
Pharmacokinetics of the CYP3A4 and CYP2B6 Inducer Carbamazepine and Its Drug–Drug Interaction Potential: A Physiologically Based Pharmacokinetic Modeling Approach



Abstract
:1. Introduction
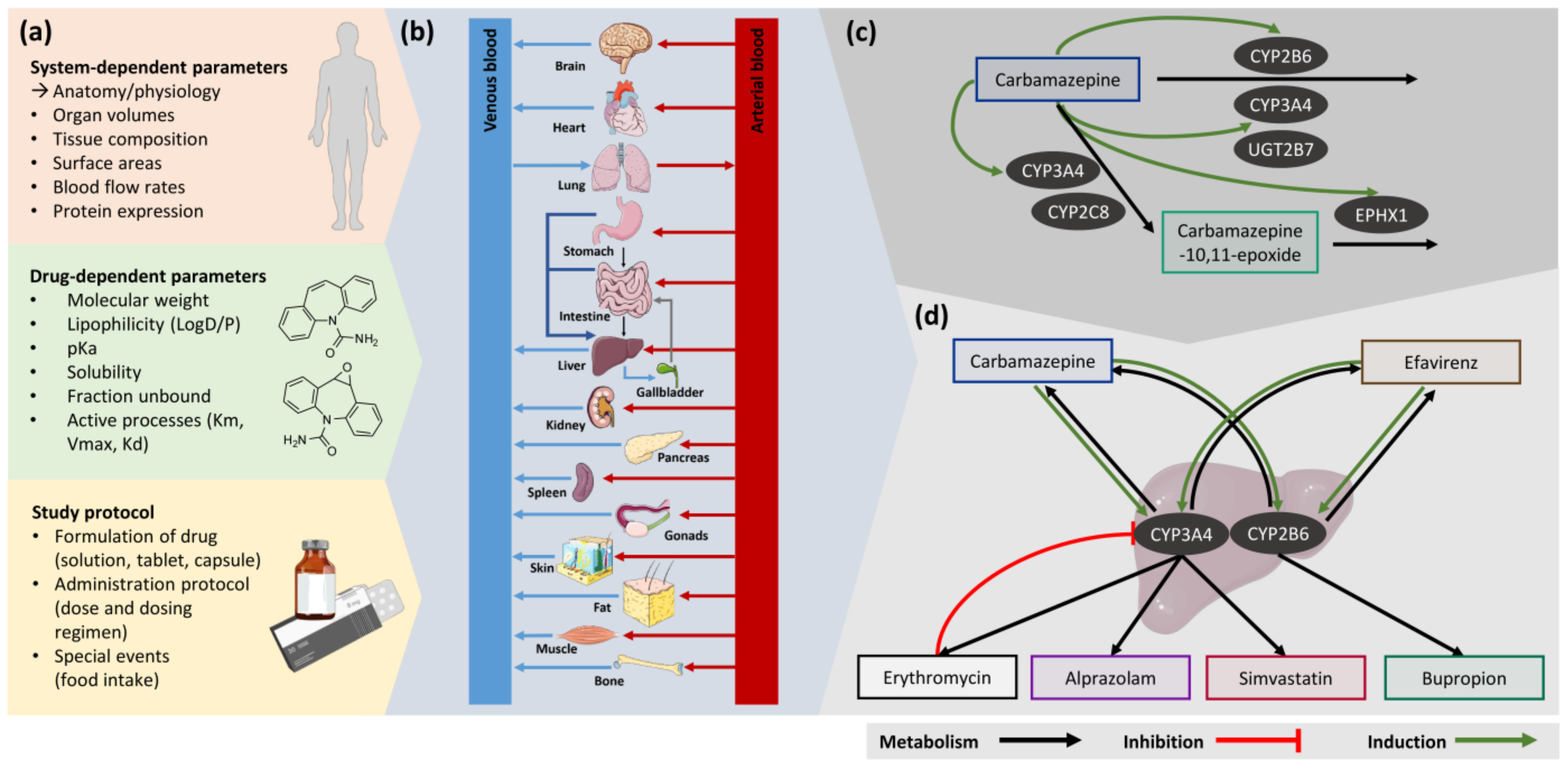
2. Materials and Methods
2.1. Software
2.2. Clinical Data
2.3. Model Building
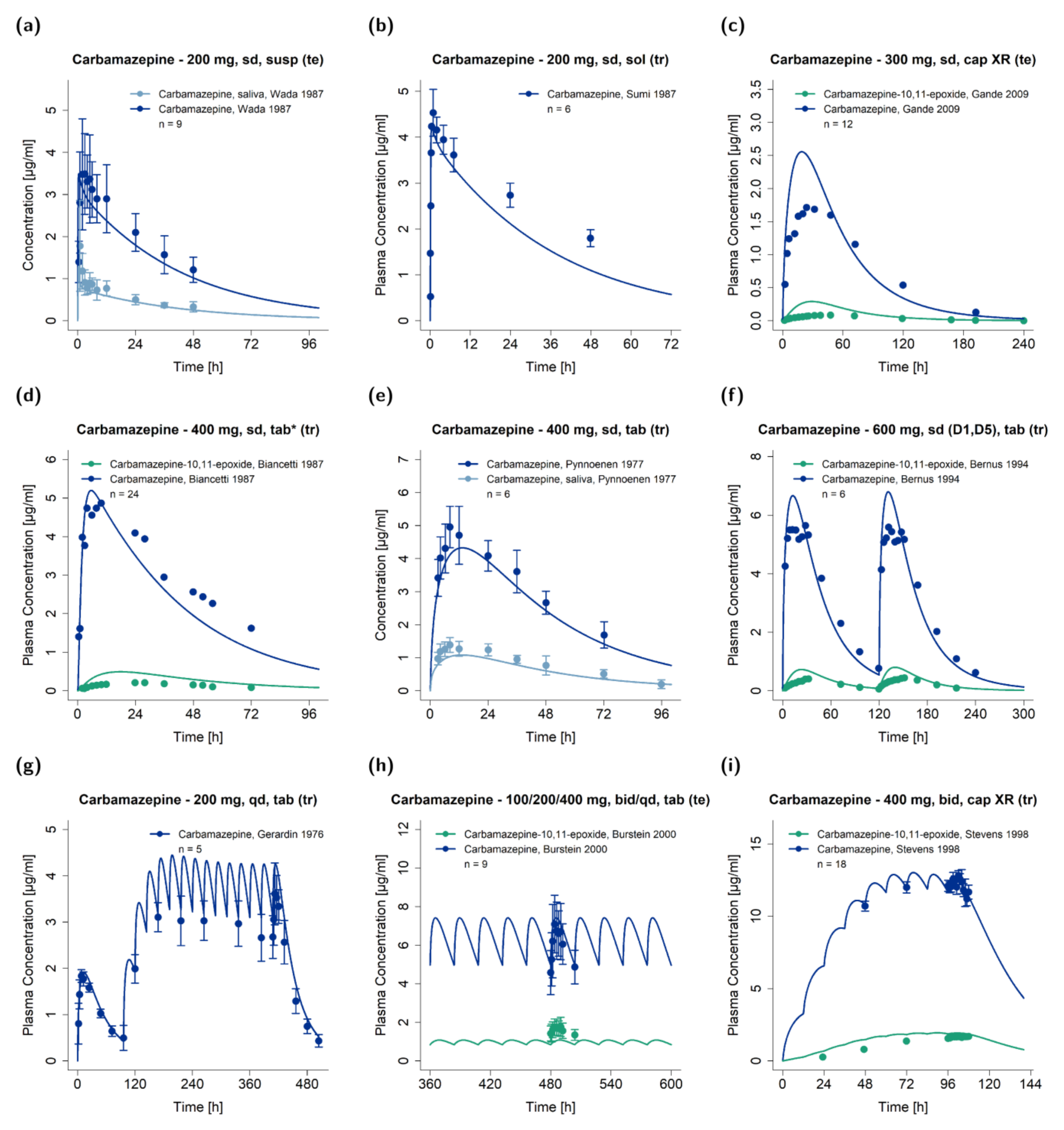
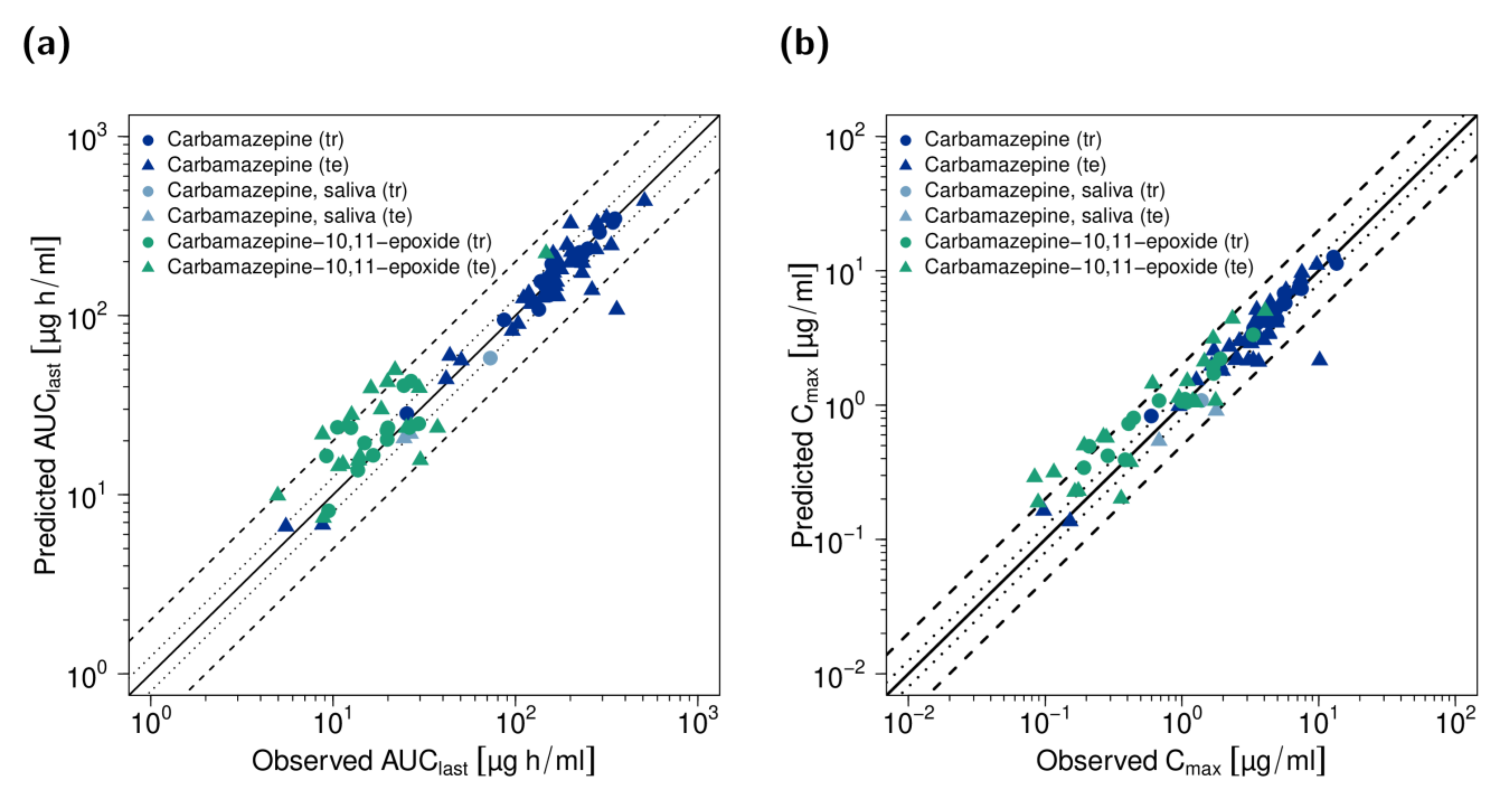
2.4. PBPK Model Evaluation
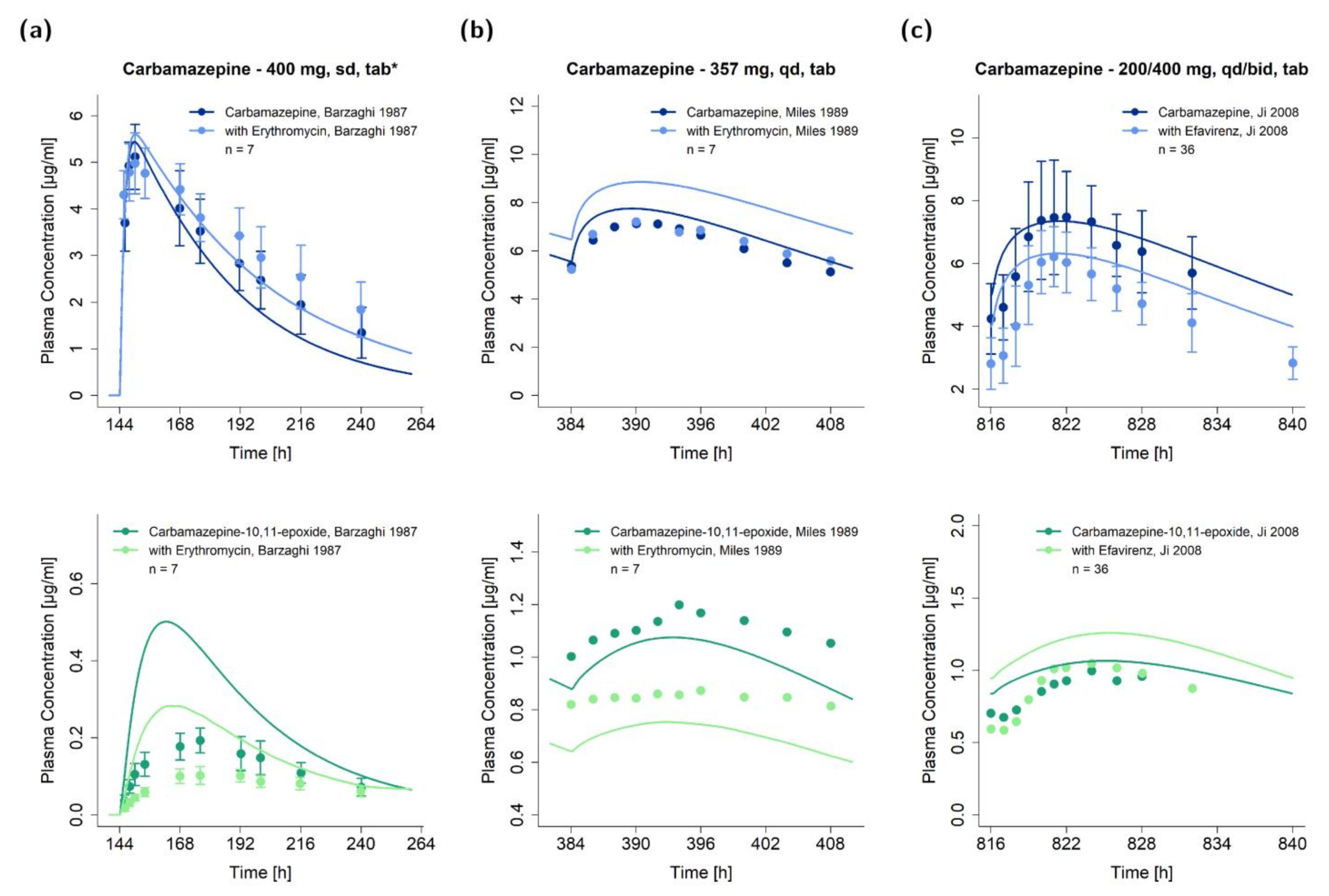
2.5. DDI Modeling
2.6. DDI Model Evaluation
3. Results
3.1. PBPK Model Building
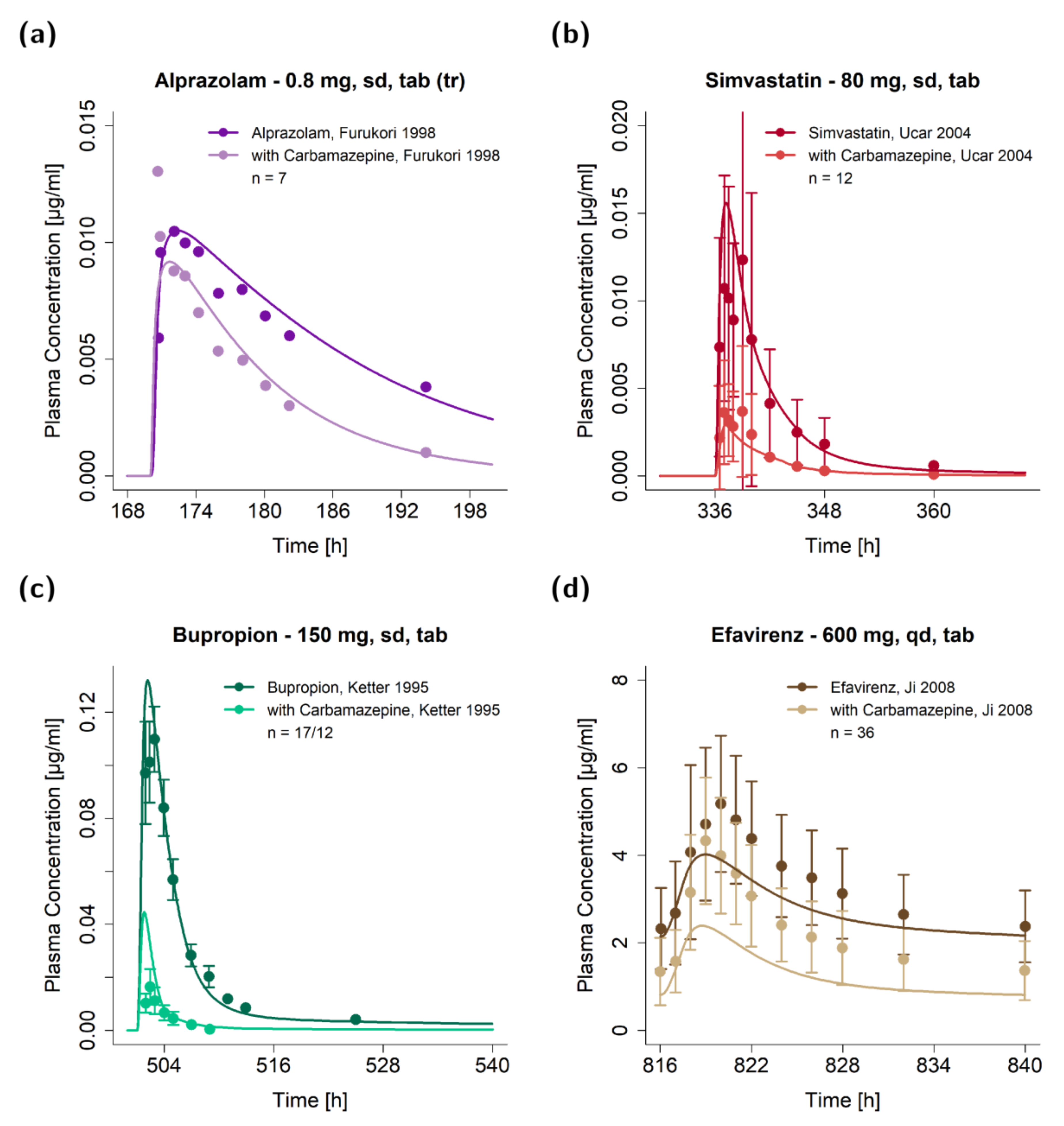
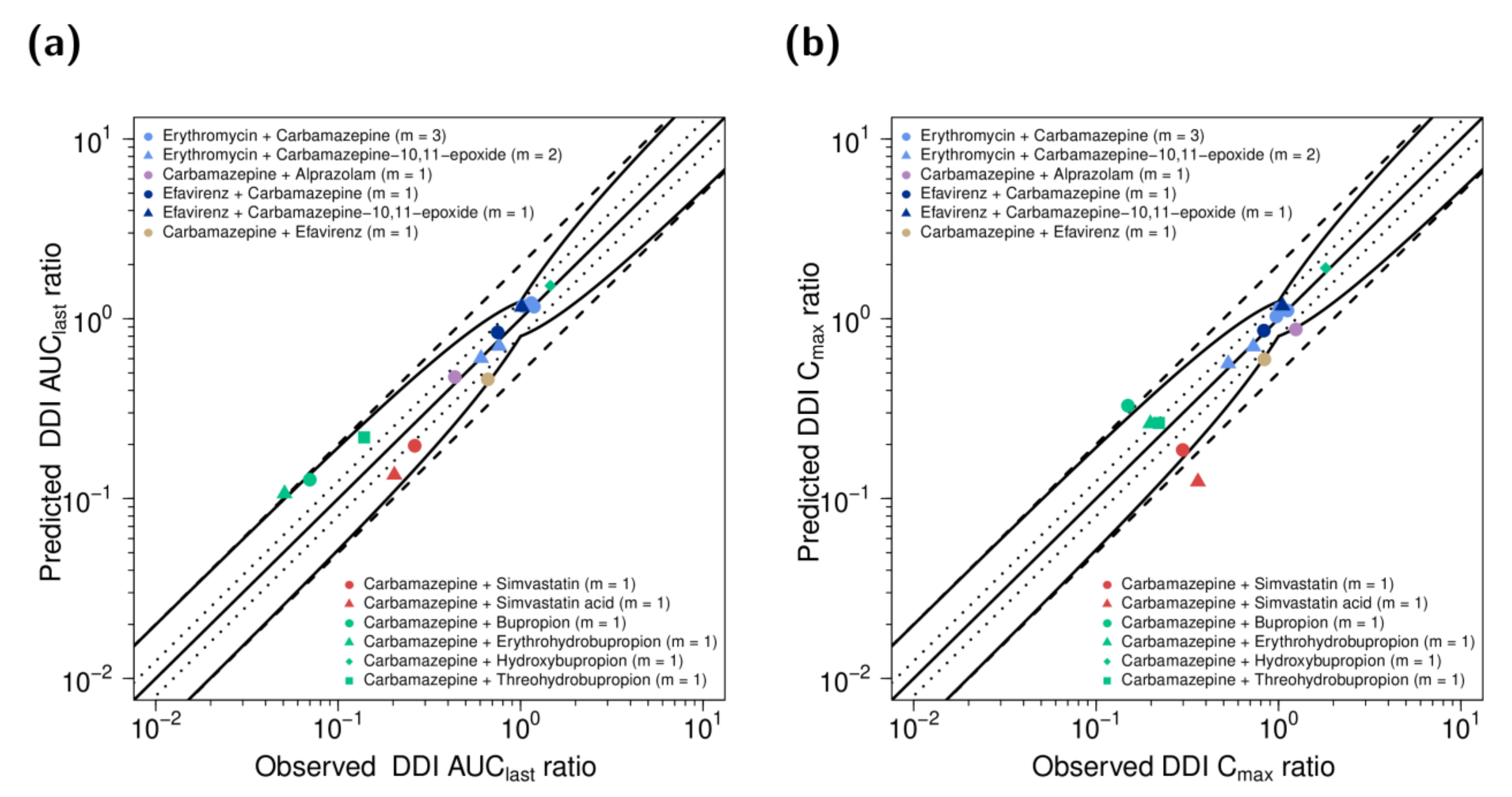
3.2. DDI Modeling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- U.S. Food and Drug Administration. Drug Development and Drug Interactions. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions (accessed on 3 March 2020).
- Kerr, B.M.; Thummel, K.E.; Wurden, C.J.; Klein, S.M.; Kroetz, D.L.; Gonzalez, F.J.; Levy, R.H. Human liver carbamazepine metabolism. Role of CYP3A4 and CYP2C8 in 10,11-epoxide formation. Biochem. Pharmacol. 1994, 47, 1969–1979. [Google Scholar] [CrossRef]
