
Giving Oncolytic Viruses a Free Ride: Carrier Cells for Oncolytic Virotherapy



Abstract
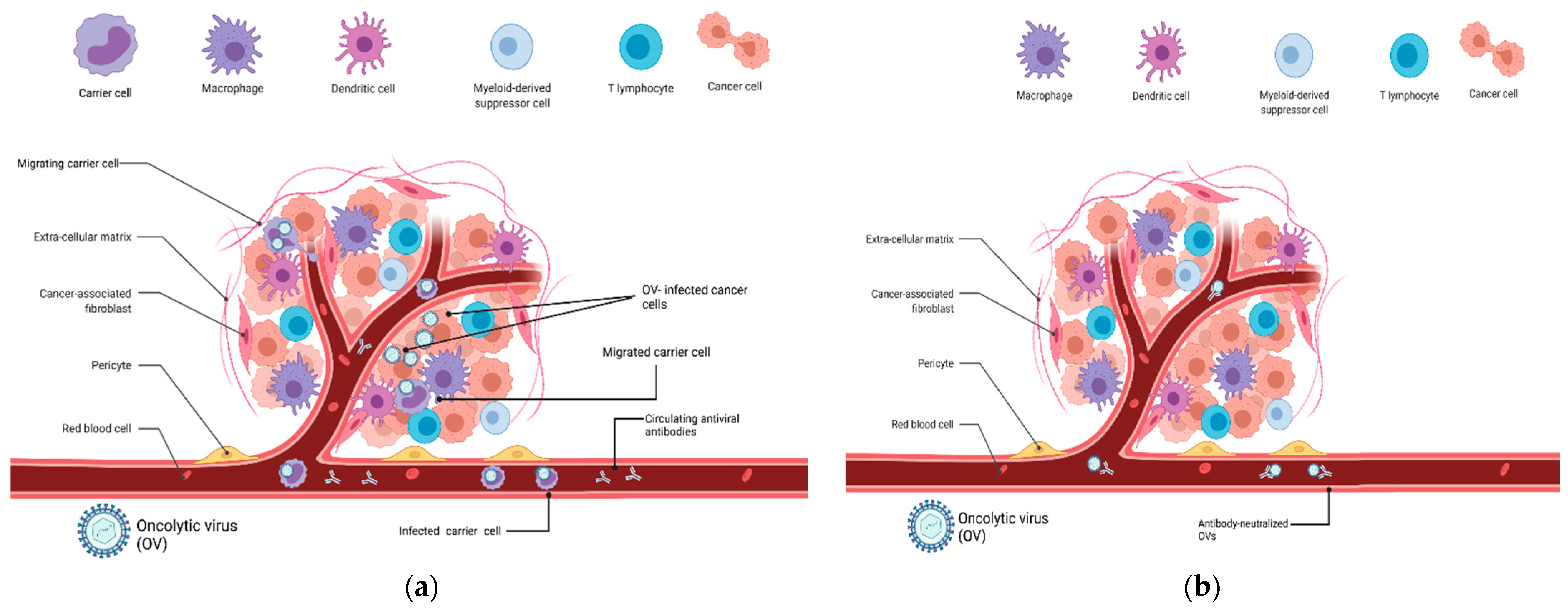
:1. Introduction
2. Carrier Cells for Oncolytic Virus Delivery
2.1. T Lymphocytes
2.2. Myeloid Cells
2.3. Neural Stem Cells
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends—An Update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maver, P.; Poljak, M. Primary HPV-Based Cervical Cancer Screening in Europe: Implementation Status, Challenges, and Future Plans. Clin. Microbiol. Infect. 2020, 26, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Nattinger, A.B.; Mitchell, J.L. Breast Cancer Screening and Prevention. Ann. Intern. Med. 2016, 164, ITC81–ITC94. [Google Scholar] [CrossRef]
- Issa, I.A.; Noureddine, M. Colorectal Cancer Screening: An Updated Review of the Available Options. World J. Gastroenterol. 2017, 23, 5086–5096. [Google Scholar] [CrossRef]
- Lawrence, W.; Lopez, M.J. Radical Surgery for Cancer: A Historical Perspective. Surg. Oncol. Clin. N. Am. 2005, 14, 441–446. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Mitchell, T.C.; Feld, E. Immunotherapy in Melanoma. Immunotherapy 2018, 10, 987–998. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Previously Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbour, K.C.; Riely, G.J. Systemic Therapy for Locally Advanced and Metastatic Non-Small Cell Lung Cancer: A Review. J. Am. Med. Assoc. 2019, 322, 764–774. [Google Scholar] [CrossRef]
- Xu, W.; Atkins, M.B.; McDermott, D.F. Checkpoint Inhibitor Immunotherapy in Kidney Cancer. Nat. Rev. Urol. 2020, 17, 137–150. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.-Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.-C.; Vlahovic, G.; et al. Durvalumab with or without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T Cell Exclusion, Immune Privilege, and the Tumor Microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Reale, A.; Vitiello, A.; Conciatori, V.; Parolin, C.; Calistri, A.; Palù, G. Perspectives on Immunotherapy via Oncolytic Viruses. Infect. Agents Cancer 2019, 14, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, T.; Karube, H.; Aruga, A. A Comparative Safety Profile Assessment of Oncolytic Virus Therapy Based on Clinical Trials. Ther. Innov. Regul. Sci. 2018, 52, 430–437. [Google Scholar] [CrossRef]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene Laherparepvec: First in Class Oncolytic Virotherapy. Hum. Vaccines Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Haines, B.B.; Denslow, A.; Grzesik, P.; Lee, J.S.; Farkaly, T.; Hewett, J.; Wambua, D.; Kong, L.; Behera, P.; Jacques, J.; et al. ONCR-177, an Oncolytic HSV-1 Designed to Potently Activate Systemic Antitumor Immunity. Cancer Immunol. Res. 2021, 9, 291–308. [Google Scholar] [CrossRef] [PubMed]
