
Novel Polymorphic Cocrystals of the Non-Steroidal Anti-Inflammatory Drug Niflumic Acid: Expanding the Pharmaceutical Landscape

 ,
,  ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
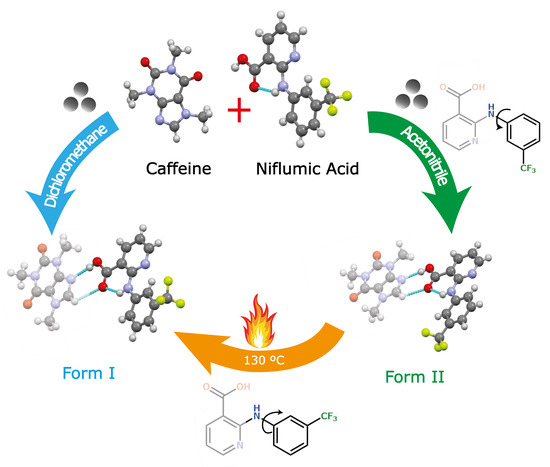
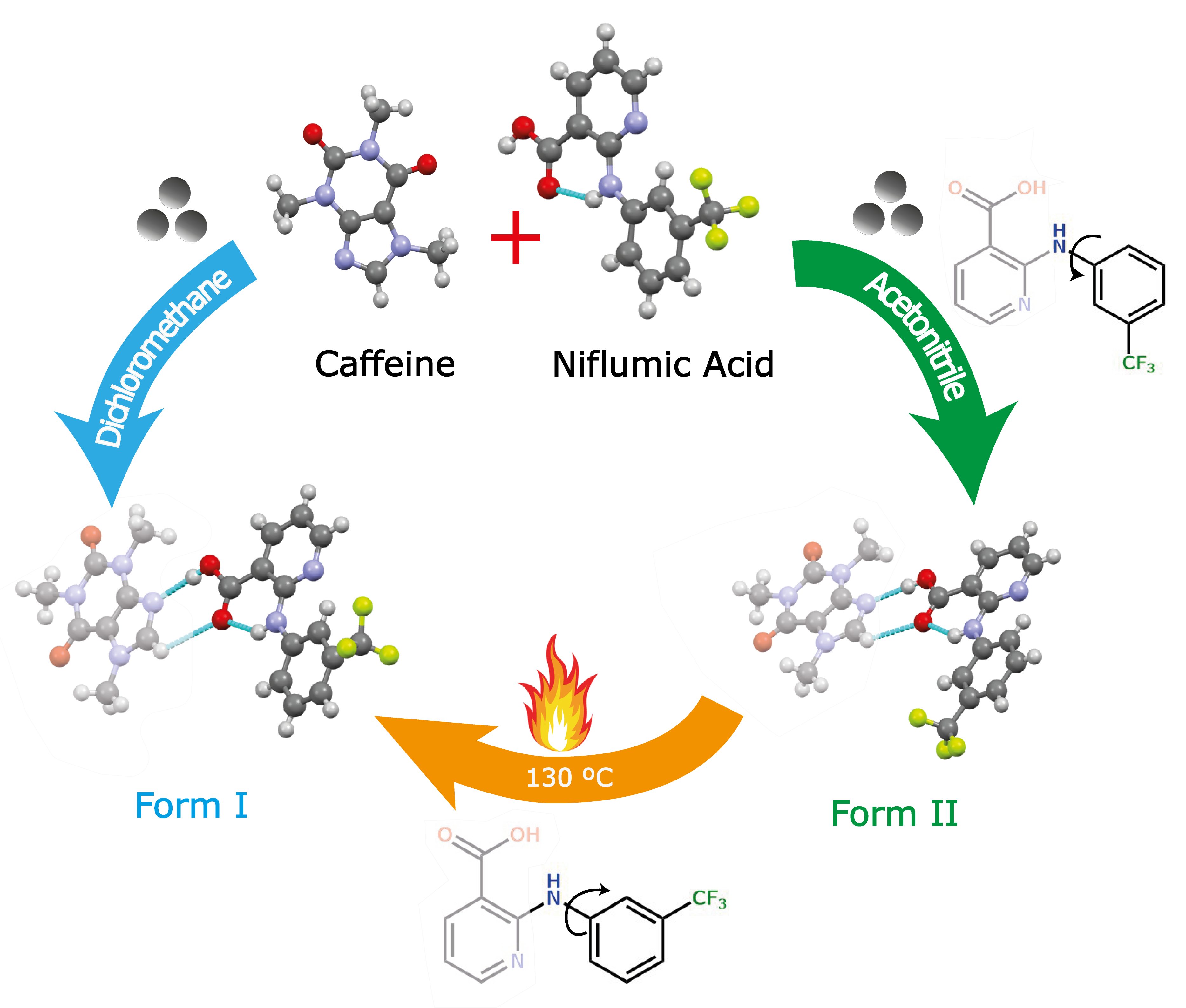