- Pelkonen, O.; Myllynen, P.; Taavitsainen, P.; Boobis, A.R.; Watts, P.; Lake, B.G.; Price, R.J.; Renwick, A.B.; Gómez-Lechón, M.J.; Castell, J.V.; et al. Carbamazepine: A “blind” assessment of CYP-associated metabolism and interactions in human liver-derived in vitro systems. Xenobiotica 2001, 31, 321–343. [Google Scholar] [CrossRef]
- Wong, Y.Y.; Ludden, T.M.; Bell, R.D. Effect of erythromycin on carbamazepine kinetics. Clin. Pharmacol. Ther. 1983, 33, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Shahzadi, A.; Javed, I.; Aslam, B.; Muhammad, F.; Asi, M.R.; Ashraf, M.Y.; Ur-Rahman, Z. Therapeutic effects of ciprofloxacin on the pharmacokinetics of carbamazepine in healthy adult male volunteers. Pak. J. Pharm. Sci. 2011, 24, 63–68. [Google Scholar]
- Kim, K.A.; Sae, O.O.; Park, P.W.; Park, J.Y. Effect of probenecid on the pharmacokinetics of carbamazepine in healthy subjects. Eur. J. Clin. Pharmacol. 2005, 61, 275–280. [Google Scholar] [CrossRef]
