
Quality by Design Applied Development of Immediate-Release Rabeprazole Sodium Dry-Coated Tablet


Abstract
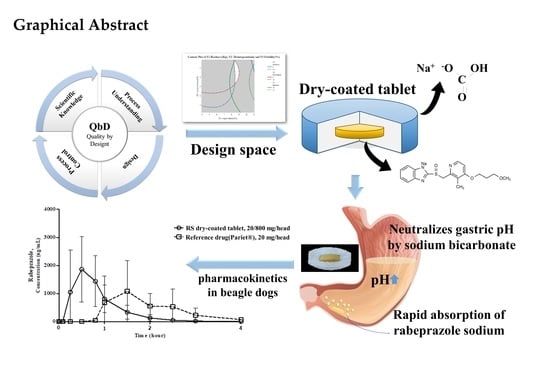
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Physicochemical Properties of APIs
2.2.1. Solubility Studies for Rabeprazole Sodium and Sodium Bicarbonate
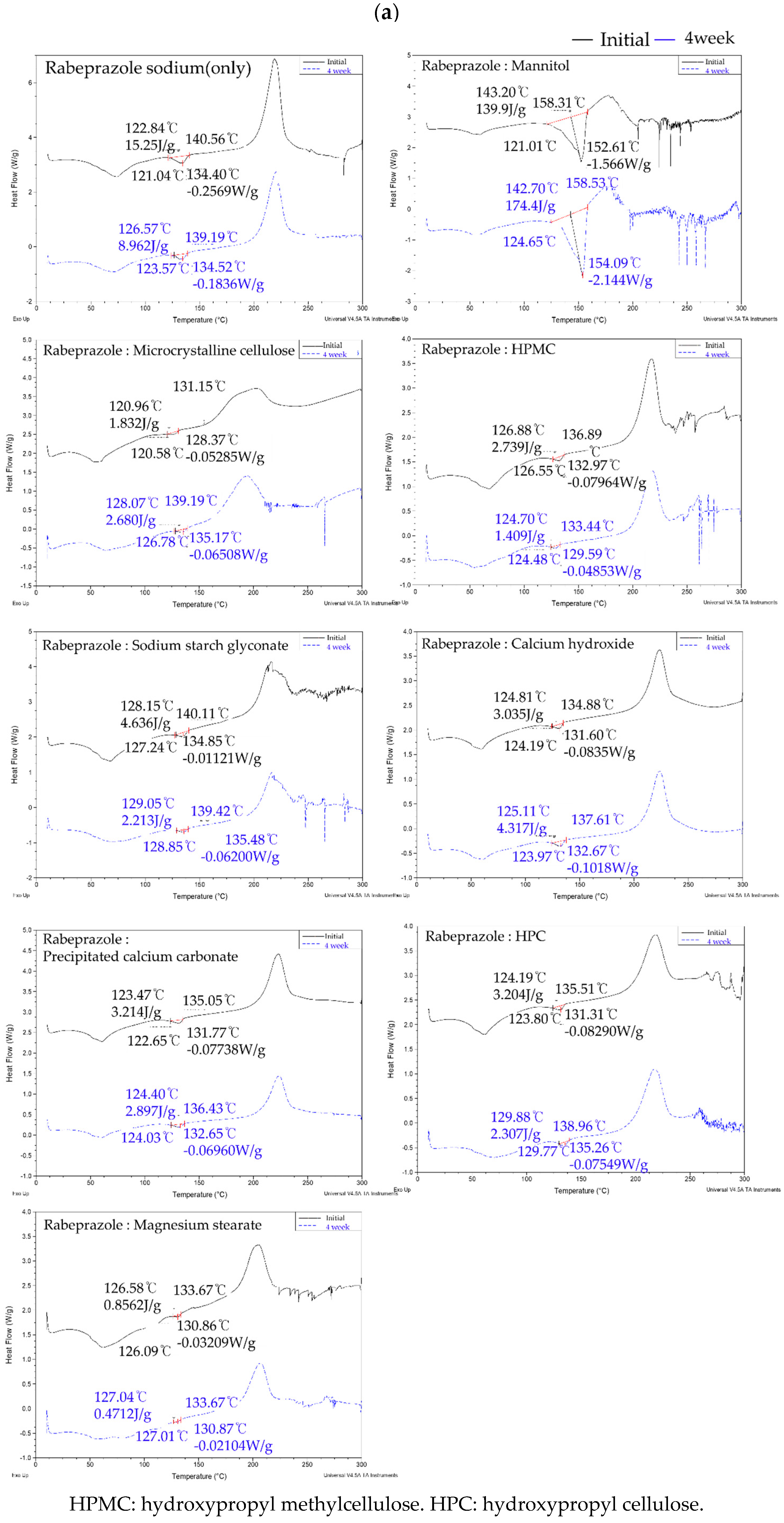
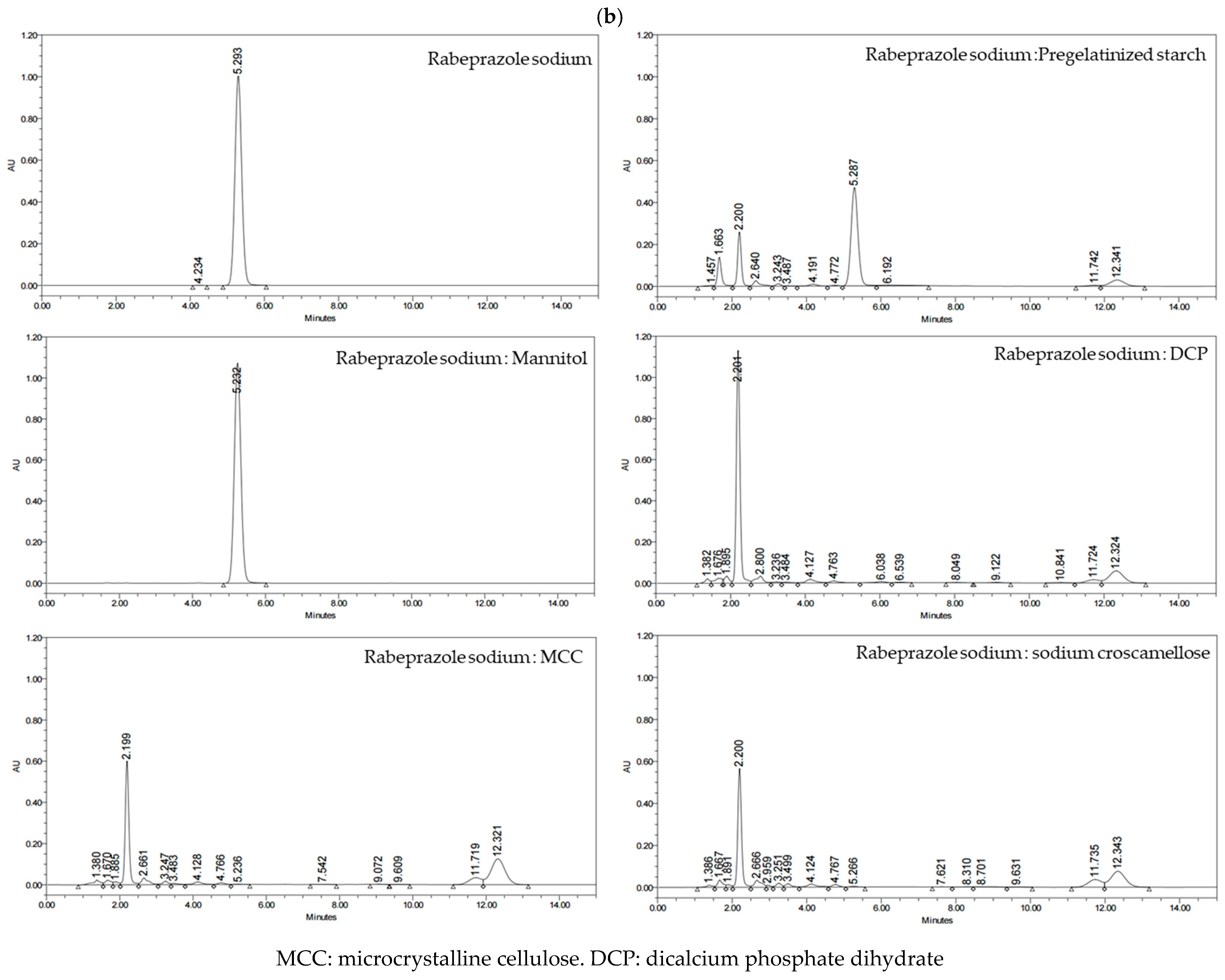
2.2.2. Compatibility Studies for the Selection of APIs and Excipients
2.3. Analysis of Acid-Neutralizing Capacity Based Sodium Bicarbonate Dose
2.4. Quality Target Product Profile (QTPP), Critical Quality Attributes (CQAs), and Risk Assessment of CMAs and CPPs (Preliminary Hazard Analysis (PHA) and Failure Mode Effect Analysis (FMEA))
2.5. Formulation Studies on RS Dry-Coated Tablets
2.6. In Vitro Dissolution Studies
2.7. Stability Studies
2.8. Analytical Methods
2.8.1. HPLC
2.8.2. Ion Chromatography
2.9. Pharmacokinetic Studies
2.10. Statistical Analysis
3. Results and Discussion
3.1. Physicochemical Properties of APIs
3.1.1. Solubility Studies for Rabeprazole Sodium and Sodium Bicarbonate
3.1.2. Compatibility Studies for the Selection of APIs and Excipients
3.2. Studies for Acid-Neutralizing Capacity by Sodium Bicarbonate Dose
3.3. QTPP, CQAs, and RA of CMAs and CPPs (PHA and FMEA)
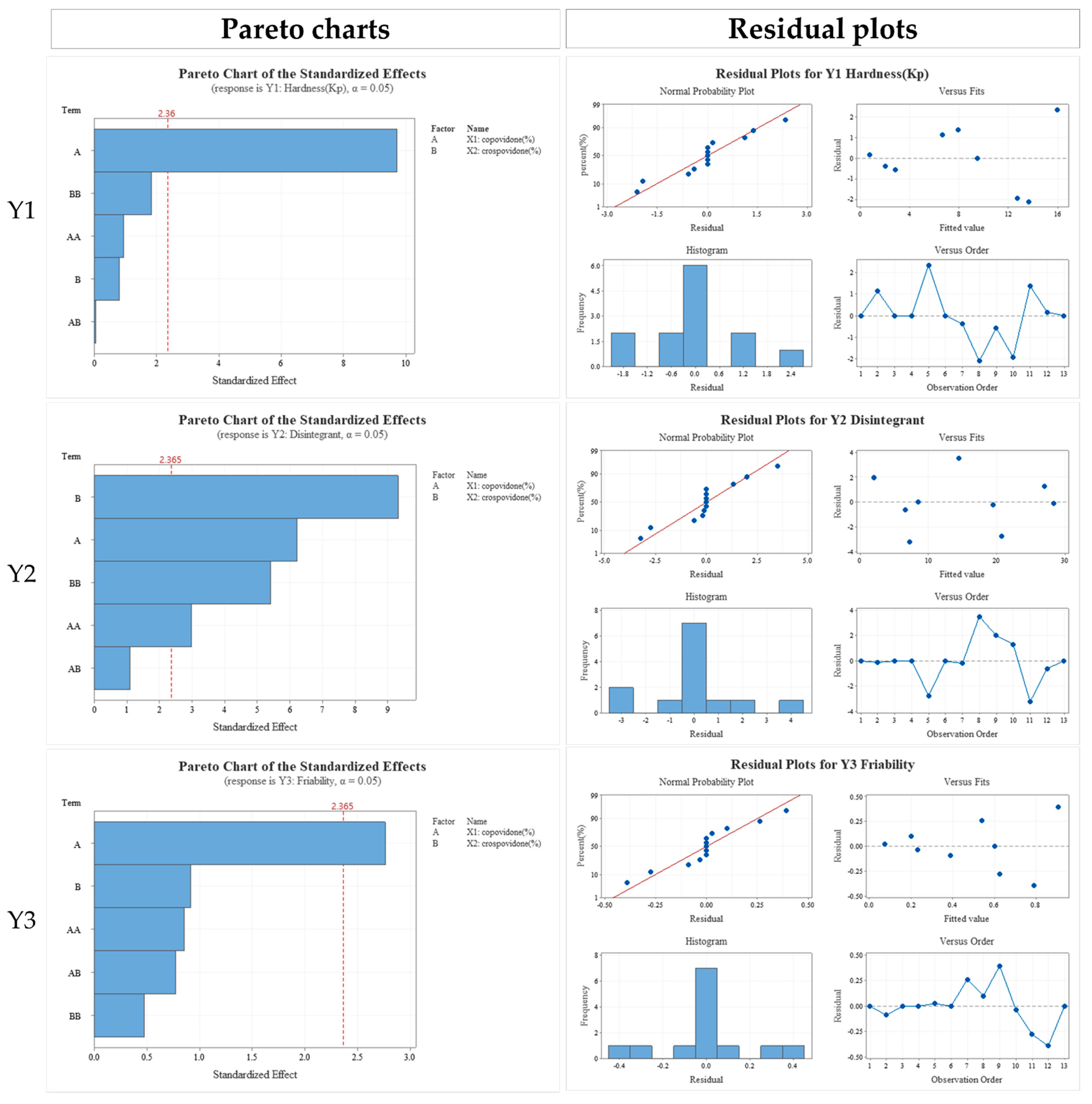
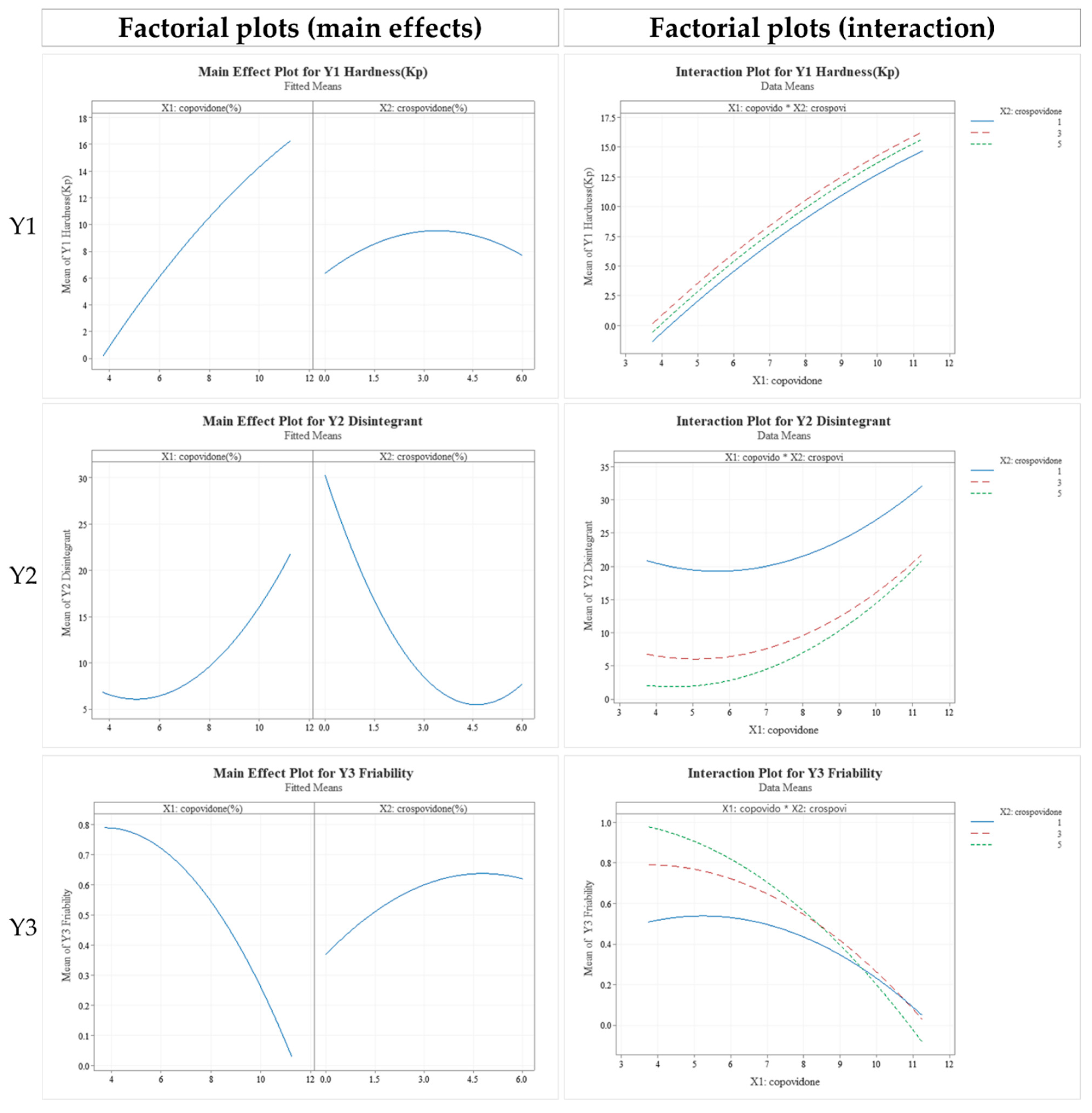
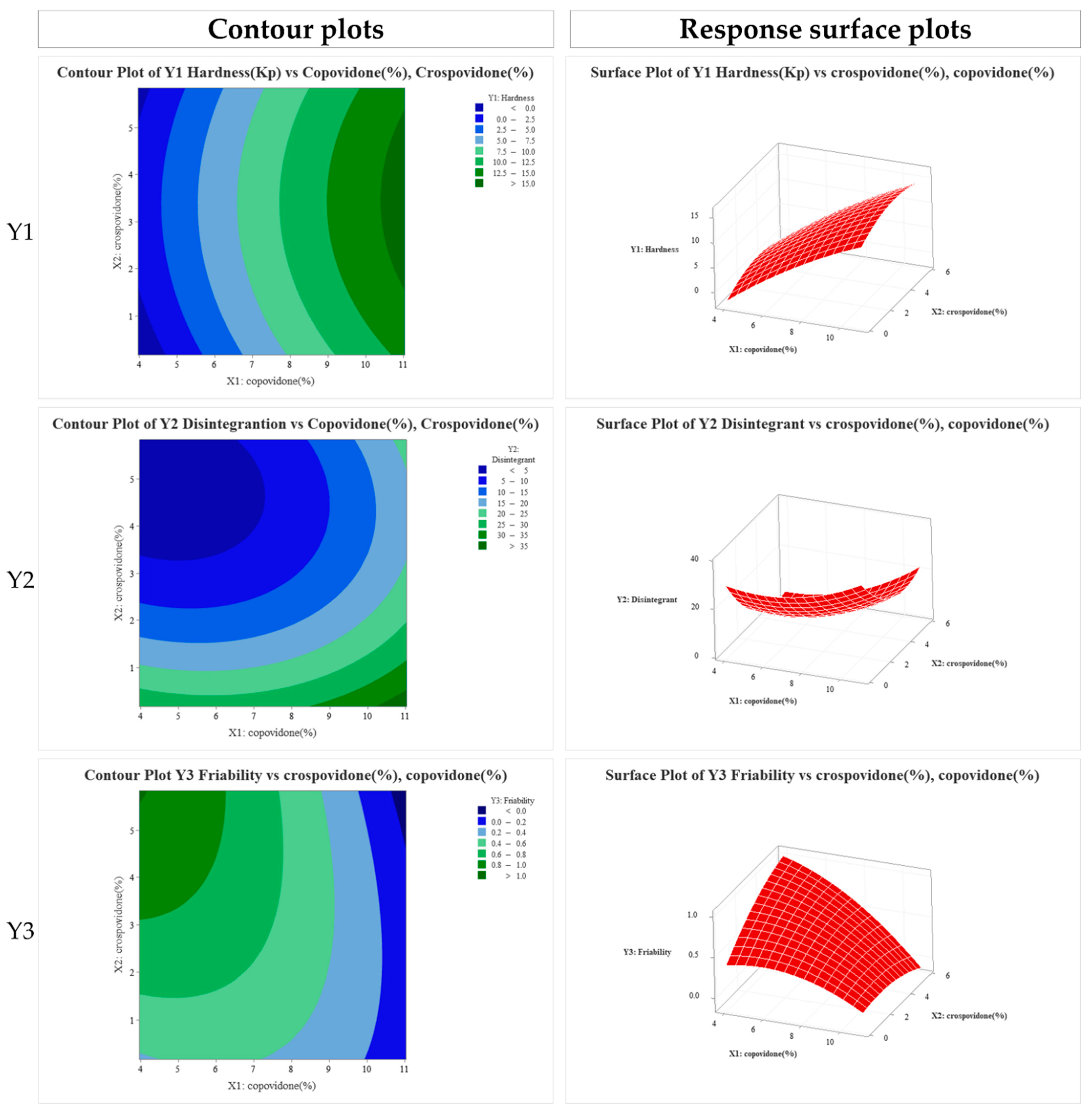
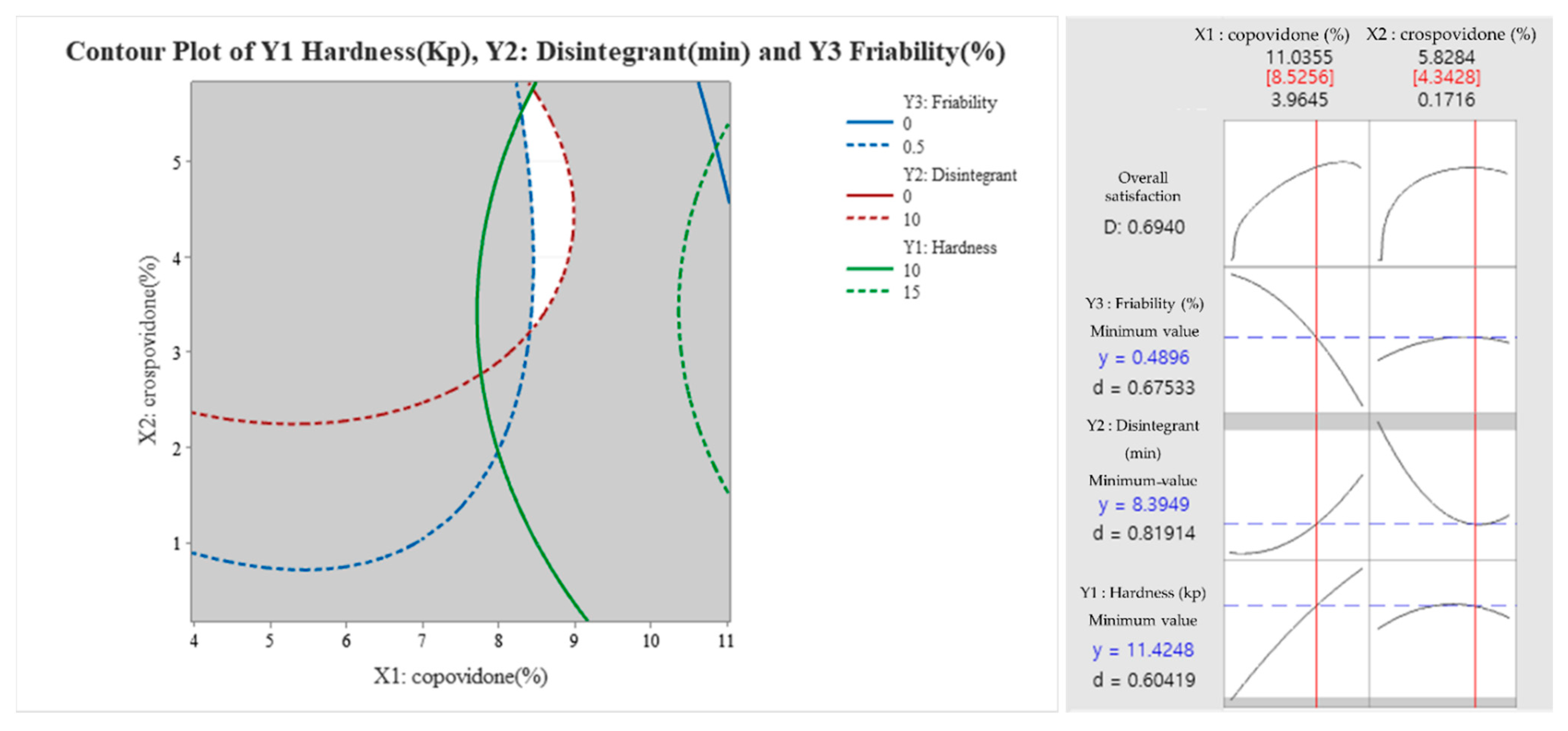
3.4. Formulation Studies
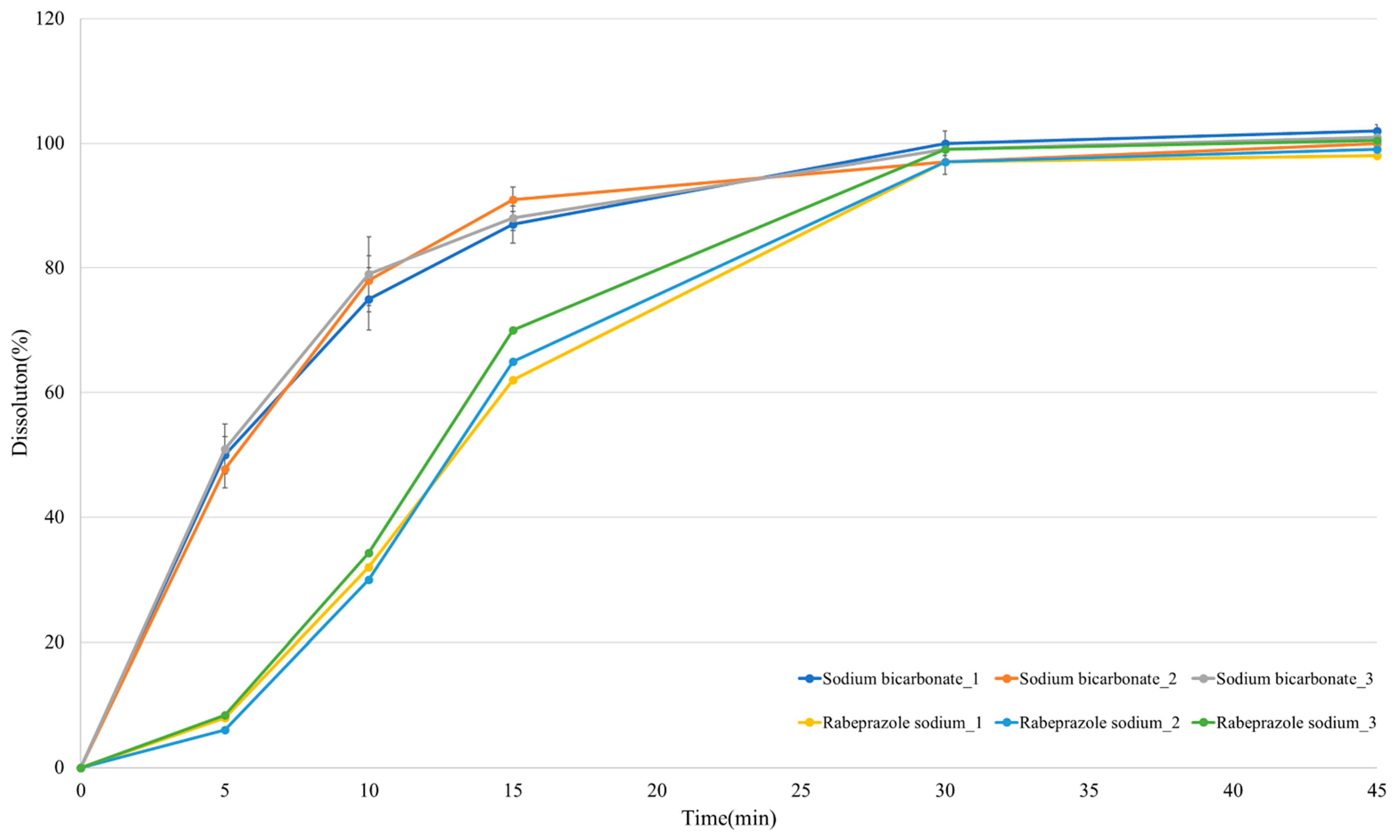
3.5. In Vitro Dissolution Profile
3.6. Stability Studies
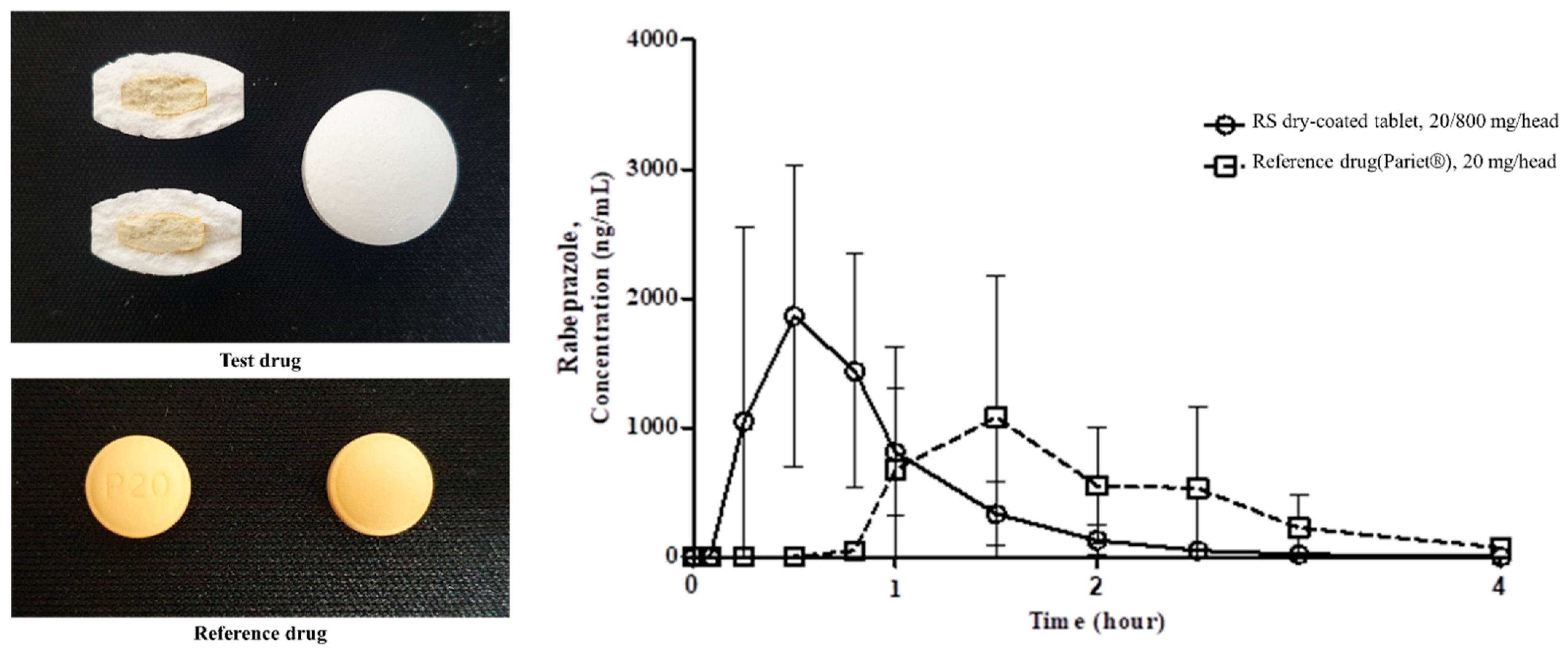
3.7. Pharmacokinetic Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lind, T.; Cederberg, C.; Ekenved, G.; Haglund, U.; Olbe, L. Effect of omeprazole—A gastric proton pump inhibitor—On pentagastrin stimulated acid secretion in man. Gut 1983, 24, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Prakash, A.; Faulds, D. Rabeprazole. Drugs 1998, 55, 261–267. [Google Scholar] [CrossRef]
- Naik, R.D.; Vaezi, M.F. Extra-esophageal manifestations of GERD: Who responds to GERD therapy? Curr. Gastroenterol. Rep. 2013, 15, 318. [Google Scholar] [CrossRef]