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. General Procedure for Synthesis of Cocrystal Polymorphs
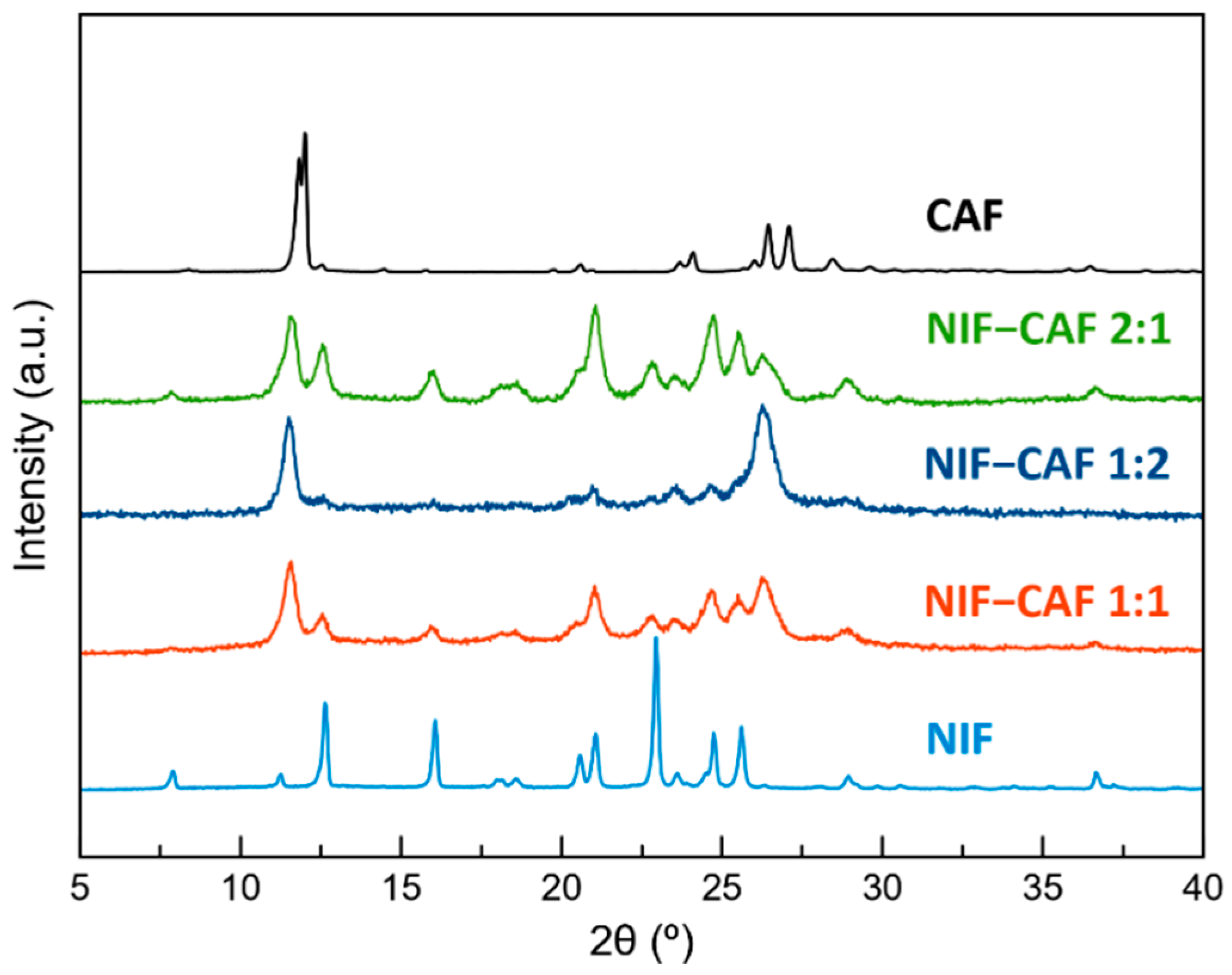
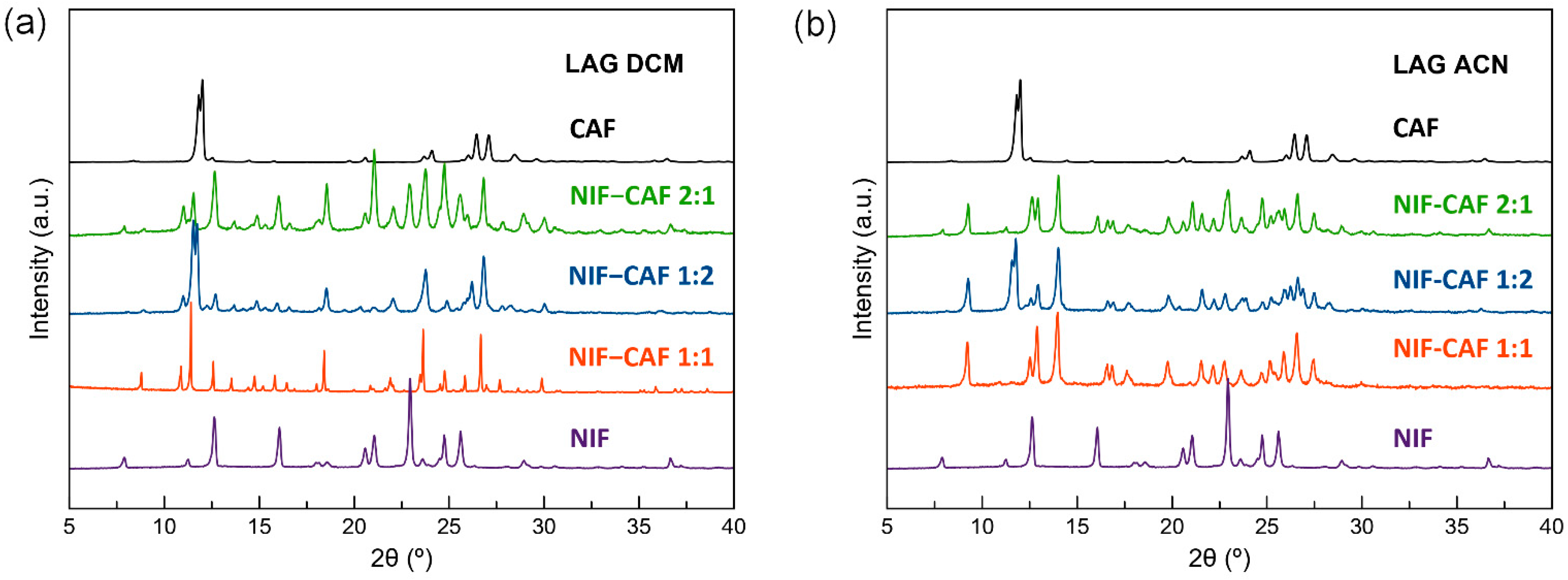
2.3. Powder X-ray Diffraction (PXRD)
2.4. Preparation of Single Crystals