- Pelin, A.; Boulton, S.; Tamming, L.A.; Bell, J.C.; Singaravelu, R. Engineering Vaccinia Virus as an Immunotherapeutic Battleship to Overcome Tumor Heterogeneity. Expert Opin. Biol. Ther. 2020, 20, 1083–1097. [Google Scholar] [CrossRef] [PubMed]
- Niemann, J.; Kühnel, F. Oncolytic Viruses: Adenoviruses. Virus Genes 2017, 53, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Durham, N.M.; Mulgrew, K.; McGlinchey, K.; Monks, N.R.; Ji, H.; Herbst, R.; Suzich, J.A.; Hammond, S.A.; Kelly, E.J. Oncolytic VSV Primes Differential Responses to Immuno-Oncology Therapy. Mol. Ther. 2017, 25, 1917–1932. [Google Scholar] [CrossRef] [PubMed]
- McGray, A.J.R.; Huang, R.Y.; Battaglia, S.; Eppolito, C.; Miliotto, A.; Stephenson, K.B.; Lugade, A.A.; Webster, G.; Lichty, B.D.; Seshadri, M.; et al. Oncolytic Maraba Virus Armed with Tumor Antigen Boosts Vaccine Priming and Reveals Diverse Therapeutic Response Patterns When Combined with Checkpoint Blockade in Ovarian Cancer. J. Immunother. Cancer 2019, 7, 189. [Google Scholar] [CrossRef] [Green Version]
- Leber, M.F.; Neault, S.; Jirovec, E.; Barkley, R.; Said, A.; Bell, J.C.; Ungerechts, G. Engineering and Combining Oncolytic Measles Virus for Cancer Therapy. Cytokine Growth Factor Rev. 2020, 56, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Kemp, V.; van den Wollenberg, D.J.M.; Camps, M.G.M.; van Hall, T.; Kinderman, P.; Pronk-van Montfoort, N.; Hoeben, R.C. Arming Oncolytic Reovirus with GM-CSF Gene to Enhance Immunity. Cancer Gene Ther. 2019, 26, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Penghui, Y.; Fang, S.; Ruilan, W.; Guanglin, L.; Hongjing, G.; Yueqiang, D.; Zhongpeng, Z.; Xiaolan, Y.; Zhaohai, W.; Shaogeng, Z.; et al. Oncolytic Activity of a Novel Influenza a Virus Carrying Granulocyte-Macrophage Colony-Stimulating Factor in Hepatocellular Carcinoma. Hum. Gene Ther. 2019, 30, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic Virus Therapy: A New Era of Cancer Treatment at Dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef]
- Ferguson, M.S.; Lemoine, N.R.; Wang, Y. Systemic Delivery of Oncolytic Viruses: Hopes and Hurdles. Adv. Virol. 2012, 2012, 805629. [Google Scholar] [CrossRef]
- Kulu, Y.; Dorfman, J.D.; Kuruppu, D.; Fuchs, B.C.; Goodwin, J.M.; Fujii, T.; Kuroda, T.; Lanuti, M.; Tanabe, K.K. Comparison of Intravenous versus Intraperitoneal Administration of Oncolytic Herpes Simplex Virus 1 for Peritoneal Carcinomatosis in Mice. Cancer Gene Ther. 2009, 16, 291–297. [Google Scholar] [CrossRef] [Green Version]