- Ucar, M.; Neuvonen, M.; Luurila, H.; Dahlqvist, R.; Neuvonen, P.J.; Mjörndal, T. Carbamazepine markedly reduces serum concentrations of simvastatin and simvastatin acid. Eur. J. Clin. Pharmacol. 2004, 59, 879–882. [Google Scholar] [CrossRef]
- Ketter, T.A.; Jenkins, J.B.; Schroeder, D.H.; Pazzaglia, P.J.; Marangell, L.B.; George, M.S.; Callahan, A.M.; Hinton, M.L.; Chao, J.; Post, R.M. Carbamazepine but not valproate induces bupropion metabolism. J. Clin. Psychopharmacol. 1995, 15, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Tirona, R.; Kim, R. Introduction to clinical pharmacology. In Clinical and Translational Science: Principles of Human Research; Robertson, G., Williams, G., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 365–388. ISBN 978-0-12802-101-9. [Google Scholar]
- Ji, P.; Damle, B.; Xie, J.; Unger, S.E.; Grasela, D.M.; Kaul, S. Pharmacokinetic interaction between efavirenz and carbamazepine after multiple-dose administration in healthy subjects. J. Clin. Pharmacol. 2008, 48, 948–956. [Google Scholar] [CrossRef]
- Chochrance. Antiepileptic Drug Monotherapy (Single Drug Treatment) for Epilepsy. Available online: https://www.cochrane.org/CD011412/EPILEPSY_antiepileptic-drug-monotherapy-single-drug-treatment-epilepsy (accessed on 25 November 2020).
- World Health Organization. World Health Organization Model List of Essential Medicines; 21st List. Available online: https://www.who.int/publications/i/item/WHOMVPEMPIAU2019.06 (accessed on 25 November 2020).
- Adiwidjaja, J.; Boddy, A.V.; McLachlan, A.J. Implementation of a physiologically based pharmacokinetic modeling approach to guide optimal dosing regimens for imatinib and potential drug interactions in paediatrics. Front. Pharmacol. 2020, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Guideline on the Reporting of Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulation; European Medicines Agency: Amsterdam, The Netherlands, 2018.
- Physiologically Based Pharmacokinetic Analyses—Format and Content; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2018.