- Des Varannes, S.B.; Coron, E.; Galmiche, J.-P. Short and long-term PPI treatment for GERD. Do we need more-potent anti-secretory drugs? Best Pract. Res. Clin. Gastroenterol. 2010, 24, 905–921. [Google Scholar] [CrossRef]
- Dadabhai, A.; Friedenberg, F.K. Rabeprazole: A Pharmacologic and Clinical Review for Acid-Related Disorders. Expert Opin. Drug Saf. 2009, 8, 119–126. [Google Scholar] [CrossRef]
- James, L.P.; Walson, P.; Lomax, K.; Kao, R.; Varughese, S.; Reyes, J. Pharmacokinetics and tolerability of rabeprazole sodium in subjects aged 12 to 16 years with gastroesophageal reflux disease: An open-label, single-and multiple-dose study. Clin. Ther. 2007, 29, 2082–2092. [Google Scholar] [CrossRef]
- Williams, M.; Pounder, R. The pharmacology of rabeprazole. Aliment. Pharmacol. Ther. 1999, 13, 3–10. [Google Scholar] [CrossRef]
- LIM, P.Y.; Goh, K.; Wong, B. CYP2C19 genotype and the PPIs–focus on rabeprazole. J. Gastroenterol. Hepatol. 2005, 20, S22–S28. [Google Scholar] [CrossRef] [PubMed]
- Shirai, N.; Furuta, T.; Moriyama, Y.; Okochi, H.; Kobayashi, K.; Takashima, M.; Xiao, F.; Kosuge, K.; Nakagawa, K.; Hanai, H. Effects of CYP2C19 genotypic differences in the metabolism of omeprazole and rabeprazole on intragastric pH. Aliment. Pharmacol. Ther. 2001, 15, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Katsube, T.; Kawamura, A.; Takashima, T.; Yuki, M.; Amano, K.; Ishihara, S.; Fukuda, R.; Watanabe, M.; Kinoshita, Y. CYP2C19 genotype status and intragastric pH during dosing with lansoprazole or rabeprazole. Aliment. Pharmacol. Ther. 2000, 14, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Ife, R.J.; Dyke, C.A.; Keeling, D.J.; Meenan, E.; Meeson, M.L.; Parsons, M.E.; Price, C.A.; Theobald, C.J.; Underwood, A.H. 2-[[(4-Amino-2-pyridyl) methyl] sulfinyl] benzimidazole H+/K+-ATPase inhibitors. The relationship between pyridine basicity, stability, and activity. J. Med. Chem. 1989, 32, 1970–1977. [Google Scholar] [CrossRef]