- Tsai, V.; Johnson, D.E.; Rahman, A.; Wen, S.F.; LaFace, D.; Philopena, J.; Nery, J.; Zepeda, M.; Maneval, D.C.; Demers, G.W.; et al. Impact of Human Neutralizing Antibodies on Antitumor Efficacy of an Oncolytic Adenovirus in a Murine Model. Clin. Cancer Res. 2004, 10, 7199–7206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alemany, R.; Suzuki, K.; Curiel, D.T. Blood Clearance Rates of Adenovirus Type 5 in Mice. J. Gen. Virol. 2000, 81, 2605–2609. [Google Scholar] [CrossRef]
- Luo, Y.; Lin, C.; Ren, W.; Ju, F.; Xu, Z.; Liu, H.; Yu, Z.; Chen, J.; Zhang, J.; Liu, P.; et al. Intravenous Injections of a Rationally Selected Oncolytic Herpes Virus as a Potent Virotherapy for Hepatocellular Carcinoma. Mol. Ther. Oncolytics 2019, 15, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Bradley, H.; Markowitz, L.E.; Gibson, T.; McQuillan, G.M. Seroprevalence of Herpes Simplex Virus Types 1 and 2—United States, 1999–2010. J. Infect. Dis. 2014, 209, 325–333. [Google Scholar] [CrossRef]
- Russell, S.J.; Barber, G.N. Oncolytic Viruses as Antigen-Agnostic Cancer Vaccines. Cancer Cell 2018, 33, 599–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andtbacka, R.H.I.; Ross, M.; Puzanov, I.; Milhem, M.; Collichio, F.; Delman, K.A.; Amatruda, T.; Zager, J.S.; Cranmer, L.; Hsueh, E.; et al. Patterns of Clinical Response with Talimogene Laherparepvec (T-VEC) in Patients with Melanoma Treated in the OPTiM Phase III Clinical Trial. Ann. Surg. Oncol. 2016, 23, 4169–4177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadryś, A.; Sochanik, A.; McFadden, G.; Jazowiecka-Rakus, J. Mesenchymal Stem Cells as Carriers for Systemic Delivery of Oncolytic Viruses. Eur. J. Pharmacol. 2020, 874, 172991. [Google Scholar] [CrossRef] [PubMed]
- Chastkofsky, M.I.; Pituch, K.C.; Katagi, H.; Zannikou, M.; Ilut, L.; Xiao, T.; Han, Y.; Sonabend, A.M.; Curiel, D.T.; Bonner, E.R.; et al. Mesenchymal Stem Cells Successfully Deliver Oncolytic Virotherapy to Diffuse Intrinsic Pontine Glioma. Clin. Cancer Res. 2020, 27, 1766–1777. [Google Scholar] [CrossRef]
- Leoni, V.; Gatta, V.; Palladini, A.; Nicoletti, G.; Ranieri, D.; Dall’Ora, M.; Grosso, V.; Rossi, M.; Alviano, F.; Bonsi, L.; et al. Systemic Delivery of HER2-Retargeted Oncolytic-HSV by Mesenchymal Stromal Cells Protects from Lung and Brain Metastases. Oncotarget 2015, 6, 34774–34787. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic Viruses: A New Class of Immunotherapy Drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Hammad, M.; Cornejo, Y.R.; Batalla-Covello, J.; Majid, A.A.; Burke, C.; Liu, Z.; Yuan, Y.C.; Li, M.; Dellinger, T.H.; Lu, J.; et al. Neural Stem Cells Improve the Delivery of Oncolytic Chimeric Orthopoxvirus in a Metastatic Ovarian Cancer Model. Mol. Ther. Oncolytics 2020, 18, 326–334. [Google Scholar] [CrossRef]