- Conner, T.M.; Nikolian, V.C.; Georgoff, P.E.; Pai, M.P.; Alam, H.B.; Sun, D.; Reed, R.C.; Zhang, T. Physiologically based pharmacokinetic modeling of disposition and drug-drug interactions for valproic acid and divalproex. Eur. J. Pharm. Sci. 2018, 111, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.N.; Zhou, D.; Bui, K.H. Development of physiologically based pharmacokinetic model to evaluate the relative systemic exposure to quetiapine after administration of IR and XR formulations to adults, children and adolescents. Biopharm. Drug Dispos. 2014, 35, 341–352. [Google Scholar] [CrossRef]
- Schuck, E.; Ferry, J.; Gidal, B.; Hussein, Z. Changes in perampanel levels during de-induction: Simulations following carbamazepine discontinuation. Acta Neurol. Scand. 2020, 142, 131–138. [Google Scholar] [CrossRef]
- Almond, L.M.; Mukadam, S.; Gardner, I.; Okialda, K.; Wong, S.; Hatley, O.; Tay, S.; Rowland-Yeo, K.; Jamei, M.; Rostami-Hodjegan, A.; et al. Prediction of drug-drug interactions arising from CYP3A induction using a physiologically based dynamic model. Drug Metab. Dispos. 2016, 44, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, B.M.; Muftakhidinov, T.W.; Jędrzejewski-Szmek, Z. Engauge Digitizer Software. Available online: https://merkummitchell.github.io/engauge-digitizer (accessed on 15 February 2021).
- Wojtyniak, J.G.; Britz, H.; Selzer, D.; Schwab, M.; Lehr, T. Data Digitizing: Accurate and Precise Data Extraction for Quantitative Systems Pharmacology and Physiologically-Based Pharmacokinetic Modeling. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Open Systems Pharmacology Suite Community PK-Sim® Ontogeny Database Documentation, Version 7.3; Available online: Github.com/Open-Systems-Pharmacology/OSPSuite.Documentation/blob/master/PK-Sim%Ontogeny%20Database%20Version%207.3.pdf (accessed on 15 February 2021).
- Vree, T.B.; Janssen, T.J.; Hekster, Y.A.; Termond, E.F.; van de Dries, A.C.; Wijnands, W.J. Clinical pharmacokinetics of carbamazepine and its epoxy and hydroxy metabolites in humans after an overdose. Ther. Drug Monit. 1986, 8, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Kerr, B.M.; Rettie, A.E.; Eddy, A.C.; Loiseau, P.; Guyot, M.; Wilensky, A.J.; Levy, R.H. Inhibition of human liver microsomal epoxide hydrolase by valproate and valpromide: In vitro/in vivo correlation. Clin. Pharmacol. Ther. 1989, 46, 82–93. [Google Scholar] [CrossRef]
- Kitteringham, N.R.; Davis, C.; Howard, N.; Pirmohamed, M.; Park, B.K. Interindividual and interspecies variation in hepatic microsomal epoxide hydrolase activity: Studies with cis-stilbene oxide, carbamazepine 10,11- epoxide and naphthalene. J. Pharmacol. Exp. Ther. 1996, 278, 1018–1027. [Google Scholar] [PubMed]
- Levy, R.H.; Pitlick, W.H.; Troupin, A.S.; Green, J.R.; Neal, J.M. Pharmacokinetics of carbamazepine in normal man. Clin. Pharmacol. Ther. 1975, 17, 657–668. [Google Scholar] [CrossRef]
- McLean, A.; Browne, S.; Zhang, Y.; Slaughter, E.; Halstenson, C.; Couch, R. The influence of food on the bioavailability of a twice-daily controlled release carbamazepine formulation. J. Clin. Pharmacol. 2001, 41, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Frechen, S.; Wendel, T.; Solodenko, J. Building and Evaluation of a PBPK Model for Efavirenz in Healthy Adults. Available online: https://github.com/Open-Systems-Pharmacology/OSP-PBPK-Model-Library/blob/v9.1/Efavirenz/efavirenz_evaluation_report.pdf (accessed on 13 November 2020).
- Frechen, S.; Dallmann, A. Building and Evaluation of a PBPK Model for Alprazolam in Healthy Adults. Available online: https://github.com/Open-Systems-Pharmacology/OSP-PBPK-Model-Library/blob/v9.1/Alprazolam/Alprazolam_evaluation_report.pdf (accessed on 13 November 2020).
- Frechen, S.; Dallmann, A. Building and Evaluation of a PBPK Model for Erythromycin in Healthy Adults. Available online: https://github.com/Open-Systems-Pharmacology/OSP-PBPK-Model-Library/blob/v9.1/Erythromycin/Erythromycin_evaluation_report.pdf (accessed on 13 November 2020).
- Marok, F.Z.; Fuhr, L.; Hanke, N.; Selzer, D.; Lehr, T. Physiologically Based Pharmacokinetic Modeling of Bupropion and its Metabolites in a CYP2B6 Drug-Drug-Gene Interaction Network. Pharmaceutics 2020. submitted for publication. [Google Scholar]
- Wojtyniak, J.; Selzer, D.; Schwab, M.; Lehr, T. Physiologically Based Precision Dosing Approach for Drug-Drug-Gene Interactions: A Simvastatin Network Analysis. Clin. Pharmacol. Ther. 2020, 109, 201–211. [Google Scholar] [CrossRef]
- Guest, E.J.; Aarons, L.; Houston, J.B.; Rostami-Hodjegan, A.; Galetin, A. Critique of the Two-Fold Measure of Prediction Success for Ratios: Application for the Assessment of Drug-Drug Interactions. Drug Metab. Dispos. 2011, 39, 170–173. [Google Scholar] [CrossRef] [Green Version]
- Les Laboratoires Servier. Servier Medical Art. Available online: https://smart.servier.com/ (accessed on 11 November 2020).
- Pearce, R.E.; Vakkalagadda, G.R.; Steven Leeder, J. Pathways of carbamazepine bioactivation in vitro I. Characterization of human cytochromes P450 responsible for the formation of 2- and 3-hydroxylated metabolites. Drug Metab. Dispos. 2002, 30, 1170–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faucette, S.R.; Zhang, T.C.; Moore, R.; Sueyoshi, T.; Omiecinski, C.J.; LeCluyse, E.L.; Negishi, M.; Wang, H. Relative activation of human pregnane X receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. J. Pharmacol. Exp. Ther. 2007, 320, 72–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichelbaum, M.; Tomson, T.; Tybring, G.; Bertilsson, L. Carbamazepine metabolism in man. Induction and pharmacogenetic aspects. Clin. Pharmacokinet. 1985, 10, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Drugbank. Carbamazepine. Available online: https://www.drugbank.ca/drugs/DB00564 (accessed on 9 February 2019).