- Mathew, M.; Gupta, V.D.; Bailey, R.E. Stability of omeprazole solutions at various pH values as determined by high-performance liquid chromatography. Drug Dev. Ind. Pharm. 1995, 21, 965–971. [Google Scholar] [CrossRef]
- Heese, G.-U.; Jünger, H.; Laicher, A.; Lorck, C.; Profitlich, T.; Weiss, G. Stable Drug form for Oral Administration with Benzimidazole Derivatives as Active Ingredient and Process for the Preparation Thereof. 0947166 U.S. Patents, 23 September 2003. [Google Scholar]
- Aravind, P.; Rathnanand, M.; Kumar, C.M. Stability enhancement of proton pump inhibitor in stomach: Formulation and in vitro evaluation of stabilized proton pump inhibitor. Asian J. Pharm. Clin. Res. 2017, 10, 88–92. [Google Scholar]
- Chen, J.; Jiang, W.M.; Gao, X.L.; Jiang, X.; Zhang, Q.Z.; Zheng, Z.H. Bioequivalence evaluation of two rabeprazole enteric coated formulations in healthy Chinese volunteers. Eur. J. Drug Metab. Pharmacokinet. 2004, 29, 103–106. [Google Scholar] [CrossRef]
- Park, C.-W.; Rhee, Y.-S.; Go, B.-W.; Kam, S.-H.; Lee, K.-H.; Lee, H.-S.; Park, E.-S. High performance liquid chromatographic analysis of rabeprazole in human plasma and its pharmacokinetic application. Arch. Pharm. Res. 2008, 31, 1195–1199. [Google Scholar] [CrossRef]
- Singh, S.S.; Jain, M.; Shah, H.; Gupta, S.; Thakker, P.; Shah, R.; Lohray, B.B. Direct injection, column switching–liquid chromatographic technique for the estimation of rabeprazole in bioequivalence study. J. Chromatogr. B 2004, 813, 247–254. [Google Scholar] [CrossRef]
- Mondal, U.; Ganesan, M.; Pal, T.; Jayakumar, M.; Chattaraj, T.; Roy, K.; Banerjee, S. Bioequivalence study of rabeprazole sodium on healthy human volunteers. J. Indian Med. Assoc. 2004, 102, 26, 28, 30. [Google Scholar]
- Sachs, G. Improving on PPI-based therapy of GORD. Eur. J. Gastroenterol. Hepatol. 2001, 13, S35. [Google Scholar]
- Ozturk, S.S.; Palsson, B.O.; Donohoe, B.; Dressman, J.B. Kinetics of release from enteric-coated tablets. Pharm. Res. 1988, 5, 550–565. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.M.; Sachs, G. Pharmacology of proton pump inhibitors. Curr. Gastroenterol. Rep. 2008, 10, 528–534. [Google Scholar] [CrossRef] [Green Version]