2.5. Single-Crystal X-ray Diffraction (SCXRD)
2.6. Computational Studies
2.7. FT-IR Analysis
2.8. Thermal Analysis
2.9. Stability Test
2.10. Powder Dissolution Profile
3. Results and Discussion
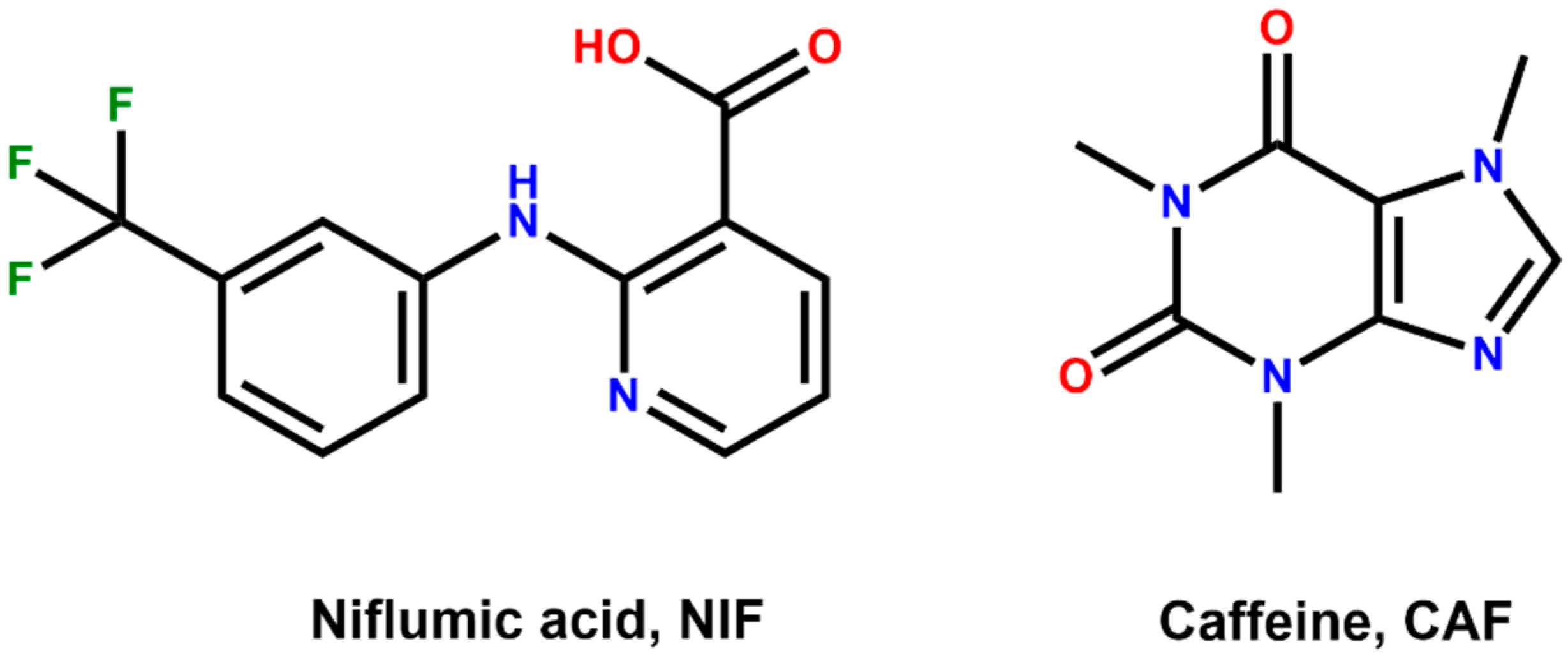
3.1. Preparation of Cocrystal Polymorphs
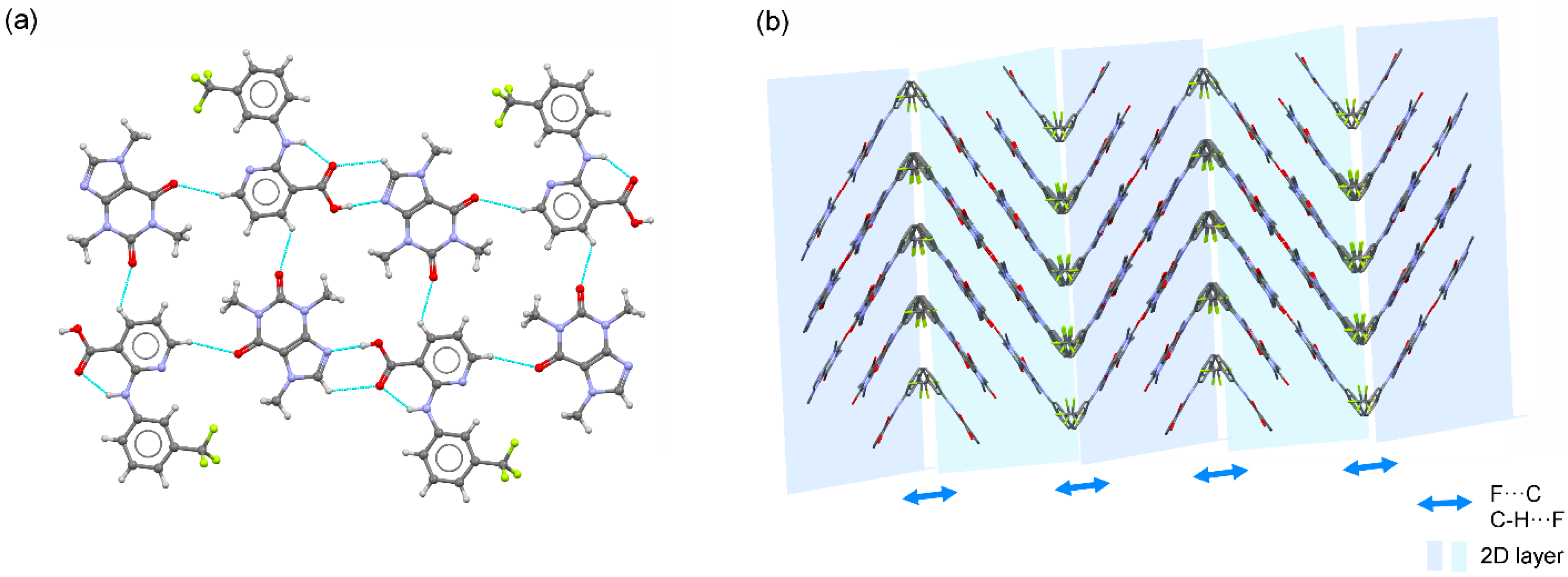