- Austin, R.P.; Barton, P.; Cockroft, S.L.; Wenlock, M.C.; Riley, R.J. The influence of nonspecific microsomal binding on apparent intrinsic clearance, and its prediction from physicochemical properties. Drug Metab. Dispos. 2002, 30, 1497–1503. [Google Scholar] [CrossRef] [Green Version]
- Avdeef, A. Absorption and Drug Development; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; ISBN 978-1-11828-606-7. [Google Scholar]
- Annaert, P.; Brouwers, J.; Bijnens, A.; Lammert, F.; Tack, J.; Augustijns, P. Ex vivo permeability experiments in excised rat intestinal tissue and in vitro solubility measurements in aspirated human intestinal fluids support age-dependent oral drug absorption. Eur. J. Pharm. Sci. 2010, 39, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Clarysse, S.; Brouwers, J.; Tack, J.; Annaert, P.; Augustijns, P. Intestinal drug solubility estimation based on simulated intestinal fluids: Comparison with solubility in human intestinal fluids. Eur. J. Pharm. Sci. 2011, 43, 260–269. [Google Scholar] [CrossRef]
- Heikkilä, T.; Karjalainen, M.; Ojala, K.; Partola, K.; Lammert, F.; Augustijns, P.; Urtti, A.; Yliperttula, M.; Peltonen, L.; Hirvonen, J. Equilibrium drug solubility measurements in 96-well plates reveal similar drug solubilities in phosphate buffer pH 6.8 and human intestinal fluid. Int. J. Pharm. 2011, 405, 132–136. [Google Scholar] [CrossRef]
- Söderlind, E.; Karlsson, E.; Carlsson, A.; Kong, R.; Lenz, A.; Lindborg, S.; Sheng, J.J. Simulating fasted human intestinal fluids: Understanding the roles of lecithin and bile acids. Mol. Pharm. 2010, 7, 1498–1507. [Google Scholar] [CrossRef] [PubMed]
- Heumann Pharma GmbH & Co. Generica KG Fachinformation—Carbamazepin 200/400 Heumann. Available online: https://www.fachinfo.de/suche/fi/006667 (accessed on 30 November 2020).
- Novartis. Tegretol® Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/016608s101,018281s048lbl.pdf (accessed on 30 November 2020).
- Bertilsson, L. Clinical pharmacokinetics of carbamazepine. Clin. Pharmacokinet. 1978, 3, 128–1473. [Google Scholar] [CrossRef]
- Pynnönen, S. The Pharmacokinetics of Carbamazepine in Plasma and Saliva of Man. Acta Pharmacol. Toxicol. (Copenh.) 1977, 41, 465–471. [Google Scholar]
- Henshall, J.; Galetin, A.; Harrison, A.; Houston, J.B. Comparative analysis of CYP3A heteroactivation by steroid hormones and flavonoids in different in vitro systems and potential in vivo implications. Drug Metab. Dispos. 2008, 36, 1332–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazali, N.; Tran, A.; Treluyer, J.M.; Rey, E.; Athis, P.; Vincent, J.; Pons, G. Inhibitory effect of stiripentol on carbamazepine and saquinavir metabolism in human. Br. J. Clin. Pharmacol. 2003, 56, 526. [Google Scholar] [CrossRef]
- Huang, W.; Lin, Y.S.; McConn, D.J.; Calamia, J.C.; Totah, R.A.; Isoherranen, N.; Glodowski, M.; Thummel, K.E. Evidence of significant contribution from CYP3A5 to hepatic drug metabolism. Drug Metab. Dispos. 2004, 32, 1434–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staines, A.G.; Coughtrie, M.W.H.; Burchell, B. N-glucuronidation of carbamazepine in human tissues is mediated by UGT2B7. J. Pharmacol. Exp. Ther. 2004, 311, 1131–1137. [Google Scholar] [CrossRef] [Green Version]
- Shou, M.; Hayashi, M.; Pan, Y.; Xu, Y.; Morrissey, K.; Xu, L.; Skiles, G.L. Modeling, prediction, and in vitro in vivo correlation of CYP3A4 induction. Drug Metab. Dispos. 2008, 36, 2355–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGinnity, D.F.; Zhang, G.; Kenny, J.R.; Hamilton, G.A.; Otmani, S.; Stams, K.R.; Haney, S.; Brassil, P.; Stresser, D.M.; Riley, R.J. Evaluation of multiple in vitro systems for assessment of CYP3A4 induction in drug discovery: Human hepatocytes, pregnane X receptor reporter gene, and Fa2N-4 and HepaRG cells. Drug Metab. Dispos. 2009, 37, 1259–1268. [Google Scholar] [CrossRef] [Green Version]
- Fahmi, O.A.; Raucy, J.L.; Ponce, E.; Hassanali, S.; Lasker, J.M. Utility of DPX2 cells for predicting CYP3A induction-mediated drug-drug interactions and associated structure-activity relationships. Drug Metab. Dispos. 2012, 40, 2204–2211. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.G.; Ho, T.; Callendrello, A.L.; Clark, R.J.; Santone, E.A.; Kinsman, S.; Xiao, D.; Fox, L.G.; Einolf, H.J.; Stresser, D.M. Evaluation of calibration curve-based approaches to predict clinical inducers and noninducers of CYP3A4 with plated human hepatocytes. Drug Metab. Dispos. 2014, 42, 1379–1391. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.; Chothe, P.P.; Tsao, H.; Hariparsad, N. Evaluation of the interplay between uptake transport and CYP3A4 induction in micropatterned cocultured hepatocytes. Drug Metab. Dispos. 2016, 44, 1910–1919. [Google Scholar] [CrossRef] [Green Version]
- Fahmi, O.A.; Kish, M.; Boldt, S.; Scott Obach, R. Cytochrome P450 3A4 mRNA is a more reliable marker than CYP3A4 activity for detecting pregnane X receptor-activated induction of drug-metabolizing enzymes. Drug Metab. Dispos. 2010, 38, 1605–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, R.; Li, F.; Parikh, S.; Cao, L.; Cooper, K.L.; Hong, Y.; Liu, J.; Faris, R.A.; Li, D.; Wang, H. Evaluation of a novel renewable hepatic cell model for prediction of clinical CYP3A4 induction using a correlation-based relative induction score approach. Drug Metab. Dispos. 2017, 45, 198–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.G.; Patel, R.; Clark, R.J.; Ho, T.; Trisdale, S.K.; Fang, Y.; Stresser, D.M. Effect of Fifteen CYP3A4 in vitro Inducers on the Induction of Hepatocytes: A Trend Analysis. In Proceedings of the 20th North American ISSX Meeting, Orlando, FL, USA, 18–22 October 2015. [Google Scholar]
- Fahmi, O.A.; Shebley, M.; Palamanda, J.; Sinz, M.W.; Ramsden, D.; Einolf, H.J.; Chen, L.; Wang, H. Evaluation of CYP2B6 induction and prediction of clinical drug-drug interactions: Considerations from the iq consortium induction working group—An industry perspective. Drug Metab. Dispos. 2016, 44, 1720–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickmann, L.J.; Isoherranen, N. Quantitative prediction of CYP2B6 induction by estradiol during pregnancy: Potential explanation for increased methadone clearance during pregnancy. Drug Metab. Dispos. 2013, 41, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Lennernäs, H. Intestinal permeability and its relevance for absorption and elimination. Xenobiotica 2007, 37, 1015–1051. [Google Scholar] [CrossRef]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically based pharmacokinetic modeling 1: Predicting the tissue distribution of moderate-to strong bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef]
- Taylor, M.J.; Tanna, S.; Sahota, T. In vivo study of a polymeric glucose-sensitive insulin delivery system using a rat model. J. Pharm. Sci. 2010, 99, 4215–4227. [Google Scholar] [CrossRef]
- Kawai, R.; Lemaire, M.; Steimer, J.L.; Bruelisauer, A.; Niederberger, W.; Rowland, M. Physiologically based pharmacokinetic study on a cyclosporin derivative, SDZ IMM 125. J. Pharmacokinet. Biopharm. 1994, 22, 327–365. [Google Scholar] [CrossRef]
- Drugbank. Metabolite 10,11-Epoxycarbamazepine. Available online: https://www.drugbank.ca/metabolites/DBMET00291 (accessed on 16 December 2020).