- Lerner, E.I.; Flashner, M.; Penhasi, A. Immediate Release Gastrointestinal Drug Delivery System. 6531152B1 U.S. Patents, 11 March 2003. [Google Scholar]
- Evans, D.; Pye, G.; Bramley, R.; Clark, A.; Dyson, T.; Hardcastle, J. Measurement of gastrointestinal pH profiles in normal ambulant human subjects. Gut 1988, 29, 1035–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewawaduge, C.; Senevirathne, A.; Lee, J.H. Enhancement of host infectivity, immunity, and protective efficacy by addition of sodium bicarbonate antacid to oral vaccine formulation of live attenuated Salmonella secreting Brucella antigens. Microb. Pathog. 2020, 138, 103857. [Google Scholar] [CrossRef]
- Massarrat, S.; Eisenmann, A. Factors affecting the healing rate of duodenal and pyloric ulcers with low-dose antacid treatment. Gut 1981, 22, 97–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasil’ev, I.; Noskova, K.; Zelenikin, S. Effectiveness of pariet (rabeprazole) in the treatment of gastroesophageal reflux disease (at the reflux-esophagitis stage). Exp. Clin. Gastroenterol. 2002, 55–60, 190. [Google Scholar]
- Mathews, H.; Moore, J. Sodium bicarbonate as a single dose antacid in obstetric anaesthesia. Anaesthesia 1989, 44, 590–591. [Google Scholar] [CrossRef]
- Lee, J.; Kim, J. QbD Platform Design Based on Edge Computing for Bioequivalent Drugs. In Proceedings of the Korea Information Processing Society Conference; Korea Information Processing Society: Seoul, Korea, 2018; pp. 89–92. [Google Scholar]
- Butler, J.N. Ionic Equilibrium: Solubility and pH Calculations; John Wiley & Sons: Hoboken, NJ, USA, 1998. [Google Scholar]
- Mosharraf, M.; Nyström, C. Apparent solubility of drugs in partially crystalline systems. Drug Dev. Ind. Pharm. 2003, 29, 603–622. [Google Scholar] [CrossRef] [PubMed]
- U.S.Pharmacopeia <1236> Solubility Study, USP43-NF38. Available online: https://uspnf.com (accessed on 18 January 2020).
- Kumari, M.; Jain, N.P. Formulation Development & Evaluation of Buffered Tablet of Proton Pump Inhibitors Drug Rabeprazole Sodium. J. Drug Deliv. Ther. 2019, 9, 315–321. [Google Scholar]
