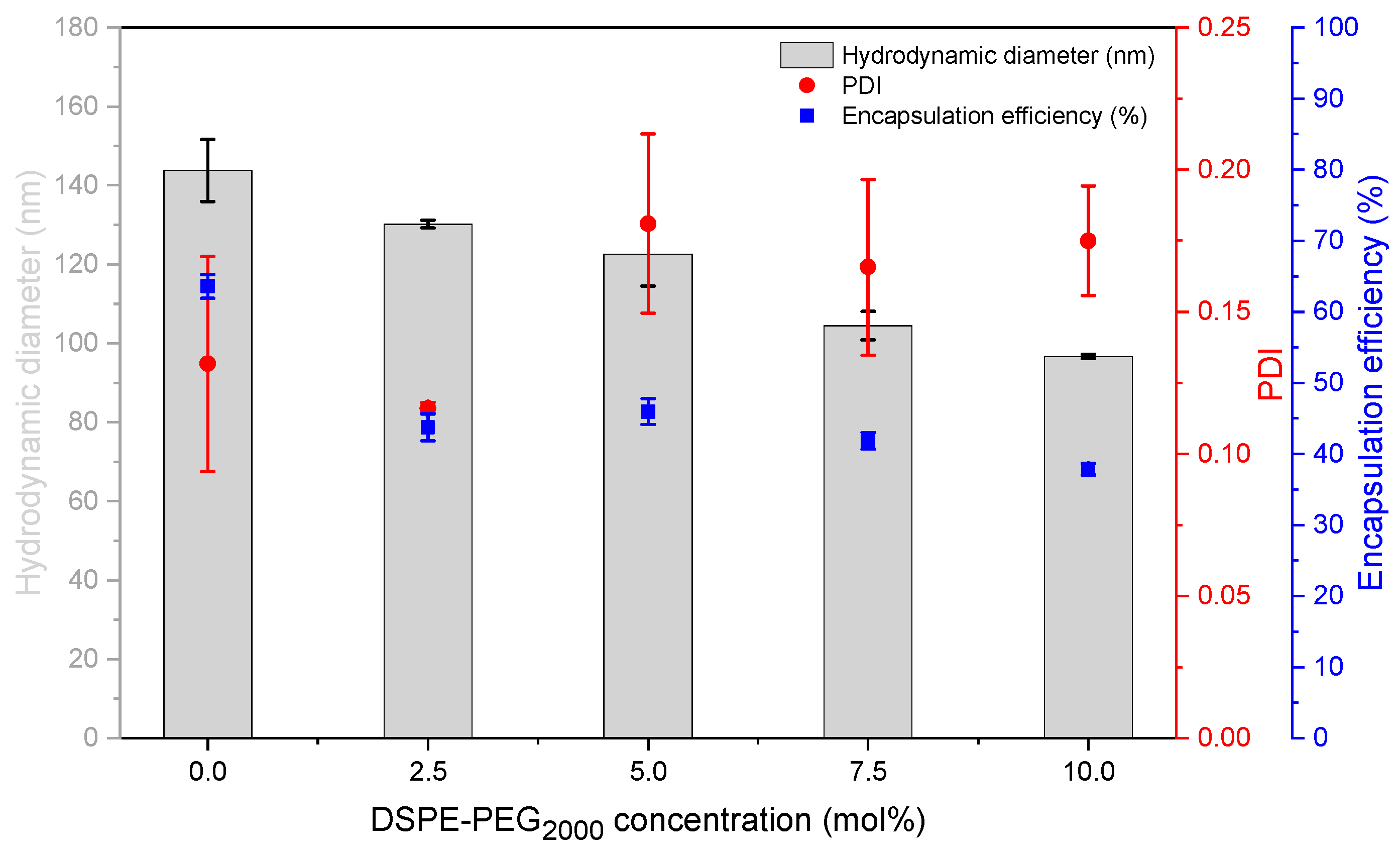
The process of reformation to larger vesicles is likely supported by the close vicinity of the bilayer fragments within the highly concentrated VPGs, and by the higher energy demand which is necessary to form smaller vesicles with a narrower curvature [
2]. This theory is supported by the finding that liposome size will not further decrease with increasing lipid concentrations, but the PDI will, showing that there is still an ongoing disruption of the small particles, although the average size remains the same. Previous studies have shown that liposome sizes decrease with increasing ratios of DSPE-PEG
2000 [
34,
35,
36] and that PEG-containing liposomes are more spherical and unilamellar [
34,
37]. It was discussed that the large hydrophilic head group of PEG lipids might cause a steric repulsion between the different lipid layers and thus results in liposomes with smaller size and reduced numbers of lamellae [
34]. In line with that, we also showed a decrease in the size of DC-made EPC/Chol liposomes with increasing amounts of PEG lipids (
Figure 4). Furthermore, the interval of minimal vesicle sizes in the liposome screening diagrams (
Figure 3) shifted toward lower lipid concentrations when adding DSPE-PEG
2000 to the lipid blends (compare
Figure 3A–D), which can be explained by the higher demand of water for the hydration of the PEG headgroups.