- Bunuales, M.; Garcia-Aragoncillo, E.; Casado, R.; Quetglas, J.I.; Hervas-Stubbs, S.; Bortolanza, S.; Benavides-Vallve, C.; Ortiz-De-Solorzano, C.; Prieto, J.; Hernandez-Alcoceba, R. Evaluation of Monocytes as Carriers for Armed Oncolytic Adenoviruses in Murine and Syrian Hamster Models of Cancer. Hum. Gene Ther. 2012, 23, 1258–1268. [Google Scholar] [CrossRef] [Green Version]
- Ong, H.; Hasegawa, K.; Dietz, A.; Russell, S.; Peng, K.-W. Evaluation of T Cells as Carriers for Systemic Measles Virotherapy in the Presence of Antiviral Antibodies. Gene Ther. 2007, 14, 324–333. [Google Scholar] [CrossRef]
- Ilett, E.; Prestwich, R.; Kottke, T.; Errington, F.; Thompson, J.M.; Harrington, K.J.K.; Pandha, H.A.; Coffey, M.; Selby, P.J.; Vile, R.G.; et al. Dendritic Cells and T Cells Deliver Oncolytic Reovirus for Tumour Killing despite Pre-Existing Anti-Viral Immunity. Gene Ther. 2009, 16, 689–699. [Google Scholar] [CrossRef] [Green Version]
- Pfirschke, C.; Schirrmacher, V. Cross-infection of tumor cells by contact with T lymphocytes loaded with Newcastle disease virus. Int. J. Oncol. 2009, 34, 951–962. [Google Scholar] [CrossRef] [Green Version]
- Qiao, J.; Kottke, T.; Willmon, C.; Galivo, F.; Wongthida, P.; Diaz, R.M.; Thompson, J.; Ryno, P.; Barber, G.N.; Chester, J.; et al. Purging Metastases in Lymphoid Organs Using a Combination of Antigen-Nonspecific Adoptive T Cell Therapy, Oncolytic Virotherapy and Immunotherapy. Nat. Med. 2008, 14, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Wang, H.; Kottke, T.; Diaz, R.M.; Willmon, C.; Hudacek, A.; Thompson, J.; Parato, K.; Bell, J.; Naik, J.; et al. Loading of Oncolytic Vesicular Stomatitis Virus onto Antigen-Specific T Cells Enhances the Efficacy of Adoptive T-Cell Therapy of Tumors. Gene Ther. 2008, 15, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.; Qiao, J.; Kottke, T.; Diaz, R.M.; Ahmed, A.; Sanchez-Perez, L.; Brunn, G.; Thompson, J.; Chester, J.; Vile, R.G. Tumor-Targeted, Systemic Delivery of Therapeutic Viral Vectors Using Hitchhiking on Antigen-Specific T Cells. Nat. Med. 2005, 11, 1073–1081. [Google Scholar] [CrossRef]
- Melzer, M.K.; Zeitlinger, L.; Mall, S.; Steiger, K.; Schmid, R.M.; Ebert, O.; Krackhardt, A.; Altomonte, J. Enhanced Safety and Efficacy of Oncolytic VSV Therapy by Combination with T Cell Receptor Transgenic T Cells as Carriers. Mol. Ther. Oncolytics 2019, 12, 26–40. [Google Scholar] [CrossRef] [Green Version]
- VanSeggelen, H.; Tantalo, D.; Afsahi, A.-M.T. Chimeric Antigen Receptor-Engineered T Cells as Oncolytic Virus Carriers; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Santos, J.; Heiniö, C.; Quixabeira, D.; Zafar, S.; Cells, J.C. Systemic Delivery of Oncolytic Adenovirus to Tumors Using Tumor-Infiltrating Lymphocytes as Carriers. Cells 2021, 10, 978. [Google Scholar] [CrossRef]