- Kuu, W.-Y.; Chilamkurti, R.; Chen, C. Effect of relative humidity and temperature on moisture sorption and stability of sodium bicarbonate powder. Int. J. Pharm. 1998, 166, 167–175. [Google Scholar] [CrossRef]
- Johnson, C.E.; Cober, M.P.; Ludwig, J.L. Stability of partial doses of omeprazole–sodium bicarbonate oral suspension. Ann. Pharmacother. 2007, 41, 1954–1961. [Google Scholar] [CrossRef]
- Mattsson, S.; Nyström, C. Evaluation of strength-enhancing factors of a ductile binder in direct compression of sodium bicarbonate and calcium carbonate powders. Eur. J. Pharm. Sci. 2000, 10, 53–66. [Google Scholar] [CrossRef]
- Oh, G.-H.; Park, J.-H.; Shin, H.-W.; Kim, J.-E.; Park, Y.-J. Quality-by-design approach for the development of telmisartan potassium tablets. Drug Dev. Ind. Pharm. 2018, 44, 837–848. [Google Scholar] [CrossRef]
- Mishra, S.M.; Rohera, B.D. An integrated, quality by design (QbD) approach for design, development and optimization of orally disintegrating tablet formulation of carbamazepine. Pharm. Dev. Technol. 2017, 22, 889–903. [Google Scholar] [CrossRef]
- Freeman, T.; Birkmire, A.; Armstrong, B. A QbD approach to continuous tablet manufacture. Procedia Eng. 2015, 102, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Park, M.-J.; Sah, H.; Lee, B.-J. Effect of pharmaceutical excipients on aqueous stability of rabeprazole sodium. Int. J. Pharm. 2008, 350, 197–204. [Google Scholar] [CrossRef]
- Kim, J.-E.; Park, Y.-J. High paclitaxel-loaded and tumor cell-targeting hyaluronan-coated nanoemulsions. Colloids Surf. B Biointerfaces 2017, 150, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Park, Y.-J. Improved antitumor efficacy of hyaluronic acid-complexed paclitaxel nanoemulsions in treating non-small cell lung cancer. Biomol. Ther. 2017, 25, 411. [Google Scholar] [CrossRef] [Green Version]
- Jenke, D. Application of ion chromatography in pharmaceutical and drug analysis. J. Chromatogr. Sci. 2011, 49, 524–539. [Google Scholar] [CrossRef] [Green Version]
- U.S.Pharmacopeia <1150> Pharmaceutical Stability, USP43-NF38. Available online: https://uspnf.com (accessed on 18 January 2020).