4.4. Liposomal Packing Accounts for 3 Characteristic Regions of EE as a Function of Lipid Content upon DC
The lipid concentration used for DC preparation of the VPGs and subsequently liposomes has great influence on the
EE values (
Figure 5). The EEs share a principal behavior including three characteristic regions: a quasi-linear increase up to ≈20% (Ⅰ), a plateau region up to roughly 70% (Ⅱ), and a region with decreasing
EE above ≈70% (Ⅲ).
At low lipid concentrations, the volume fraction occupied by liposomes is low, so that liposome–liposome interactions are small. Each liposome entraps its core volume of drug solution and thus, the EE increases proportionally with the lipid concentration (region Ⅰ). Internal vesicles in oligovesicular or oligolamellar liposomes reduce the slope of this relationship, since they do not contribute to further entrapment, but rather fill space within the outermost liposome core with lipid.
It is important to recall that the mathematical limit for the densest packing of perfect, monodisperse spheres fills a volume fraction of 74%. Of course, the real system differs from this calculation in a number of ways. On the one hand, aqueous solutes are entrapped only in the aqueous core of the liposome, which is smaller than the outer volume by the volume filled by the lipids. This should limit
EE of monodisperse, spherical liposomes to a value well below 74%. On the other hand, real VPGs comprise liposomes of different sizes and higher lamellarity, which increases the theoretical dense-packing limit. In line with these considerations,
EEs of non-pegylated liposomes (cf.
Figure 3A,B) approached maximal
EE values of roughly 60% at about a lipid concentration of 20% (region Ⅰ), followed by a plateau with only minor changes of
EE up to about 70% lipid (region Ⅱ). The larger volume required by PEG-lipids may explain the lower plateau values of
EE in
Figure 3C,D.
The decreasing
EE in region Ⅲ, i.e., above ≈70% lipid, can be explained by an insufficient amount of water in the VPG to form stable, closed liposomes. With insufficient water, stacks of bilayers are likely to be formed that do not exhibit enclosed cores, and hence, cannot entrap a cargo. Only upon addition of additional buffer to the VPG, closed liposomal vesicles will be formed and thus enclose part of the aqueous medium added. It has been demonstrated that adding a small amount of calcein solution to a VPG of 80% lipid prepared without calcein leads to the partial entrapment of the “reconstitution medium” (
EE > 50%). By contrast, VPGs produced at lipid contents below ≈70% (plateau region, Ⅱ) retain their original content and hardly take up any solution added upon reconstitution (
Table 2). On the other hand, the liposome reconstitution process in region Ⅲ must reduce
EE as reconstitution is performed with calcein-free buffer.
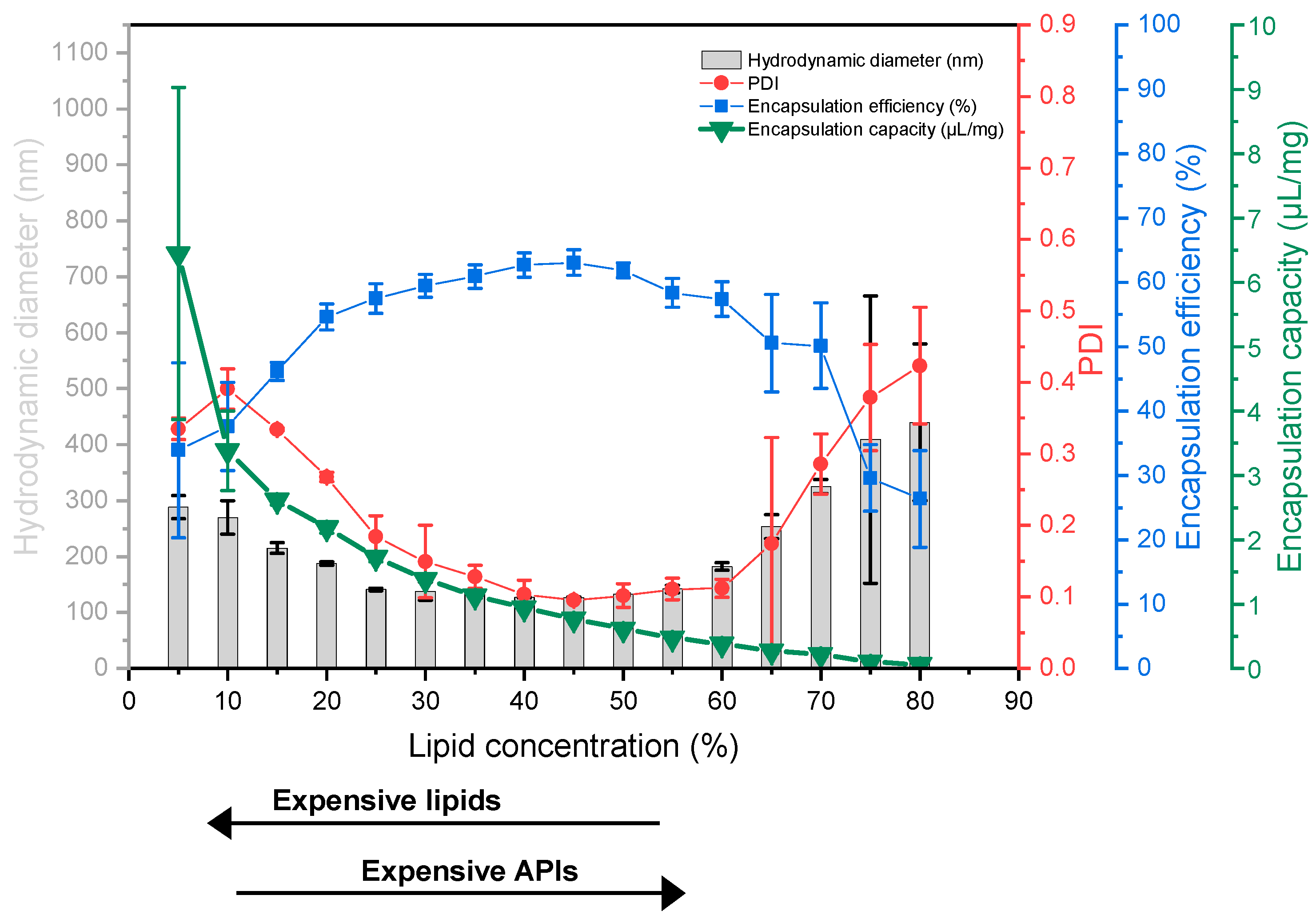
4.5. The Economic Aspect of DC-Homogenization—Introducing “Encapsulation Capacity”
At a first glance, the lipid concentration resulting in the lowest liposome sizes and narrow size distributions in combination with the highest
EE values appears optimal and can typically be found around lipid concentrations of 40–50% used for DC homogenization. However,