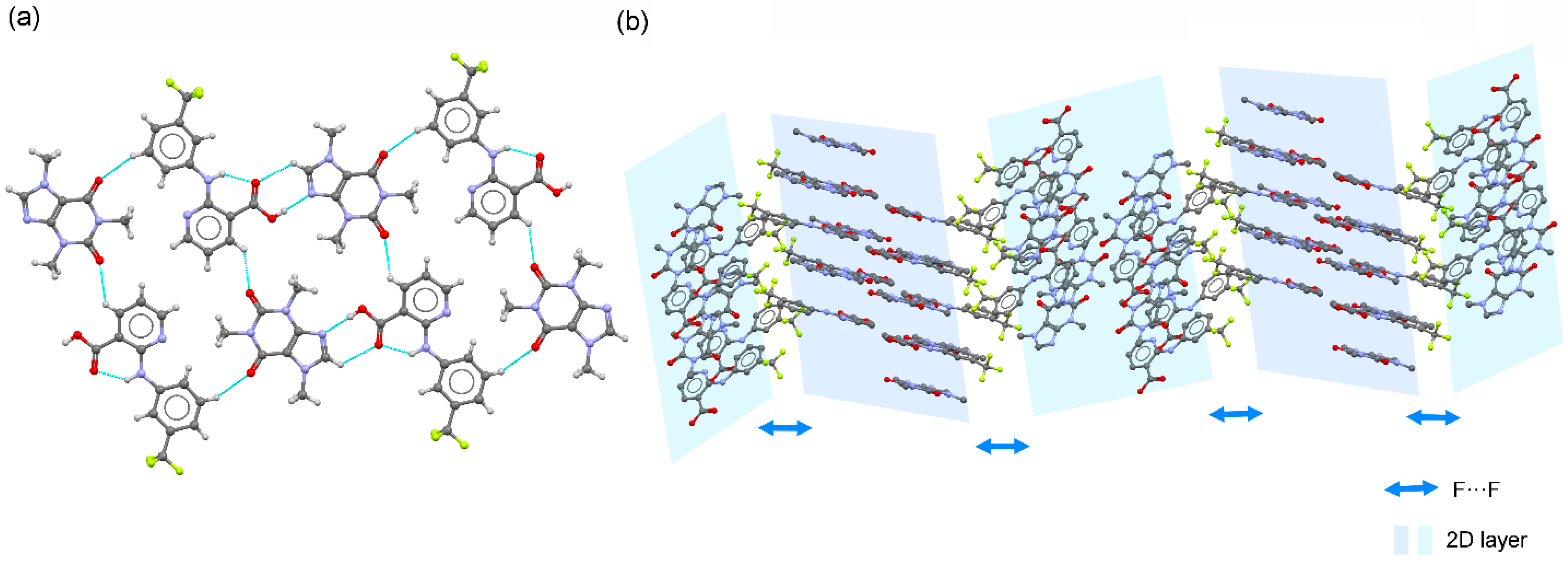
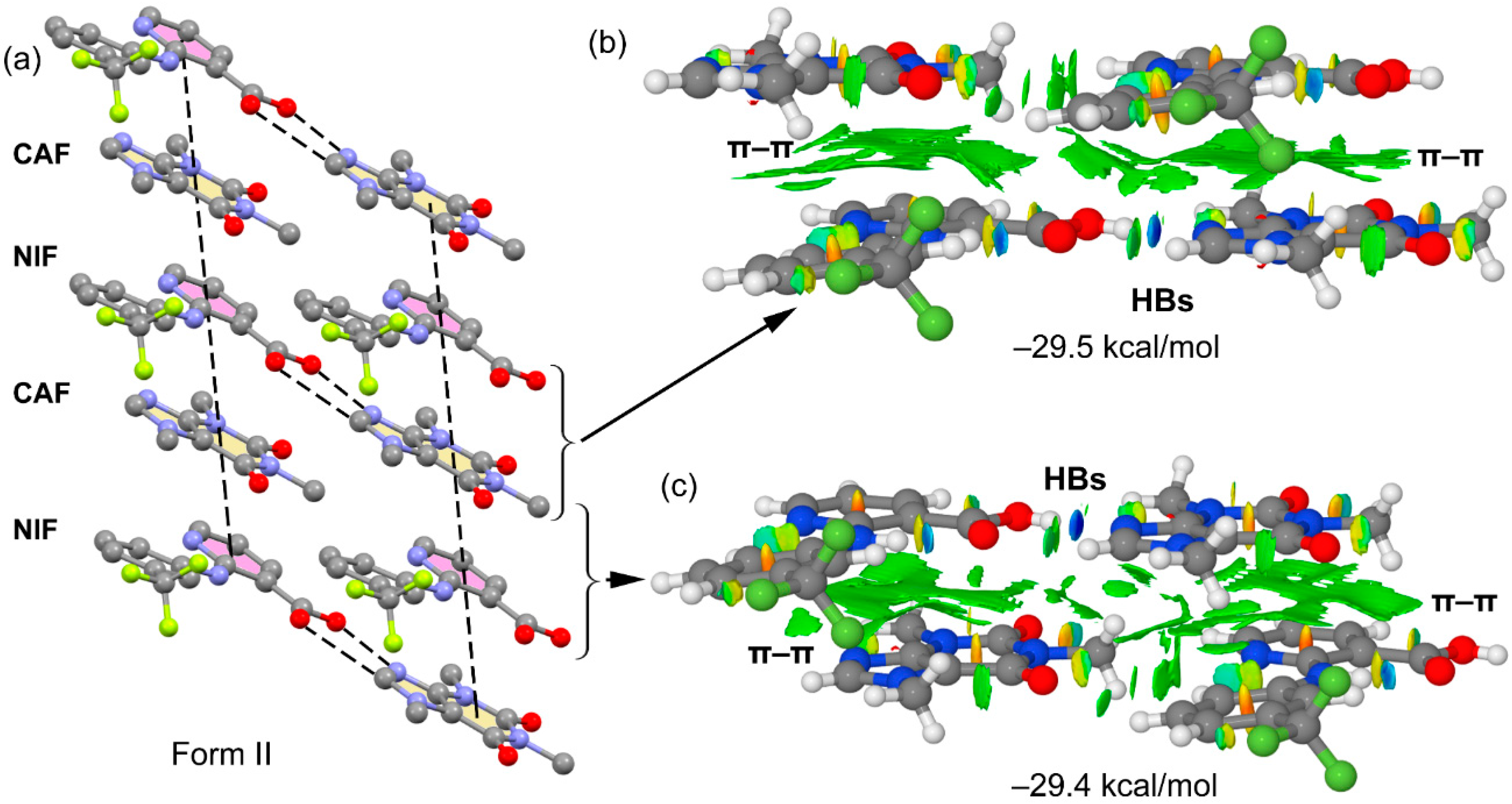
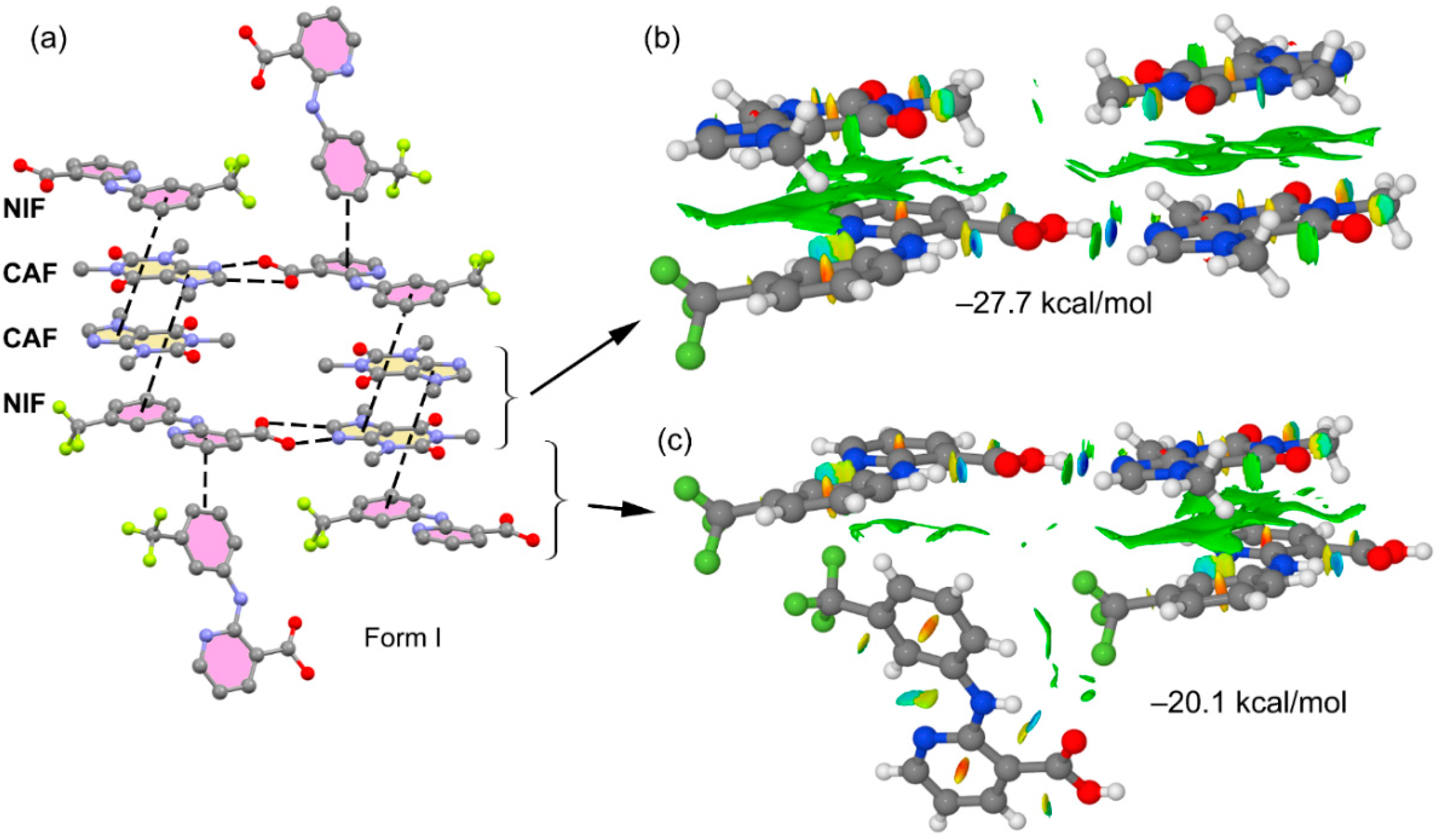
3.2. Crystal Structure Analysis
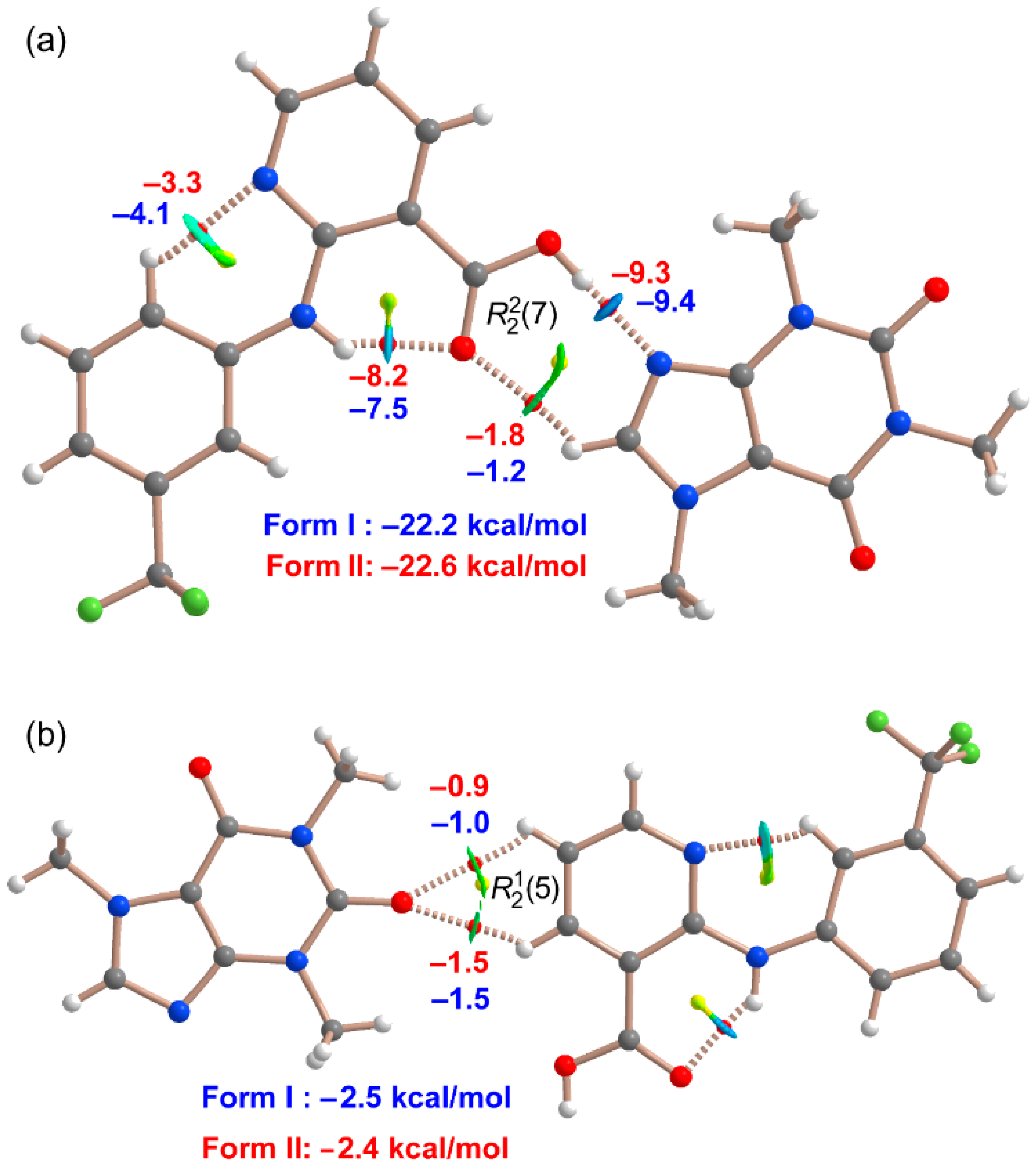
3.3. DFT Study of Noncovalent Interactions
3.4. FT-IR Analysis