- Garcia, C.; Sippel, J.; Steppe, M.; Schapoval, E. Development and validation of derivative spectrophotometric method for determination of rabeprazole sodium in pharmaceutical formulation. Anal. Lett. 2006, 39, 341–348. [Google Scholar] [CrossRef]
- Heda, A.; Gadade, D.; Kathiriya, J.; Puranik, P. HPLC Method development and validation for simultaneous analysis of diclofenac sodium and rabeprazole sodium. J. Chem. 2010, 7, S386–S390. [Google Scholar] [CrossRef] [Green Version]
- Battu, P.R.; Reddy, M. Development and validation of RP-HPLC for the rabeprazole sodium in pharmaceutical formulations and human plasma. Asian J. Res. Chem. 2009, 2, 49–51. [Google Scholar]
- Treigerman, Z.; Sasson, Y. Generation and Quantification of Formate Ion Produced from Aqueous Sodium Bicarbonate in the Presence of Homogeneous Ruthenium Catalyst. ACS Omega 2018, 3, 12797–12801. [Google Scholar] [CrossRef] [PubMed]
- Cao, N.; Liu, L.; Hao, Y.-B.; Sun, L.-L.; Zou, Q.-G.; Ma, X.-L.; Xiong, K.-H. Simultaneous determination of rabeprazole enantiomers and their four metabolites after intravenous administration in beagle dogs by a stereoselective HPLC-MS/MS method and its application in pharmacokinetic studies. Anal. Methods 2016, 8, 1405–1414. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| QTPP 1 | Target | Risk | Justification |
|---|---|---|---|
| Indication | Reflux esophagitis | Yes | Inconsistent indications for patients taking this drug as a therapeutic drug may cause unnecessary drug misuse and side effects to the patient. |
| Dosage design | Immediate-release dry-coated tablets of less than 1100 mg | Yes | This drug is a dry-coated tablet formulation. Sodium bicarbonate in the outer layer is dissolved to act as an antacid in the stomach. After the antacid action is sufficiently completed, rabeprazole sodium in the inner core is released from the upper part of the small intestine to exert its medicinal effect. If the outer layer does not properly exert the antacid action, rabeprazole sodium can be decomposed in gastric acid, thereby reducing the patient’s therapeutic effect. |
| Route of administration | Oral administration | No | The oral route allows the administration of the highest drug dose and ensures high patient compliance. |
| Dosage strength | 20 mg/800 mg once a day (Rabeprazole sodium 20 mg/Sodium bicarbonate 800 mg) | Yes | If the patient fails to take a certain dose, the patient’s treatment may deteriorate, and if the number of administration once a day increases to more than two times a day in consideration of long-term administration, the timing of administration may be missed. |
| Pharmacokinetics | The AUC is the same as the reference drug, but the Tmax is about 3 times faster. | Yes | Maximum concentration (Cmax) and AUC can affect clinical trials such as safety and efficacy. |
| Stability and shelf life | Stable for at least 24 months at room temperature | Yes | If the quality characteristics of the drug are not suitable for the set period of use, the drug cannot be properly effective for the patients taking the drug. |
| Appearance | White circular shaped tablets | Yes | Changes in product appearance can lead to errors in the patient group’s selection of products for treatment. |
| Identification | Same equivalence requirement of peak retention time | No | When the purity of each active pharmaceutical ingredient is secured, there is no difficulty in identification. |
| Assay | 90.0–110.0% of the label claim (as Rabeprazole sodium%, Sodium bicarbonate%) | Yes | If the content is high, it may cause side effects, and if the content is low it may affect the lack of efficacy. |
| Weight variation/ Content uniformity | Conforms to USP 5 <905> Uniformity of Dosage Units: 90.0–110.0% of labeled claim with AV 6: NMT 15.0; RSD: NMT 5.0% | Yes | Variability in content uniformity can affect adverse drug reactions and clinical response. |
| Dissolution | Rabeprazole sodium: NLT 2 80% of labeled amount of drug is dissolved in 30 min in pH 8.0 buffer, Paddle speed: 75 rpm Sodium bicarbonate: NLT 90% of labeled amount of drug is dissolved in 30 min in water, Paddle speed: 75 rpm | Yes | The dissolution properties of the active pharmaceutical ingredients are important for bioavailability. (Especially, the salt of the main ingredient is important in evaluating the improvement of bioavailability by improving the solubility.) |
| Impurities | Unknown impurities: NMT 3 0.2%, Total impurities: NMT 3.5% (As per ICH 4 Q3A and Q3B) | Yes | Related substances (unknown and total related substances) of the main active pharmaceutical ingredient must be managed. |
| Residual solvent | NMT 5000 ppm of ethanol | Yes | Ethanol, the residual solvent, must be managed. |
| Primary packaging | Packaging and container suitable for maintaining the physicochemical stability of pharmaceuticals | Yes | Packaging materials increase drug stability by protecting drugs from the surrounding environment. In addition, packaging materials in direct contact may react with drugs and promote their degradation. |
| Quality Attributes of Dry Coated Tablet | Objective | CQA | Justification |
|---|---|---|---|
| Appearance | It should be in a shape and color for patients’ convenience in taking, and as a tablet, no defects should be observed. | Yes | Color, shape, and appearance are not directly linked to safety and effectiveness. However, in the case of sodium bicarbonate, which is used as the active pharmaceutical ingredient of the target tablet, the binding force is weak, and defects may be observed as a tablet. Therefore, this CQA should be studied through formulation research and process development. |
| Tablet size for convenience in taking | No | To ensure patient adherence to therapy and to facilitate swallowing, the target of tablet size is minimized as long as no defects in the tablet are observed. | |
| Identification | The main active pharmaceutical ingredient should be identified. | No | While the identification test is an important factor for safety and efficacy, this CQA can be effectively controlled by quality control systems and easily monitored in pharmaceuticals. Formulation studies and process parameters do not affect the identification test. Therefore, this CQA does not have to be discussed in formulation development and process development. |
| Assay | Rabeprazole sodium: 90~110% sodium bicarbonate: 90~110% | Yes | Variations in the assay can affect safety and effectiveness. Process variables can affect the content of the drug product. Therefore, the content should be evaluated through formulation research and process development. |
| Weight variation/ Content uniformity | Conforms to USP <905> Content uniformity: NMT 15.0%; RSD: NMT 5.0% Inner layer (Rabeprazole sodium): Content uniformity Outer layer (Sodium bicarbonate): Weight variation | Yes |
|
| Moisture content | Management in house spec according to stability test (less than 2.0%) | No | If the active ingredient is sensitive to moisture, stability, safety and efficacy may be affected. However, if the active ingredient is not sensitive to moisture or if appropriate packaging is used, the stability of the tablet will not be affected. |