EE values take only the drug compounds into account, but not the amount of lipids which are part of the formulation. When it comes to liposomes made by DC or HPH, the lipid concentrations are rather high. Those high lipid concentrations automatically limit the amount of water (as well as the water-soluble drug compound) which can be entrapped even at high
EE values. Thus, the “capacity” of DC-made liposomes to entrap a water-soluble drug is intrinsically low. Following that, we also considered the ratio of the water-soluble drug compound (here: calcein) to the amount of lipid used for entrapping the drug, defined as encapsulation capacity (
EC), and indicated this with the unit (µL/mg).
EC is calculated from the data obtained from the liposome screening experiments using Equation (3).
EC is typically high at low lipid concentrations, but also depends on the
EE values reached (
Figure 5, green triangles).
Using
EC allows the selection of the optimal lipid concentration from a more economic point of view, including the costs of the lipids and APIs. For example, if very expensive or rare APIs will be entrapped or only a very small number of liposomes is needed, one would select the lipid concentration resulting in the highest
EE values in combination with the optimal particle sizes. Examples of these APIs are mRNA, siRNA, peptides, or rare natural products. If the lipid is the most expensive ingredient, or if higher amounts of liposomes are needed, one would select a lipid concentration which results in a higher
EC value. Since the resulting liposomes tend to be rather large with a broader size distribution, those formulations can potentially be used for oral applications or as depot formulations (intraperitoneal injection, intramuscular injection, subcutaneous injection). A third scenario is an expensive lipid mixture, or the need for high amounts of liposomes, but with the necessity of small and uniform liposomes. Here, the optimal lipid-concentration for DC homogenization will be a compromise and will be found in between the previous examples, at the lowest possible lipid concentration which produces acceptable liposomes.
5. Conclusions
We present a fast, economic, and convenient screening procedure to optimize liposomes obtained from vesicular phospholipid gels (VPGs) with respect to encapsulation efficiency (EE), encapsulation capacity of the lipid (EC), and the size and size distribution (PDI) of the liposomes. Therefore, we developed a new method for the direct determination of the EE by using time-resolved fluorescence measurements, without the need to remove the non-entrapped calcein. Furthermore, we were able to show that lipid film preparation is not necessary for liposome preparation by DC, which additionally accelerates and simplifies the screening process.
Using this approach, favorable protocols were identified to produce VPGs of different lipid compositions via dual centrifugation (DC) and to optimize the preparation for the specific needs of a selected API (including si- or mRNA, hydrophilic and hydrophobic drugs), application route (intravenous, subcutaneous injection, oral, etc.), or release profile (including temperature-sensitive liposomes, etc.).
This renders DC a very promising technique to produce small batches of liposomal formulations as needed for personalized bedside production, small studies, or the development of VPGs that can later be scaled up by switching from DC to HPH technology.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}