
An In Vivo Predictive Dissolution Methodology (iPD Methodology) with a BCS Class IIb Drug Can Predict the In Vivo Bioequivalence Results: Etoricoxib Products

 ,
,  ,
,  ,
,  , ,
, ,
Abstract
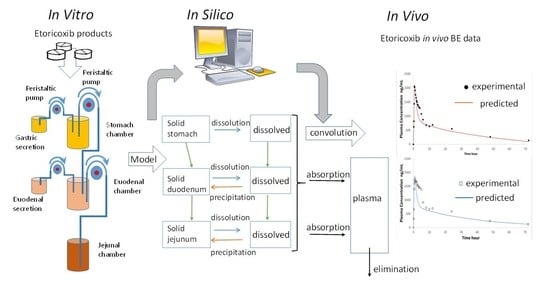
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. In Vivo Studies
2.3. Dissolution Experiments in GIS
2.4. HPLC Analytical Method
2.5. Analysis of the Mass Transport of ETO throughout the GIS
2.6. In Silico Simulations to Predict the Pharmacokinetic (PK) Profiles of ETO
3. Results
3.1. Performance of the ETO Products in the GIS
3.2. In Silico PK Model to Forecast the Systemic Performance of Oral Products
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FDA. FDA Guidance for Industry. Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs—General Considerations; FDA: Silver Spring, MD, USA, 2014. [Google Scholar]
- ICH Harmonissed Guideline—Biopharmaceutics Classification System-Based Biowaivers M9. Available online: https://database.ich.org/sites/default/files/M9_Guideline_Step4_2019_1116.pdf (accessed on 29 December 2020).
- Carino, S.R.; Sperry, D.C.; Hawley, M. Relative bioavailability estimation of carbamazepine crystal forms using an artificial stomach-duodenum model. J. Pharm. Sci. 2006, 95, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Tsume, Y.; Amidon, G.L.; Takeuchi, S. Dissolution effect of gastric and intestinal pH fora BCS class II drug, pioglitazone: New in vitro dissolution system to predict in vivo dissolution. J. Bioequiv. Bioavailab. 2013, 5, 224–227. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, S.; Tsume, Y.; Amidon, G.E.; Amidon, G.L. Evaluation of a three compartment in Vitro gastrointestinal simulator dissolution apparatus to predict in Vivo dissolution. J. Pharm. Sci. 2014, 103, 3416–3422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, A.; Kesisoglou, F.; Dogterom, P. Application of Absorption Modeling to Predict Bioequivalence Outcome of Two Batches of Etoricoxib Tablets. AAPS PharmSciTech 2014, 16, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Okumu, A.; DiMaso, M.; Löbenberg, R. Computer simulations using GastroPlusTM to justify a biowaiver for etoricoxib solid oral drug products. Eur. J. Pharm. Biopharm. 2009, 72, 91–98. [Google Scholar] [CrossRef]
- Agrawal, N.G.B.; Porras, A.G.; Matthews, C.Z.; Rose, M.J.; Woolf, E.J.; Musser, B.J.; Dynder, A.L.; Mazina, K.E.; Lasseter, K.C.; Hunt, T.L.; et al. Single- and multiple-dose pharmacokinetics of etoricoxib, a selective inhibitor of cyclooxygenase-2, in man. J. Clin. Pharmacol. 2003, 43, 268–276. [Google Scholar] [CrossRef]
- Rodrigues, A.D.; Halpin, R.A.; Geer, L.A.; Cui, D.; Woolf, E.J.; Matthews, C.Z.; Gottesdiener, K.M.; Larson, P.J.; Lasseter, K.C.; Agrawal, N.G.B. Absorption, metabolism, and excretion of etoricoxib, a potent and selective cyclooxygenase-2 inhibitor, in healthy male volunteers. Drug Metab. Dispos. 2003, 31, 224–232. [Google Scholar] [CrossRef] [Green Version]
- Takemoto, J.K.; Reynolds, J.K.; Remsberg, C.M.; Vega-Villa, K.R.; Davies, N.M. Clinical pharmacokinetic and pharmacodynamic profile of etoricoxib. Clin. Pharmacokinet. 2008, 47, 703–720. [Google Scholar] [CrossRef]
- Matsui, K.; Tsume, Y.; Amidon, G.E.; Amidon, G.L. In Vitro Dissolution of Fluconazole and Dipyridamole in Gastrointestinal Simulator (GIS), Predicting in Vivo Dissolution and Drug–Drug Interaction Caused by Acid-Reducing Agents. Mol. Pharm. 2015, 12, 2418–2428. [Google Scholar] [CrossRef]
- Tsume, Y.; Takeuchi, S.; Matsui, K.; Amidon, G.E.; Amidon, G.L. In vitro dissolution methodology, mini-Gastrointestinal Simulator (mGIS), predicts better in vivo dissolution of a weak base drug, dasatinib. Eur. J. Pharm. Sci. 2015. [Google Scholar] [CrossRef]
- Matsui, K.; Tsume, Y.; Amidon, G.E.; Amidon, G.L. The Evaluation of In Vitro Drug Dissolution of Commercially Available Oral Dosage Forms for Itraconazole in Gastrointestinal Simulator With Biorelevant Media. J. Pharm. Sci. 2016. [Google Scholar] [CrossRef] [Green Version]
- Chemicalize—Instant Cheminformatics Solutions Available online:. Available online: https://chemicalize.com/app/calculation/etoricoxib (accessed on 16 December 2020).