| Impurities | Unknown impurities: NMT 0.2% Total impurities: NMT 3.5% (As per ICH Q3A and Q3B) | Yes | Degradation products may affect safety and should be controlled based on pharmacopeia or ICH requirements to limit exposure to patients. Limits for total related substances are based on the USP43-NF38. Formulation studies and process parameters can affect degradation products. Therefore, related substances must be evaluated during product and process development. |
| Residual solvent | USP <476> Option 1: NMT 5000 ppm of ethanol. | No | Residual solvent may affect safety, but it can be sufficiently controlled by the drying methods when manufacturing drugs or pharmaceuticals. Therefore, formulation studies and process variables are unlikely to have a significant impact on this CQA. |
| Dissolution | Rabeprazole sodium: NLT 80% of labeled amount of drug is dissolved in 45 min in pH 8.0 buffer, Paddle speed: 75 rpm Sodium bicarbonate: NLT 90% of labeled amount of drug is dissolved in 45 min in water, Paddle speed: 75 rpm | No | Failure to meet the dissolution conditions may affect bioavailability (efficacy). Formulation studies and process variables affect dissolution. However, rabeprazole sodium and sodium bicarbonate have very good solubility in most solvents, so they do not significantly affect the design of immediate-release. |
| Rabeprazole Sodium | |||
|---|---|---|---|
| Chemical structure |  | CAS. NO. | 117976-90-6 |
| Chemical name | Rabeprazole sodium | ||
| Formula | C18H20N3NaO3S | ||
| Mol. Mass | 381.42 g/mol | Description | White powder |
| Melting point | 140~141 °C | Solubility | 10 mg/mL (Water) |
| Boiling point | 603.9 °C | ||
| PKa | pKa (Strongest Acidic): 9.35 pKa (Strongest Basic): 4.24 | BCS Class | BCS III |
| Storage Condition | Airtight container, storage at room temperature | ||
| Mechanism of action | It is a powerful proton pump inhibitor. Inhibits the secretion of gastric acid by inhibiting the parietal cell H+/K+ ATP pump. | ||
| Pharmacokinetics | -Action onset time: within 1 h -Duration: 24 h -Absorption: Oral: well absorbed within 1 h; Food delays absorption by up to 4 h or more. -Protein binding rate: 96.3% -Metabolism: metabolized by CYP3A and 2C19 to inactive metabolites in the liver; CYP2C19 exhibits a genetic polymorphism that slows metabolism due to deficiency in some populations (subpopulations, Caucasian 3–5%, Asian 17-20%). -Bioavailability: Tablets: ~52% -Half-life (dose dependent): adolescents: ~0.55–1 h, adults: 1–2 h; 2–3 times higher in patients with mild to moderate hepatic impairment. -Time to reach maximum plasma concentration: Adolescents: Tablets: 3.3–4.1 h Adults: Tablets: 2–5 h; Capsule: 1–6.5 h -Excretion: urine (mainly 90% of metabolites of thioether carboxylic acid); The rest is feces | ||
| Sodium Bicarbonate | |||
| Chemical structure |  | CAS. NO. | 144-55-8 |
| Chemical name | Sodium bicarbonate | ||
| Formula | NaHCO3 | ||
| Mol. Mass | 84.01 g/mol | Description | White, crystalline powder |
| Melting point | 270 °C | Solubility | 69 g/L (0 °C) 96 g/L (20 °C) 165 g/L (60 °C) |
| Boiling point | 851 °C | ||
| PKa | 10.329 6.351 (carbonic acid) | BCS Class | BCSI |
| USP | USP40-NF35 | ||
| Storage Condition | Store in a tightly closed container. Store in a cool | ||
| Mechanism of action | Separation produces bicarbonate ions that neutralize hydrogen ions and raise the pH of blood and urine. Neutralizing Additive (Dental Use): Increases the pH of Lidocaine and epinephrine solutions to improve tolerance and increase tissue absorption. | ||
| Pharmacokinetics | -Onset time of action: oral: 15 min; Intravenous (IV): fast. -Duration: Oral: 1–3 h; Intravenous (IV): 8–10 min -Absorption: Oral: Well absorbed. -Excretion: urine (<1%) | ||
| Characteristic | -As an antacid, it is used to improve symptoms caused by gastric ulcer, duodenal ulcer, gastritis, and excessive stomach acid. -As an alkalinizing agent, it is used for the purpose of reducing the acidity of blood or urine. -Widely used as a pH buffer. | ||
| Items | Initial (%) | 4 Weeks (%) | |
|---|---|---|---|
| Rabeprazole sodium | 0.06 | Room Temperature | 0.07 |
| Accelerated Condition | 1.31 | ||
| Rabeprazol sodium: Sodium bicarbonate 1:1 (w/w) | 0.57 | Room Temperature | 0.79 |
| Accelerated Condition | 2.17 | ||
| Rabeprazol sodium: Sodium bicarbonate 20:800 (w/w) | 3.41 | Room Temperature | 6.80 |
| Accelerated Condition | 9.83 | ||
| CQAs 1 | Rabeprazole Sodium | Sodium Bicarbonate | Diluent | Binder | Disintegrant | Anti-Adherent | Glidant | Lubricant |
|---|---|---|---|---|---|---|---|---|
| Identification | Low | Low | Low | High | Low | Low | Low | Low |
| Assay | Low | Low | Low | Low | High | Low | Low | Low |
| Uniformity | Low | Medium | Low | Medium | Low | Low | Low | Medium |
| Impurities | Low | Low | Low | Low | Low | Low | Low | Low |
| Dissolution | Low | Low | Low | High | High | Low | Low | Low |
| Functions | CMAs 2 | Failure Mode (Critical Event) | Effect on CQAs with Respect to QTPP 3 (Justification of Failure Mode) | P 6 | S 7 | D 8 | RPN 9 |
|---|---|---|---|---|---|---|---|
| Physical property of API 4 | Solid state form | Different PSD 5/ Different form | The solubility of the active pharmaceutical ingredient (API) may be affected, and the dissolution of the drug product is affected. Thus, this causes damage to bioavailability and efficacy. | 1 | 2 | 2 | 4 |
| Chemical property of API | Solubility | Different Salt/ Different form | May affect the dissolution of tablets. Thus, bioavailability and efficacy may be compromised. | 1 | 1 | 2 | 2 |
| Chemical stability | Unstable | Decomposition products may be affected by dry heat/oxidation/hydrolysis/UV light, thus causing quality and safety damage. | 1 | 1 | 2 | 2 | |
| Diluent | PSD | Uneven | It can affect the flow properties of blending and can affect the content uniformity. Thus, quality/safety may be compromised. | 1 | 1 | 2 | 2 |
| Moisture Content | High | May affect the impurity profile. Thus, this causes damage to safety. | 3 | 2 | 2 | 12 | |
| Binding solution (Inner layer) | Volume of binding solution | Higher than optimum | Produces hard granules, which can affect disintegration and dissolution time. Thus, bioavailability and efficacy may be compromised. | 3 | 2 | 1 | 6 |
| Lower than optimum | Loose, fragile granules can produce tablets of weaker hardness (fast disintegration). Thus, bioavailability and efficacy may be compromised. | 3 | 2 | 1 | 6 | ||
| Binder (Outer layer) | Concentration of binder | Higher than optimum | Delayed disintegration and dissolution time of tablets. Thus, bioavailability and efficacy may be compromised. | 4 | 5 | 3 | 60 |
| Lower than optimum | The friability of the tablet is high, and the desired dissolution pattern cannot be obtained. Thus, bioavailability and efficacy may be compromised. | 4 | 4 | 3 | 48 | ||
| Disintegrant | Concentration of disintegrant | Higher than optimum | The desired dissolution pattern cannot be obtained, and the hardness of the tablet may be affected. Thus, bioavailability and efficacy may be compromised. | 4 | 4 | 3 | 48 |
| Lower than optimum | The desired dissolution pattern cannot be obtained. Thus, bioavailability and efficacy may be compromised. | 4 | 3 | 3 | 36 | ||