- Yotnda, P.; Savoldo, B.; Charlet-Berguerand, N.; Blood, C.R. Targeted Delivery of Adenoviral Vectors by Cytotoxic T Cells. Blood 2004, 104, 2272–2280. [Google Scholar] [CrossRef] [PubMed]
- Lasner, T.M.; Tal-Singer, R.; Kesari, S.; Lee, V.M.-Y.; Trojanowski, J.Q.; Fraser, N.W. Toxicity and Neuronal Infection of a HSV-1 ICP34.5 Mutant in Nude Mice. J. Neurovirol. 1998, 4, 100–105. [Google Scholar] [CrossRef]
- Kanzaki, A.; Kasuya, H.; Yamamura, K.; Sahin, T.T.; Nomura, N.; Shikano, T.; Shirota, T.; Tan, G.; Fukuda, S.; Misawa, M.; et al. Antitumor efficacy of oncolytic herpes simplex virus adsorbed onto antigen-specific lymphocytes. Cancer Gene Ther 2012, 19, 292–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellone, M.; Calcinotto, A. Ways to Enhance Lymphocyte Trafficking into Tumors and Fitness of Tumor Infiltrating Lymphocytes. Front. Oncol. 2013, 3, 231. [Google Scholar] [CrossRef] [Green Version]
- Calcinotto, A.; Grioni, M.; Jachetti, E.; Curnis, F.; Mondino, A.; Parmiani, G.; Corti, A.; Bellone, M. Targeting TNF-α to Neoangiogenic Vessels Enhances Lymphocyte Infiltration in Tumors and Increases the Therapeutic Potential of Immunotherapy. J. Immunol. 2012, 188, 2687–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the Tumor Vasculature to Enhance T Cell Activity; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Mantovani, A.; Marchesi, F.; Jaillon, S.; Garlanda, C.; Allavena, P. Tumor-Associated Myeloid Cells: Diversity and Therapeutic Targeting. Cell. Mol. Immunol. 2021, 18, 566–578. [Google Scholar] [CrossRef]
- Tan, Y.; Wang, M.; Zhang, Y.; Ge, S.; Zhong, F.; Xia, G.; Sun, C. Tumor-Associated Macrophages: A Potential Target for Cancer Therapy. Front. Oncol. 2021, 11, 3517. [Google Scholar] [CrossRef]
- Cerezo-Wallis, D.; Ballesteros, I. Neutrophils in Cancer, a Love–Hate Affair. FEBS J. 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vanhaver, C.; van der Bruggen, P.; Bruger, A. MDSC in Mice and Men: Mechanisms of Immunosuppression in Cancer. J. Clin. Med. 2021, 10, 2872. [Google Scholar] [CrossRef]
- Miyoshi, I.; Hiraki, S.; Kubonishi, I.; Matsuda, Y.; Nakayama, T.; Kishimoto, H.; Masuji, H.; Kimura, I. Establishment and Characterization of Two Hamster Macrophage Cell Lines. Cancer Lett. 1978, 4, 253–257. [Google Scholar] [CrossRef]
- Peng, K.W.; Dogan, A.; Vrana, J.; Liu, C.; Ong, H.T.; Kumar, S.; Dispenzieri, A.; Dietz, A.B.; Russell, S.J. Tumor-Associated Macrophages Infiltrate Plasmacytomas and Can Serve as Cell Carriers for Oncolytic Measles Virotherapy of Disseminated Myeloma. Am. J. Hematol. 2009, 84, 401–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilett, E.J. Delivery of oncolytic reovirus by cell carriers. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2020; Volume 2058, pp. 229–236. [Google Scholar]