- Morselli, P.L.; Gerna, M.; de Maio, D.; Zanda, G.; Viani, F.; Garattini, S. Pharmacokinetic studies on carbamazepine in volunteers and in epileptic patients. In Clinical Pharmacology of Anti-Epileptic Drugs; Schneider, H., Janz, D., Gardner-Thorpe, C., Meinardi, H., Sherwin, A.L., Eds.; Springer: Berlin, Germany, 1975; pp. 166–180. ISBN 978-3-64285-923-6. [Google Scholar]
- Achour, B.; Russell, M.R.; Barber, J.; Rostami-Hodjegan, A. Simultaneous quantification of the abundance of several cytochrome P450 and uridine 5′-diphospho-glucuronosyltransferase enzymes in human liver microsomes using multiplexed targeted proteomics. Drug Metab. Dispos. 2014, 42, 500–510. [Google Scholar] [CrossRef]
- Wada, J.A.; Troupin, A.S.; Friel, P.; Remick, R.; Leal, K.; Pearmain, J. Pharmacokinetic comparison of tablet and suspension dosage forms of carbamazepine. Epilepsia 1978, 19, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Sumi, M.; Watari, N.; Umezawa, O.; Kaneniwa, N. Pharmacokinetic study of carbamazepine and its epoxide metabolite in humans. J. Pharmacobiodyn. 1987, 10, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Bianchetti, G.; Padovani, P.; Thénot, J.P.; Thiercelin, J.F.; Morselli, P.L. Pharmacokinetic interactions of progabide with other antiepileptic drugs. Epilepsia 1987, 28, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Pynnönen, S. Pharmacokinetics of Carbamazepine in man: A review. Ther. Drug Monit. 1979, 1, 409–431. [Google Scholar] [CrossRef] [PubMed]
- Bernus, I.; Dickinson, R.G.; Hooper, W.D.; Eadie, M.J. Early stage autoinduction of carbamazepine metabolism in humans. Eur. J. Clin. Pharmacol. 1994, 47, 355–360. [Google Scholar] [CrossRef]
- Gérardin, A.P.; Abadie, F.V.; Campestrini, J.A.; Theobald, W. Pharmacokinetics of carbamazepine in normal humans after single and repeated oral doses. J. Pharmacokinet. Biopharm. 1976, 4, 521–535. [Google Scholar] [CrossRef]
- Møller, S.E.; Larsen, F.; Khan, A.Z.; Rolan, P.E. Lack of effect of citalopram on the steady-state pharmacokinetics of carbamazepine in healthy male subjects. J. Clin. Psychopharmacol. 2001, 21, 493–499. [Google Scholar] [CrossRef]
- Stevens, R.E.; Limsakun, T.; Evans, G.; Mason, D.H. Controlled, multidose, pharmacokinetic evaluation of two extended-release carbamazepine formulations (Carbatrol and Tegretol-XR). J. Pharm. Sci. 1998, 87, 1531–1534. [Google Scholar] [CrossRef]
- Gande, M.; Gondalia, R.; Kothapalli, M.; Velishala, N.M.; Koppuri, V. Carbamazepine Extended Release Dosage Form. US Patent US 2009/01696.19A1, 2 July 2009. [Google Scholar]
- Barzaghi, N.; Gatti, G.; Crema, F.; Monteleone, M.; Amione, C.; Leone, L.; Perucca, E. Inhibition by erythromycin of the conversion of carbamazepine to its active 10,11-epoxide metabolite. Br. J. Clin. Pharmacol. 1987, 24, 836–838. [Google Scholar] [CrossRef] [Green Version]
- Miles, M.V.; Tennison, M.B. Erythromycin Effects on Multiple-Dose Carbamazepine Kinetics. Ther. Drug Monit. 1989, 11, 47–52. [Google Scholar] [CrossRef]
- Furukori, H. Effect of Carbamazepine on the Single Oral Dose Pharmacokinetics of Alprazolam. Neuropsychopharmacology 1998, 18, 364–369. [Google Scholar] [CrossRef] [Green Version]
- Tomson, T.; Tybring, G.; Bertilsson, L. Single-dose kinetics and metabolism of carbamazepine-10,11-epoxide. Clin. Pharmacol. Ther. 1983, 33, 58–65. [Google Scholar] [CrossRef]
- Václavíková, R.; Hughes, D.J.; Souček, P. Microsomal epoxide hydrolase 1 (EPHX1): Gene, structure, function, and role in human disease. Gene 2015, 571, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Kwan, P.; Zuo, Z.; Baum, L. The transport of antiepileptic drugs by P-glycoprotein. Adv. Drug Deliv. Rev. 2012, 64, 930–942. [Google Scholar] [CrossRef]
- Zhang, C.; Zuo, Z.; Kwan, P.; Baum, L. In vitro transport profile of carbamazepine, oxcarbazepine, eslicarbazepine acetate, and their active metabolites by human P-glycoprotein. Epilepsia 2011, 52, 1894–1904. [Google Scholar] [CrossRef]
- Luna-Tortós, C.; Fedrowitz, M.; Löscher, W. Several major antiepileptic drugs are substrates for human P-glycoprotein. Neuropharmacology 2008, 55, 1364–1375. [Google Scholar] [CrossRef]
- Owen, A.; Pirmohamed, M.; Tettey, J.N.; Morgan, P.; Chadwick, D.; Kevin Park, B. Carbamazepine is not a substrate for P-glycoprotein. Br. J. Clin. Pharmacol. 2001, 51, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.M.; Cheng, J. Effects of MDR1 (C3435T) Polymorphism on Resistance, Uptake, and Efflux to Antiepileptic Drugs. DNA Cell. Biol. 2019, 38, 250–255. [Google Scholar] [CrossRef]
- Shirasaka, Y.; Sakane, T.; Yamashita, S. Effect of P-glycoprotein expression levels on the concentration-dependent permeability of drugs to the cell membrane. J. Pharm. Sci. 2008, 97, 553–565. [Google Scholar] [CrossRef]