| Anti-adherent | Concentration of Anti-adherent | Lower than optimum | It may be difficult to discharge tablets from tooling. The excipient can be stuck on the surface of the filling die. Thus, product quality may be compromised. | 3 | 3 | 2 | 18 |
| Glidant | Concentration of glidant | Lower than optimum | By reducing the friction in the particles, it may affect the flowability of granules or powders such as die friction. May affect content uniformity. Therefore, content uniformity and product quality may be compromised. | 2 | 2 | 2 | 8 |
| Lubricant | Concentration of Lubricant | Higher than optimum | Hydrophobic lubricants can be coated on the surface of drug particles, which can delay dissolution. Thus, efficacy may be compromised. | 3 | 3 | 3 | 27 |
| Lower than optimum | The powder can stick to the surface of tooling/punch and cause picking. Thus, product quality may be compromised. | 3 | 3 | 3 | 27 |
| CQAs | Screening | Blending | Granulation | Drying | Milling | Blending and Lubrication | Compression |
|---|---|---|---|---|---|---|---|
| Identification | Low | Low | Low | Medium | Low | Low | Medium |
| Assay | Low | Low | Medium | Low | Low | Low | Medium |
| Uniformity | Low | Low | Low | Low | Low | Low | Low |
| Impurities | Low | Low | Low | Medium | Low | Low | Low |
| Dissolution | Low | Low | Low | Low | Low | Medium | Medium |
| Functions | CPPs 1 | Failure Mode (Critical Event) | Effect on CQAs 2 with Respect to QTPP3 (Justification of Failure Mode) | P | S | D | RPN |
|---|---|---|---|---|---|---|---|
| Screening | Sifting | Larger than optimum sieve size | Uneven particle size mixture could cause content non-uniformity. Thus, quality and safety may be compromised. | 1 | 1 | 1 | 1 |
| Blending | Mixing rate (Rpm and Time) | Lower mixingspeed and shorter time | Insufficient total number of revolutions leads to the inhomogeneity of the mixture. Thus, bioavailability and efficacy may be compromised. | 1 | 2 | 1 | 2 |
| Granulation | Impeller/Mixer speed | Higher mixing speed and longer time | Production of large granules (agglomerate/lumps) increases the elution time of tablets. Thus, bioavailability and efficacy may be compromised. | 3 | 2 | 2 | 12 |
| Chopper/Granulator speed | Lower mixing speed and shorter time | Production of large granules (agglomerate/lumps) increases the elution time of tablets. Thus, bioavailability and efficacy may be compromised. | 3 | 2 | 2 | 12 | |
| Granulation time | Longer than optimum time | Production of large granules (agglomerate/lumps) increases the elution time of tablets. Thus, bioavailability and efficacy may be compromised. | 3 | 2 | 2 | 12 | |
| Drying | Inlet temperature | Lower than optimum temperature | If the temperature is lower than the optimum temperature, the solution is not dried well, and the residual solvent affects the physical aspect of the tablet. | 2 | 2 | 3 | 12 |
| Higher product temperature | Degradation and impurities profile may be affected. Thus, safety and efficacy may be compromised | 3 | 2 | 3 | 18 | ||
| Milling | Mill speed | Higher than optimum speed | Poor flow and non-uniformity can occur due to the generation of fine powder. | 1 | 1 | 2 | 2 |
| Mill screen size | Larger than optimum screen size | Uneven PSD causes inhomogeneity. Larger particles increase the dissolution time. Thus, efficacy may be compromised. | 1 | 1 | 2 | 2 | |
| Blending and lubrication | Blending rate (RPM and Time) | Higher than optimum speed and longer time | Dissolution time may increase. Thus, efficacy may be compromised. | 1 | 2 | 2 | 4 |
| Compression | Speed of turret and feeder | Higher than optimum speed | Lamination and weight variation can be observed = It affects content uniformity, disintegration time, and dissolution. Thus, efficacy may be compromised. | 4 | 1 | 6 | 24 |
| Compression force (Pre-compression and compression) | Higher than optimum force | The appearance and hardness of tablets may be affected. Disintegration and dissolution profiles may be affected. Thus, efficacy may be compromised. | 3 | 2 | 2 | 12 | |
| Coating | Speed of coating pan | Higher than optimum speed | If the speed of the coating pan is high, it causes damage to the tablet. Thus, bioavailability and efficacy may be compromised. | 3 | 1 | 1 | 3 |
| Run | CMAs | CQAs | |||
|---|---|---|---|---|---|
| X1: Copovidone (%) | X2: Crospovidone (%) | Y1: Hardness (kp) | Y2: Disintegration (min) | Y3: Friability (%) | |
| 1 | 5.0 | 1.0 | 1.7 | 19.3 | 0.8 |
| 2 | 10.0 | 1.0 | 10.8 | 28.3 | 0.2 |
| 3 | 5.0 | 5.0 | 2.3 | 4.0 | 1.3 |
| 4 | 10.0 | 5.0 | 11.6 | 18.0 | 0.3 |
| 5 | 3.9 | 3.0 | 0.9 | 6.0 | 0.4 |
| 6 | 11.0 | 3.0 | 18.3 | 18.0 | 0.1 |
| 7 | 7.5 | 0.2 | 7.8 | 28.3 | 0.3 |
| 8 | 7.5 | 5.8 | 9.3 | 4.0 | 0.3 |
| 9 | 7.5 | 3.0 | 9.5 | 8.5 | 0.6 |
| 10 | 7.5 | 3.0 | 9.5 | 8.5 | 0.6 |
| 11 | 7.5 | 3.0 | 9.5 | 8.5 | 0.6 |
| 12 | 7.5 | 3.0 | 9.5 | 8.5 | 0.6 |
| 13 | 7.5 | 3.0 | 9.5 | 8.5 | 0.6 |
| APIs | Stability | Criterion | Drugs | Initial | 6 Month | 12 Month |
|---|---|---|---|---|---|---|
| Rabeprazole sodium | Assay | 90–110% | Test drug | 100.8 ± 1.2% | 102.7 ± 0.5% | 99.8 ± 1.3% |
| Reference drug | 100.2 ± 0.8% | 98.9 ± 1.3% | 98.7 ± 1.7% | |||
| Content uniformity | NMT 15% | Test drug | 3.12 ± 0.1% | 3.08 ± 2.5% | 3.30 ± 0.3% | |
| Reference drug | 5.27 ± 1.2% | 5.64 ± 1.1% | 5.71 ± 0.6% | |||
| Dissolution | NLT 80% | Test drug | 91.2 ± 2.4% | 92.8 ± 0.8% | 90.1 ± 1.1% | |
| Reference drug | 90.4 ± 1.8% | 92.1 ± 1.4% | 91.4 ± 0.9% | |||
| Total impurities | NMT 3.5% | Test drug | 0.32 ± 0.2% | 0.55 ± 0.3% | 1.01 ± 1.2% | |
| Reference drug | 0.38 ± 0.1% | 0.72 ± 0.1% | 1.28 ± 0.7% | |||
| Sodium bicarbonate | Assay | 90–110% | Test drug | 101.7 ± 1.3% | 102.2 ± 2.1% | 100.3 ± 1.7% |
| Content uniformity | NMT 15% | Test drug | 4.14 ± 0.1% | 3.98 ± 0.4% | 4.27 ± 0.7% | |
| Dissolution | NLT 90% | Test drug | 102.3 ± 0.2% | 101.4 ± 0.7% | 100.6 ± 1.4% |
| PK Parameter | RS Dry-Doated Tablet | Reference Drug |
|---|---|---|
| 20 mg/head | 20 mg/head | |
| Cmax 1 (ng/mL) | 2493.5 ± 1073.2 | 1575.4 ± 991.4 |
| Tmax 2 (hr) 1) | 0.5 (0.3–0.8) | 1.5 (1–2.5) |
| AUClast 3 (ng·hr/mL) | 1614.0 ± 793.1 | 1603.1 ± 864.6 |
| AUCinf (ng·hr/mL) | 1620.2 ± 793.8 | 1782.7 ± 886.8 |
| T1/2 (hr) | 0.4 | 1.9 ± 2.6 |
| CL/F 4 (mL·hr/kg) | 15,930.4 ± 10,034.3 | 15,822.5 ± 13,023.2 |
| Vd/F 5 (mL/kg) | 8182.0 ± 5617.7 | 47,350.3 ± 56,108.4 |
| Rsq_adjusted | 1.0 | 0.7 ± 0.5 |
| %AUCexp (%) | 0.5 ± 0.5 | 10.1 ± 19.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.-H.; Kim, J.-E. Quality by Design Applied Development of Immediate-Release Rabeprazole Sodium Dry-Coated Tablet. Pharmaceutics 2021, 13, 259. https://doi.org/10.3390/pharmaceutics13020259
Lee S-H, Kim J-E. Quality by Design Applied Development of Immediate-Release Rabeprazole Sodium Dry-Coated Tablet. Pharmaceutics. 2021; 13(2):259. https://doi.org/10.3390/pharmaceutics13020259
Chicago/Turabian StyleLee, Sang-Ho, and Joo-Eun Kim. 2021. "Quality by Design Applied Development of Immediate-Release Rabeprazole Sodium Dry-Coated Tablet" Pharmaceutics 13, no. 2: 259. https://doi.org/10.3390/pharmaceutics13020259
APA StyleLee, S. -H., & Kim, J. -E. (2021). Quality by Design Applied Development of Immediate-Release Rabeprazole Sodium Dry-Coated Tablet. Pharmaceutics, 13(2), 259. https://doi.org/10.3390/pharmaceutics13020259