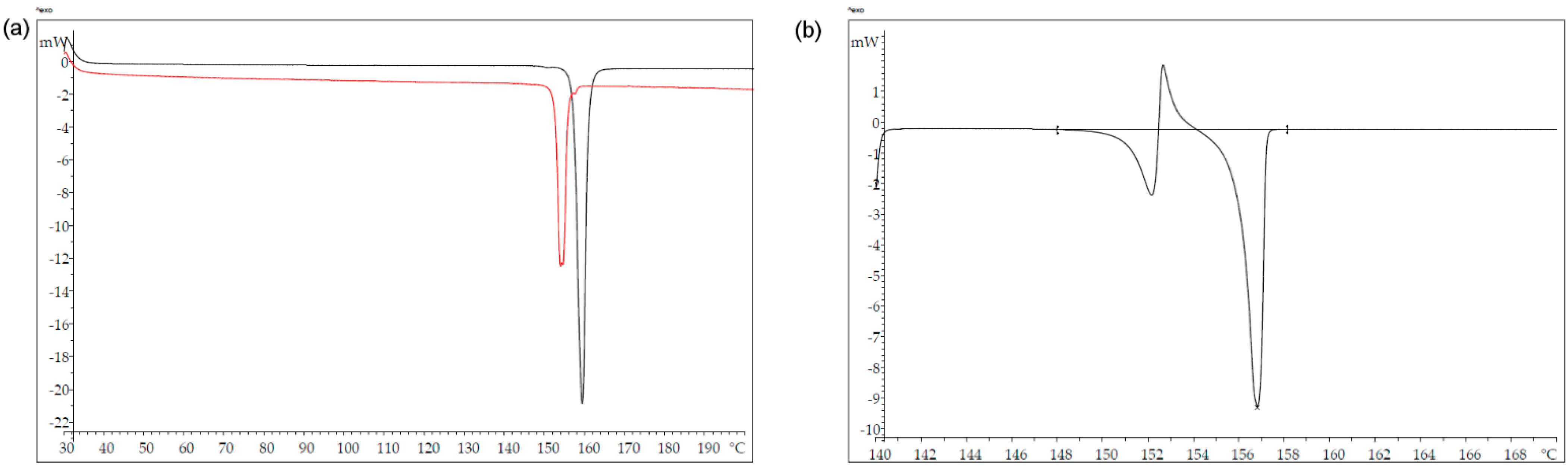
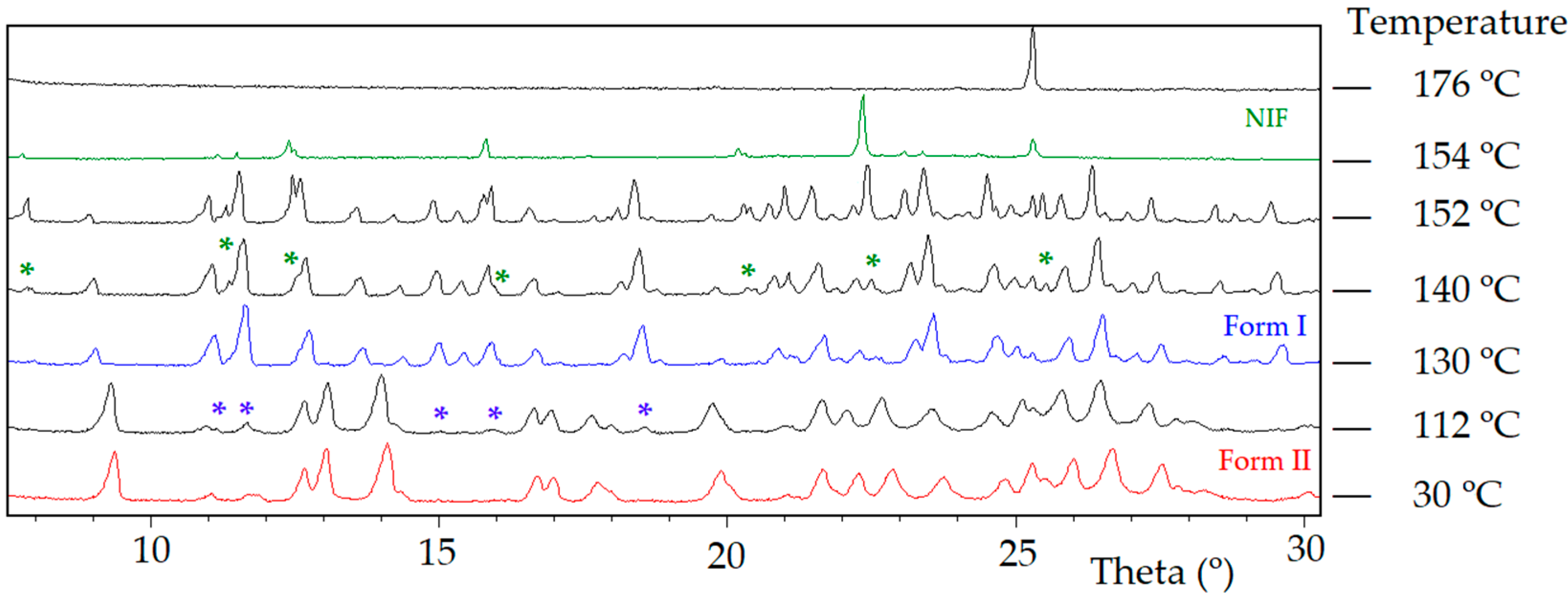
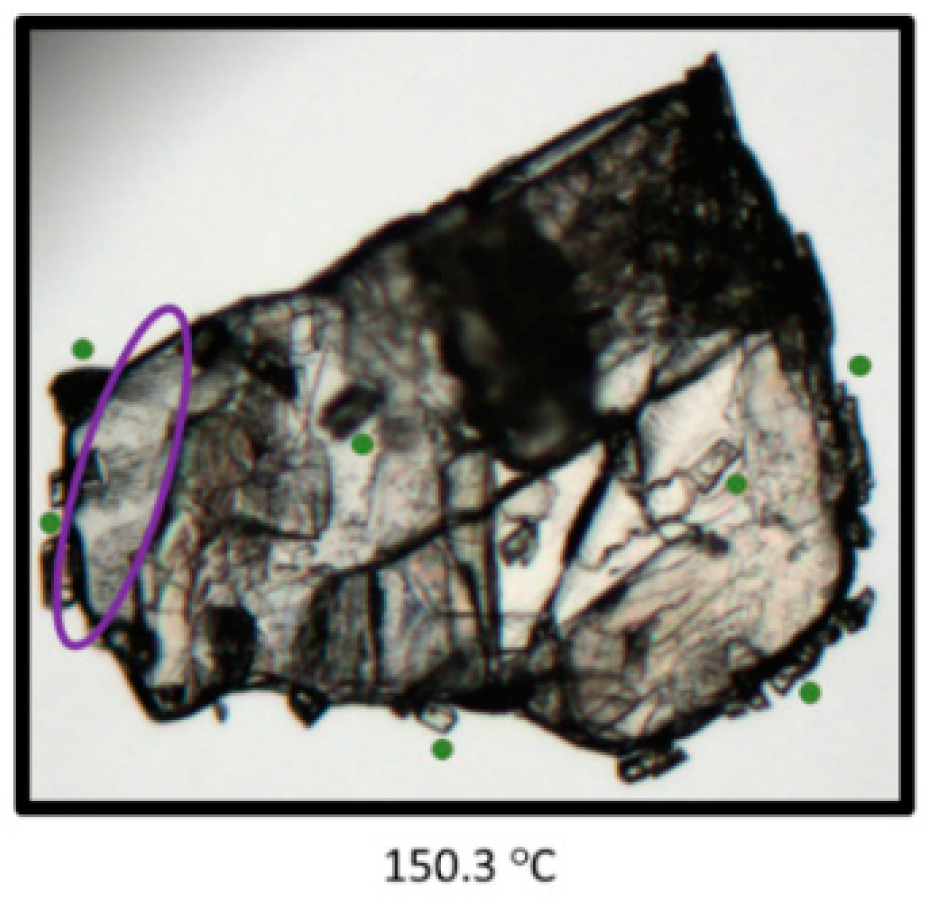
3.5. Thermal Analysis
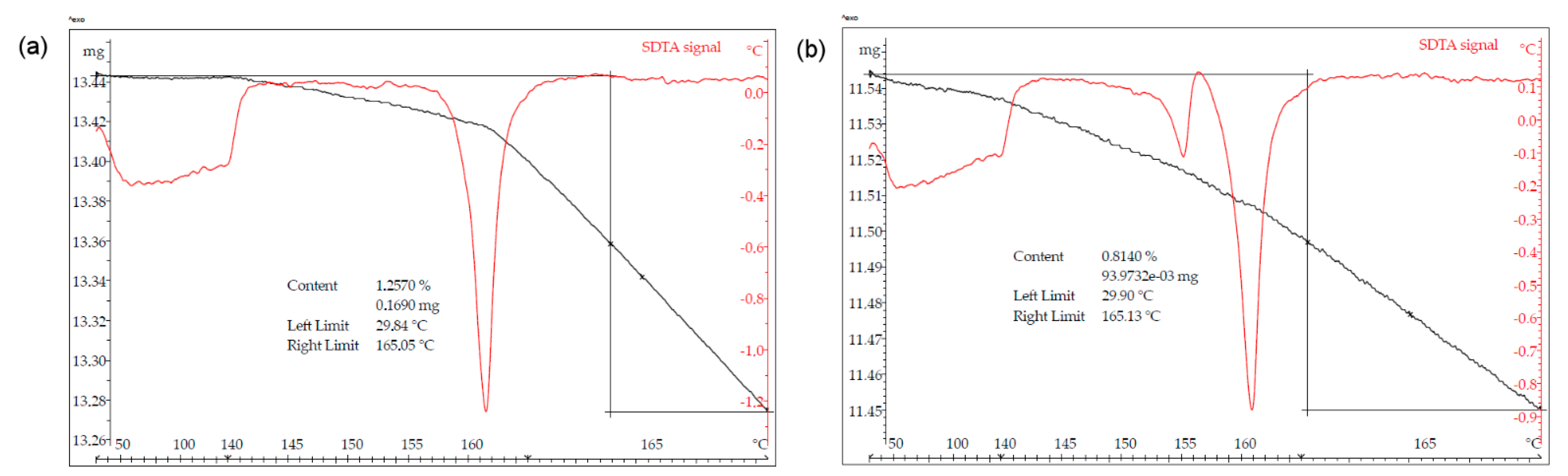
3.6. Stability Studies
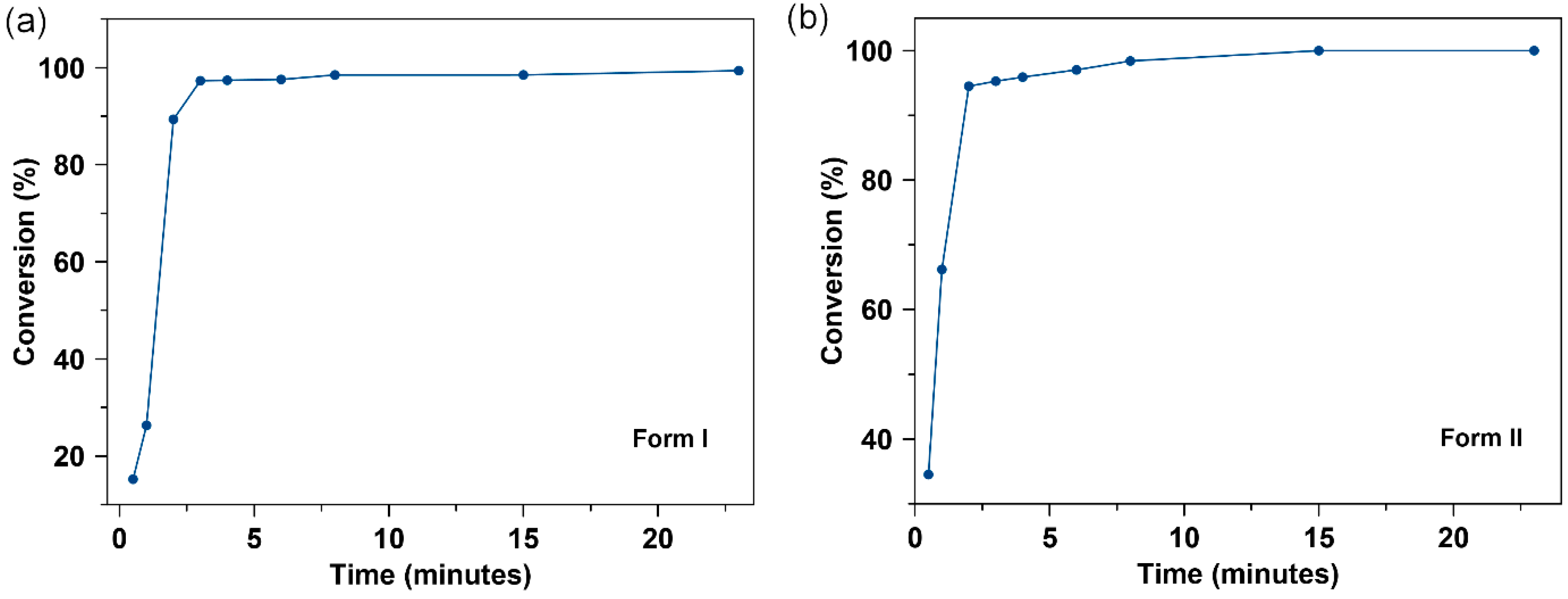
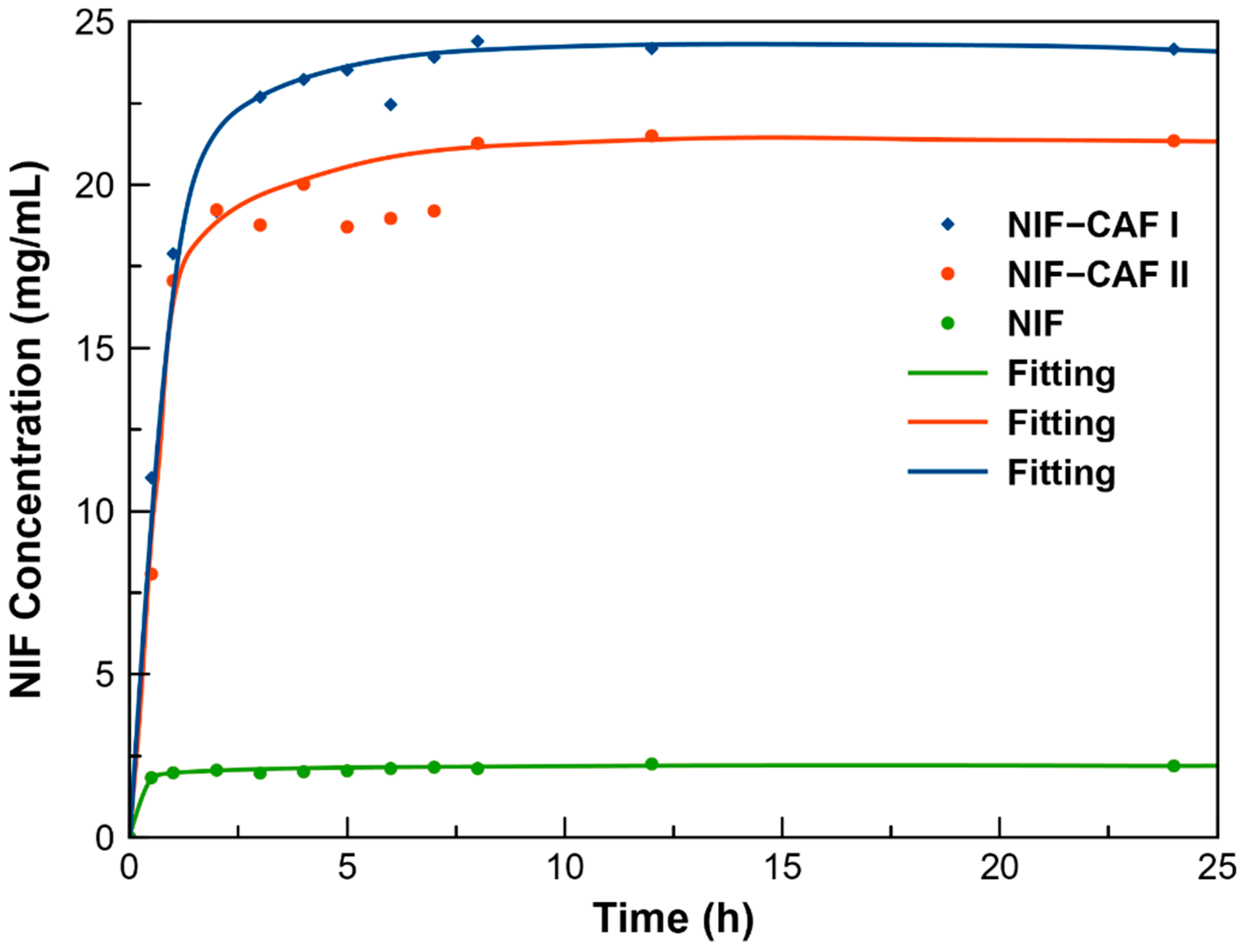
3.7. Powder Dissolution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Center for Biotechnology Information. PubChem Compound Summary for CID 4488, Niflumic Acid. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Niflumic-acid (accessed on 21 October 2021).