- Eisenstein, S.; Coakley, B.A.; Briley-Saebo, K.; Ma, G.; Chen, H.M.; Meseck, M.; Ward, S.; Divino, C.; Woo, S.; Chen, S.H.; et al. Myeloid-Derived Suppressor Cells as a Vehicle for Tumor-Specific Oncolytic Viral Therapy. Cancer Res. 2013, 73, 5003–5015. [Google Scholar] [CrossRef] [Green Version]
- Berkeley, R.A.; Steele, L.P.; Mulder, A.A.; van den Wollenberg, D.J.M.; Kottke, T.J.; Thompson, J.; Coffey, M.; Hoeben, R.C.; Vile, R.G.; Melcher, A.; et al. Antibody-Neutralized Reovirus Is Effective in Oncolytic Virotherapy. Cancer Immunol. Res. 2018, 6, 1161–1173. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic Virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morshed, R.A.; Gutova, M.; Juliano, J.; Barish, M.E.; Hawkins-Daarud, A.; Oganesyan, D.; Vazgen, K.; Yang, T.; Annala, A.; Ahmed, A.U.; et al. Analysis of Glioblastoma Tumor Coverage by Oncolytic Virus-Loaded Neural Stem Cells Using MRI-Based Tracking and Histological Reconstruction. Cancer Gene Ther. 2015, 22, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Trial | Target Disease | Oncolytic Virus | Results |
|---|---|---|---|
| EudraCT Number: 2008-000364-16 | Pediatric solid tumors | ICOVIR5 (AdV) CELYVIR | Trial ended prematurely |
| NCT 02068794 | Ovarian cancer | Measles virus | Recruiting |
| NCT 01844661 | Miscellaneous metastatic tumors | ICOVIR5 CELYVIR | Completed in 2016—results not available |
| EudraCT Number: 2019-001154-26 | Extracranial solid tumors | ICOVIR5 AloCELYVIR | Ongoing |
| NCT03896568 | Recurrent high-grade glioma | AdV, DNX-2401 | Recruiting |
| NCT05047276 (phase I/II) | Metastatic Uveal Melanoma | ICOVIR5 AloCELYVIR | Trial not yet recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reale, A.; Calistri, A.; Altomonte, J. Giving Oncolytic Viruses a Free Ride: Carrier Cells for Oncolytic Virotherapy. Pharmaceutics 2021, 13, 2192. https://doi.org/10.3390/pharmaceutics13122192
Reale A, Calistri A, Altomonte J. Giving Oncolytic Viruses a Free Ride: Carrier Cells for Oncolytic Virotherapy. Pharmaceutics. 2021; 13(12):2192. https://doi.org/10.3390/pharmaceutics13122192
Chicago/Turabian StyleReale, Alberto, Arianna Calistri, and Jennifer Altomonte. 2021. "Giving Oncolytic Viruses a Free Ride: Carrier Cells for Oncolytic Virotherapy" Pharmaceutics 13, no. 12: 2192. https://doi.org/10.3390/pharmaceutics13122192
APA StyleReale, A., Calistri, A., & Altomonte, J. (2021). Giving Oncolytic Viruses a Free Ride: Carrier Cells for Oncolytic Virotherapy. Pharmaceutics, 13(12), 2192. https://doi.org/10.3390/pharmaceutics13122192