- Ward, B.A.; Gorski, J.C.; Jones, D.R.; Hall, S.D.; Flockhart, D.A.; Desta, Z. The cytochrome P450 2B6 (CYP2B6) is the main catalyst of efavirenz primary and secondary metabolism: Implication for HIV/AIDS therapy and utility of efavirenz as a substrate marker of CYP2B6 catalytic activity. J. Pharmacol. Exp. Ther. 2003, 306, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Kohlmann, P.; Stillhart, C.; Kuentz, M.; Parrott, N. Investigating Oral Absorption of Carbamazepine in Pediatric Populations. AAPS J. 2017, 19, 1864–1877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lionberger, R.A.; Davit, B.M.; Yu, L.X. Utility of physiologically based absorption modeling in implementing quality by design in drug development. AAPS J. 2011, 13, 59–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicali, B.; Lingineni, K.; Cristofoletti, R.; Wendl, T.; Hoechel, J.; Wiesinger, H.; Chaturvedula, A.; Vozmediano, V.; Schmidt, S. Quantitative Assessment of Levonorgestrel Binding Partner Interplay and Drug-Drug Interactions Using Physiologically Based Pharmacokinetic Modeling. CPT Pharmacomet. Syst. Pharmacol. 2021, 10(1), 48–58. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Yasui-Furukori, N.; Akamine, Y.; Kaneko, S.; Uno, T. Effects of the P-glycoprotein inducer carbamazepine on fexofenadine pharmacokinetics. Ther. Drug Monit. 2009, 31, 764–768. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Unit | Model | Literature | Reference |
|---|---|---|---|---|
| Carbamazepine | ||||
| Molecular weight | g/mol | 236.27 (Lit) | 236.27 | [38] |
| Lipophilicity | Log Units | 2.00 (Fit) | 1.45; 2.1; 2.45; 2.77 | [38,39,40] |
| Solubility (FaHIF) | µg/mL | 336 (Lit) | 170; 283; 306; 336 | [41,42,43,44] |
| Fraction unbound | % | 25 (Lit) | 21; 24; 25 | [45,46,47,48] |
| Km (CYP3A4) → CBZ-E | µM | 248 (Lit) | 119; 248; 442; 630 | [2,49,50,51] |
| kcat (CYP3A4) → CBZ-E | 1/min | 0.75 (Fit) | 1.17; 1.7; 4.87; 5.3 b | [2,49,50,51] |
| Km (CYP2C8) → CBZ-E | µM | 757 (Lit) | 757 | [50] |
| kcat (CYP2C8) → CBZ-E | 1/min | 0.67 (Lit) | 0.67 b | [50] |
| Km (CYP3A4) | µM | 282 (Lit) | 282 | [35] |
| kcat (CYP3A4) | 1/min | 0.20 (Fit) | 0.16 b | [35] |
| Km (CYP2B6) | µM | 420 (Lit) | 420 | [35] |
| kcat (CYP2B6) | 1/min | 0.43 (Lit) | 0.43 b | [35] |
| Km (UGT2B7) | µM | 214 (Lit) | 214 | [52] |
| kcat (UGT2B7) | 1/min | 9.53 × 10−3 (Lit) | 9.53 × 10−3 c | [52] |
| CLhep | 1/min | 0.02 (Fit) | - | - |
| GFR fraction | - | 0.03 (Fit) | - | - |
| EC50 (CYP3A4) | µM | 20.00 a (Lit) | 4.3–137 | [53,54,55,56,57,58,59,60] |
| Emax (CYP3A4) | - | 6.00 (Fit) | 1.9–23 | [53,54,55,56,57,58,59,60] |
| EC50 (CYP2B6) | µM | 20.00 a (Asm) | 22–145 | [60,61,62] |
| Emax (CYP2B6) | - | 17.00 (Fit) | 3.1–21.5 | [60,61,62] |
| EC50 (EPHX1) | µM | 20.00 a (Asm) | - | - |
| Emax (EPHX1) | - | 3.25 (Fit) | - | - |
| Intestinal permeability | cm/s | 4.3 × 10−4 (Lit) | 4.3 × 10−4 | [63] |
| Partition coefficients | - | Rodgers and Rowlands | [64,65] | |
| Cellular permeabilities | cm/s | PK-Sim Standard | [66] | |
| Carbamazepine-10,11-epoxide | ||||
| Molecular weight | g/mol | 252.27 (Lit) | 252.27 | [67] |
| Lipophilicity | Log Units | 1.00 (Fit) | 1.58; 1.97 | [67] |
| Solubility | µg/mL | 1340 (Lit) | 1340 | [67] |
| Fraction unbound | % | 51.8 (Lit) | 46.8; 49.0; 47.0; 51.8; 50.0 | [68] |
| CLspec (EPHX1) | 1/min | 0.01 (Fit) | - | - |
| GFR fraction | - | 0.21 (Fit) | - | - |
| Intestinal permeability | cm/s | 5.0 × 10−3 (Fit) | - | - |
| Partition coefficients | - | Rodgers and Rowlands | [64,65] | |
| Cellular permeabilities | cm/s | PK-Sim Standard | [66] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuhr, L.M.; Marok, F.Z.; Hanke, N.; Selzer, D.; Lehr, T. Pharmacokinetics of the CYP3A4 and CYP2B6 Inducer Carbamazepine and Its Drug–Drug Interaction Potential: A Physiologically Based Pharmacokinetic Modeling Approach. Pharmaceutics 2021, 13, 270. https://doi.org/10.3390/pharmaceutics13020270
Fuhr LM, Marok FZ, Hanke N, Selzer D, Lehr T. Pharmacokinetics of the CYP3A4 and CYP2B6 Inducer Carbamazepine and Its Drug–Drug Interaction Potential: A Physiologically Based Pharmacokinetic Modeling Approach. Pharmaceutics. 2021; 13(2):270. https://doi.org/10.3390/pharmaceutics13020270
Chicago/Turabian StyleFuhr, Laura Maria, Fatima Zahra Marok, Nina Hanke, Dominik Selzer, and Thorsten Lehr. 2021. "Pharmacokinetics of the CYP3A4 and CYP2B6 Inducer Carbamazepine and Its Drug–Drug Interaction Potential: A Physiologically Based Pharmacokinetic Modeling Approach" Pharmaceutics 13, no. 2: 270. https://doi.org/10.3390/pharmaceutics13020270
APA StyleFuhr, L. M., Marok, F. Z., Hanke, N., Selzer, D., & Lehr, T. (2021). Pharmacokinetics of the CYP3A4 and CYP2B6 Inducer Carbamazepine and Its Drug–Drug Interaction Potential: A Physiologically Based Pharmacokinetic Modeling Approach. Pharmaceutics, 13(2), 270. https://doi.org/10.3390/pharmaceutics13020270