- Reynolds, J.E. (Ed.) Martindale the Extra Pharmacopoeia, 31st ed.; Royal Pharmaceutical Society: London, UK, 1996. [Google Scholar]
- Berry, D.J.; Steed, J.W. Pharmaceutical Cocrystals, Salts and Multicomponent Systems; Intermolecular Interactions and Property Based Design. Adv. Drug Deliv. Rev. 2017, 117, 3–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.; Grant, D.J.W. Crystal Structures of Drugs: Advances in Determination, Prediction and Engineering. Nat. Rev. Drug Discov. 2004, 3, 42–57. [Google Scholar] [CrossRef]
- Bolla, G.; Nangia, A. Pharmaceutical Cocrystals: Walking the Talk. Chem. Commun. 2016, 52, 8342–8360. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.L.; Chen, J.M.; Lu, T.B. Pharmaceutical Cocrystallization: An Effective Approach to Modulate the Physicochemical Properties of Solid-State Drugs. CrystEngComm 2018, 20, 5292–5316. [Google Scholar] [CrossRef]
- Karpinski, P.H. Polymorphism of Active Pharmaceutical Ingredients. Chem. Eng. Technol. 2006, 29, 233–237. [Google Scholar] [CrossRef]
- Coquerel, G. Thermodynamics of Polymorphs and Solvates. In Polymorphism in the Pharmaceutical Industry, 2nd ed.; Hilfiker, R., von Raumer, M., Eds.; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2018; pp. 91–132. ISBN 9783527340408. [Google Scholar]
- Aitipamula, S.; Chow, P.S.; Tan, R.B.H. Polymorphism in Cocrystals: A Review and Assessment of Its Significance. CrystEngComm 2014, 16, 3451–3465. [Google Scholar] [CrossRef]
- Sangtani, E.; Jha, K.; Munshi, P.; Gonnade, R. Co-Crystals/Salts of Furosemide: Interesting Case of Colour Co-Crystal Polymorphism. Acta Crystallogr. Sect. A 2017, 73, C724. [Google Scholar] [CrossRef] [Green Version]
- Guerain, M.; Guinet, Y.; Correia, N.T.; Paccou, L.; Danède, F.; Hédoux, A. Polymorphism and Stability of Ibuprofen/Nicotinamide Cocrystal: The Effect of the Crystalline Synthesis Method. Int. J. Pharm. 2020, 584, 119454. [Google Scholar] [CrossRef]
- Aramini, A.; Bianchini, G.; Lillini, S.; Bordignon, S.; Tomassetti, M.; Novelli, R.; Mattioli, S.; Lvova, L.; Paolesse, R.; Chierotti, M.R.; et al. Unexpected Salt/Cocrystal Polymorphism of the Ketoprofen–Lysine System: Discovery of a New Ketoprofen–l-Lysine Salt Polymorph with Different Physicochemical and Pharmacokinetic Properties. Pharmaceuticals 2021, 14, 555. [Google Scholar] [CrossRef]
- Trask, A.V. An Overview of Pharmaceutical Cocrystals as Intellectual Property. Mol. Pharm. 2007, 4, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Stolar, T.; Lukin, S.; Tireli, M.; Sović, I.; Karadeniz, B.; Kereković, I.; Matijašić, G.; Gretić, M.; Katančić, Z.; Dejanović, I.; et al. Control of Pharmaceutical Cocrystal Polymorphism on Various Scales by Mechanochemistry: Transfer from the Laboratory Batch to the Large-Scale Extrusion Processing. ACS Sustain. Chem. Eng. 2019, 7, 7102–7110. [Google Scholar] [CrossRef]
- Sun, C.C.; Hou, H. Improving Mechanical Properties of Caffeine and Methyl Gallate Crystals by Cocrystallization. Cryst. Growth Des. 2008, 8, 1575–1579. [Google Scholar] [CrossRef] [Green Version]
- Verdugo-Escamilla, C.; Alarcón-Payer, C.; Frontera, A.; Acebedo-Martínez, F.J.; Domínguez-Martín, A.; Gómez-Morales, J.; Choquesillo-Lazarte, D. Interconvertible Hydrochlorothiazide–Caffeine Multicomponent Pharmaceutical Materials: A Solvent Issue. Crystals 2020, 10, 1088. [Google Scholar] [CrossRef]
- Perlovich, G.L.; Manin, A.N.; Manin, N.G.; Surov, A.O.; Voronin, A.P. Cocrystalline Form of Niflumic Acid with Isonicotinamide or Caffeine. Russian Patent No. RU2536484C1, 24 December 2013. [Google Scholar]
- Kumar, V.; Thaimattam, R.; Dutta, S.; Munshi, P.; Ramanan, A. Structural Landscape of Multicomponent Solids Based on Sulfa Drugs. CrystEngComm 2017, 19, 2914–2924. [Google Scholar] [CrossRef]
- Bhattacharya, B.; Das, S.; Lal, G.; Soni, S.R.; Ghosh, A.; Reddy, C.M.; Ghosh, S. Screening, Crystal Structures and Solubility Studies of a Series of Multidrug Salt Hydrates and Cocrystals of Fenamic Acids with Trimethoprim and Sulfamethazine. J. Mol. Struct. 2020, 1199, 127028. [Google Scholar] [CrossRef]
- Surov, A.O.; Voronin, A.P.; Vener, M.V.; Churakov, A.V.; Perlovich, G.L. Specific Features of Supramolecular Organisation and Hydrogen Bonding in Proline Cocrystals: A Case Study of Fenamates and Diclofenac. CrystEngComm 2018, 20, 6970–6981. [Google Scholar] [CrossRef]
- Surov, A.O.; Simagina, A.A.; Manin, N.G.; Kuzmina, L.G.; Churakov, A.V.; Perlovich, G.L. Fenamate Cocrystals with 4,4′-Bipyridine: Structural and Thermodynamic Aspects. Cryst. Growth Des. 2015, 15, 228–238. [Google Scholar] [CrossRef]
- Bag, P.P.; Reddy, C.M. Screening and Selective Preparation of Polymorphs by Fast Evaporation Method: A Case Study of Aspirin, Anthranilic Acid, and Niflumic Acid. Cryst. Growth Des. 2012, 12, 2740–2743. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism in Molecular Crystals; Oxford University Press: Oxford, UK, 2007; ISBN 9780199236565. [Google Scholar]
- Coelho, A.A. TOPAS and TOPAS-Academic: An Optimization Program Integrating Computer Algebra and Crystallographic Objects Written in C++ J. Appl. Crystallogr. 2018, 51, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Bruker APEX3. In APEX3 V2019.1; Bruker-AXS: Madison, WI, USA, 2019.
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian16 (Revision A.03); Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Keith, T.A. AIMALL (Version 19.10.12); TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Delori, A.; Friščić, T.; Jones, W. The Role of Mechanochemistry and Supramolecular Design in the Development of Pharmaceutical Materials. CrystEngComm 2012, 14, 2350. [Google Scholar] [CrossRef]
- Etter, M.C.; MacDonald, J.C.; Bernstein, J. Graph-Set Analysis of Hydrogen-Bond Patterns in Organic Crystals. Acta Crystallogr. Sect. B Struct. Sci. 1990, 46, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Etter, M.C. Encoding and Decoding Hydrogen-Bond Patterns of Organic Compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Childs, S.L.; Stahly, G.P.; Park, A. The Salt-Cocrystal Continuum: The Influence of Crystal Structure on Ionization State. Mol. Pharm. 2007, 4, 323–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surov, A.O.; Perlovich, G.L. CCDC 1020379: Experimental Crystal Structure Determination; Cambridge Crystallographic Data Centre: Cambridge, UK, 2016. [Google Scholar] [CrossRef]
- Krishna Murthy, H.M.; Vijayan, M. 2{[3-(Trifluoromethyl)Phenyl]Amino}-3-Pyridinecarboxylic Acid (Niflumic Acid). Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1979, 35, 262–263. [Google Scholar] [CrossRef]
- Heinz, A.; Strachan, C.J.; Gordon, K.C.; Rades, T. Analysis of Solid-State Transformations of Pharmaceutical Compounds Using Vibrational Spectroscopy. J. Pharm. Pharmacol. 2009, 61, 971–988. [Google Scholar] [CrossRef]
- Mukherjee, A.; Tothadi, S.; Chakraborty, S.; Ganguly, S.; Desiraju, G.R. Synthon Identification in Co-Crystals and Polymorphs with IR Spectroscopy. Primary Amides as a Case Study. CrystEngComm 2013, 15, 4640–4654. [Google Scholar] [CrossRef]
- Burger, A.; Ramberger, R. On the Polymorphism of Pharmaceuticals and Other Molecular Crystals. I. Mikrochim. Acta 1979, 72, 259–271. [Google Scholar] [CrossRef]
- Yu, L. Inferring Thermodynamic Stability Relationship of Polymorphs from Melting Data. J. Pharm. Sci. 1995, 84, 966–974. [Google Scholar] [CrossRef]
- Erickson, K.L. Thermal Decomposition Mechanisms Common to Polyurethane, Epoxy, Poly(Diallyl Phthalate), Polycarbonate and Poly(Phenylene Sulfide). J. Therm. Anal. Calorim. 2007, 89, 427–440. [Google Scholar] [CrossRef]
- Cesaro, A.; Starec, G. Thermodynamic Properties of Caffeine Crystal Forms. J. Phys. Chem. 1980, 84, 1345–1346. [Google Scholar] [CrossRef]
- Alsirawan, M.B.; Lai, X.; Prohens, R.; Vangala, V.R.; Pagire, S.K.; Shelley, P.; Bannan, T.J.; Topping, D.O.; Paradkar, A. Solid-State Competitive Destabilization of Caffeine Malonic Acid Cocrystal: Mechanistic and Kinetic Investigations. Cryst. Growth Des. 2020, 20, 7598–7605. [Google Scholar] [CrossRef]
- Kacso, I.; Rus, L.; Pop, M.; Borodi, G.; Bratu, I. Structural Characterization of Ambazone Salt with Niflumic Acid. Spectroscopy 2012, 27, 49–58. [Google Scholar] [CrossRef]
- Docherty, R.; Clydesdale, G.; Roberts, K.J.; Bennema, P. Application of Bravais-Friedel-Donnay-Harker, Attachment Energy and Ising Models to Predicting and Understanding the Morphology of Molecular Crystals. J. Phys. D Appl. Phys. 1991, 24, 89. [Google Scholar] [CrossRef]
- Kitamura, M.; Abe, T.; Kishida, M. Mechanism for the Release of the Industrial Biocide CMI from Clathrate Crystal. Chem. Eng. Res. Des. 2008, 86, 1053–1058. [Google Scholar] [CrossRef]
- Kitamura, M.; Tanaka, T. Crystallization Behavior of Polymorphous Ni-Complex Clathrate in the Presence of 2-Methylnaphthalene. J. Cryst. Growth 1994, 142, 165–170. [Google Scholar] [CrossRef]
- Aitipamula, S.; Cadden, J.; Chow, P.S. Cocrystals of Zonisamide: Physicochemical Characterization and Sustained Release Solid Forms. CrystEngComm 2018, 20, 2923–2931. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | NIF–CAF Form I | NIF–CAF Form II |
|---|---|---|
| Formula | C21H19F3N6O4 | C21H19F3N6O4 |
| Formula weight | 476.42 | 476.42 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21/n |
| a/Å | 8.618(3) | 6.9667(4) |
| b/Å | 23.908(9) | 8.1222(6) |
| c/Å | 10.872(4) | 37.438(3) |
| α/° | 90 | 90 |
| β/° | 104.568(12) | 94.400(4) |
| γ/° | 90 | 90 |
| V/Å3 | 2168.1(14) | 2112.2(2) |
| Z | 4 | 4 |
| Dc/g cm−3 | 1.460 | 1.498 |
| µ/mm−1 | 1.042 | 1.070 |
| F(000) | 984 | 984 |
| Reflections collected | 15737 | 19086 |
| Unique reflections | 3763 | 3710 |
| Rint | 0.0796 | 0.0435 |
| Data/restraints/parameters | 3763/93/340 | 3710/81/340 |
| Goodness-of-fit (F2) | 0.974 | 1.111 |
| R1 (I > 2σ(I)) | 0.0637 | 0.0467 |
| wR2 (I > 2σ(I)) | 0.1633 | 0.1282 |
| CCDC | 2116472 | 2116473 |
| Compound | ν(C-H) Aromatic | νas(CH3) | ν(C=O) Carboxyl | ν(C=O) Keto | δ(N-H) | δ(O-H) In-Plane | ν(C-O) Carboxyl | δ(O-H) Out-Of-Plane |
|---|---|---|---|---|---|---|---|---|
| NIF | 1665 | 1529 | 1448 | 1241 | 935 | |||
| CAF | 3118 | 2957 | 1701 1655 | |||||
| NIF–CAF Form I | 3134 | 2955 | 1662 | 1710 | 1530 | 1461 | 1244 | 947 |
| NIF–CAF Form II | 3131 | 2955 | 1662 | 1709 | 1533 | 1446 | 1242 | 947 |
| Polymorph | Melting Point (K) | Enthalpy of Fusion (J/g) |
|---|---|---|
| I | 430.83 | 145.70 |
| II | 425.32 | 161.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acebedo-Martínez, F.J.; Alarcón-Payer, C.; Frontera, A.; Barbas, R.; Prohens, R.; Di Crisci, M.; Domínguez-Martín, A.; Gómez-Morales, J.; Choquesillo-Lazarte, D. Novel Polymorphic Cocrystals of the Non-Steroidal Anti-Inflammatory Drug Niflumic Acid: Expanding the Pharmaceutical Landscape. Pharmaceutics 2021, 13, 2140. https://doi.org/10.3390/pharmaceutics13122140
Acebedo-Martínez FJ, Alarcón-Payer C, Frontera A, Barbas R, Prohens R, Di Crisci M, Domínguez-Martín A, Gómez-Morales J, Choquesillo-Lazarte D. Novel Polymorphic Cocrystals of the Non-Steroidal Anti-Inflammatory Drug Niflumic Acid: Expanding the Pharmaceutical Landscape. Pharmaceutics. 2021; 13(12):2140. https://doi.org/10.3390/pharmaceutics13122140
Chicago/Turabian StyleAcebedo-Martínez, Francisco Javier, Carolina Alarcón-Payer, Antonio Frontera, Rafael Barbas, Rafel Prohens, Milena Di Crisci, Alicia Domínguez-Martín, Jaime Gómez-Morales, and Duane Choquesillo-Lazarte. 2021. "Novel Polymorphic Cocrystals of the Non-Steroidal Anti-Inflammatory Drug Niflumic Acid: Expanding the Pharmaceutical Landscape" Pharmaceutics 13, no. 12: 2140. https://doi.org/10.3390/pharmaceutics13122140
APA StyleAcebedo-Martínez, F. J., Alarcón-Payer, C., Frontera, A., Barbas, R., Prohens, R., Di Crisci, M., Domínguez-Martín, A., Gómez-Morales, J., & Choquesillo-Lazarte, D. (2021). Novel Polymorphic Cocrystals of the Non-Steroidal Anti-Inflammatory Drug Niflumic Acid: Expanding the Pharmaceutical Landscape. Pharmaceutics, 13(12), 2140. https://doi.org/10.3390/pharmaceutics13122140