- González-García, I.; Mangas-Sanjuan, V.; Merino-Sanjuán, M.; Álvarez-Álvarez, C.; Díaz-Garzón Marco, J.; Rodrí-Guez-Bonnín, M.A.; Langguth, T.; Torrado-Durán, J.J.; Langguth, P.; García-Arieta, A.; et al. IVIVC approach based on carbamazepine bioequivalence studies combination. Pharmazie 2017, 72, 449–455. [Google Scholar] [CrossRef]
- Ruiz Picazo, A.; Martinez-Martinez, M.T.T.; Colón-Useche, S.; Iriarte, R.; Sánchez-Dengra, B.; González-Álvarez, M.; García-Arieta, A.; González-Álvarez, I.; Bermejo, M. In Vitro Dissolution as a Tool for Formulation Selection: Telmisartan Two-Step IVIVC. Mol. Pharm. 2018, 15, 2307–2315. [Google Scholar] [CrossRef]
- Hens, B.; Bermejo, M.; Tsume, Y.; Gonzalez-Alvarez, I.; Ruan, H.; Matsui, K.; Amidon, G.E.; Cavanagh, K.L.; Kuminek, G.; Benninghoff, G.; et al. Evaluation and optimized selection of supersaturating drug delivery systems of posaconazole (BCS class 2b) in the gastrointestinal simulator (GIS): An in vitro-in silico-in vivo approach. Eur. J. Pharm. Sci. 2018, 115, 258–269. [Google Scholar] [CrossRef]
- Hens, B.; Brouwers, J.; Anneveld, B.; Corsetti, M.; Symillides, M.; Vertzoni, M.; Reppas, C.; Turner, D.B.; Augustijns, P. Gastrointestinal transfer: In vivo evaluation and implementation in in vitro and in silico predictive tools. Eur. J. Pharm. Sci. 2014, 63, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Matsui, K.; Tsume, Y.; Takeuchi, S.; Searls, A.; Amidon, G.L. Utilization of Gastrointestinal Simulator, an in Vivo Predictive Dissolution Methodology, Coupled with Computational Approach To Forecast Oral Absorption of Dipyridamole. Mol. Pharm. 2017, 14, 1181–1189. [Google Scholar] [CrossRef]
- Bermejo, M.; Kuminek, G.; Al-Gousous, J.; Ruiz-Picazo, A.; Tsume, Y.; Garcia-Arieta, A.; González-Alvarez, I.; Hens, B.; Amidon, G.E.; Rodriguez-Hornedo, N.; et al. Exploring bioequivalence of dexketoprofen trometamol drug products with the gastrointestinal simulator (GIS) and precipitation pathways analyses. Pharmaceutics 2019, 11, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, M.B.; Fisher, M.B.; Dabir-Vaziri, N.; Schaffer, M. Effect of protein intake and dialysis on the abnormal growth hormone, glucose, and insulin homeostasis in uremia. Metabolism 1976, 25, 455–464. [Google Scholar] [CrossRef]
- Matsumura, N.; Ono, A.; Akiyama, Y.; Fujita, T.; Sugano, K. Bottom-up physiologically based oral absorption modeling of free weak base drugs. Pharmaceutics 2020, 12, 844. [Google Scholar] [CrossRef]
- Schwartz, J.I.; Agrawal, N.G.B.; Kher, U.A.; DeSmet, M.; Cavanaugh, P.F.; Guillaume, M.; Ebel, D.L.; Merschman, S.A.; Wagner, J.A. Lack of effect of antacids on single-dose pharmacokinetics of etoricoxib. J. Clin. Pharmacol. 2007, 47, 1342–1346. [Google Scholar] [CrossRef]
- Warren, D.B.; Bergström, C.A.S.; Benameur, H.; Porter, C.J.H.; Pouton, C.W. Evaluation of the structural determinants of polymeric precipitation inhibitors using solvent shift methods and principle component analysis. Mol. Pharm. 2013, 10, 2823–2848. [Google Scholar] [CrossRef] [PubMed]
- Mudie, D.M.; Samiei, N.; Marshall, D.J.; Amidon, G.E.; Bergström, C.A.S. Selection of In Vivo Predictive Dissolution Media Using Drug Substance and Physiological Properties. AAPS J. 2020, 22, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Study 1 Failed to Conclude BE | Study 2 BE Concluded | ||
|---|---|---|---|---|
| Parameter | Ratio Test/Reference | 90% Confidence Interval | Ratio Test/Reference | 90% Confidence Interval |
| Cmax | 118.20 | 111.26–125.57 | 112.57 | 104.27–121.54 |
| AUC0–72h | 100.48 | 97.54–103.50 | 102.96 | 99.10–106.97 |
| Fasted State Test Conditions | GISStomach | GISDuodenum | GISJejunum |
|---|---|---|---|
| Dissolution Media | Simulated Gastric Fluid (SGF), pH 2.0, 0.01 M HCl + 34.2 mM NaCL | Phosphate Buffer 5 mM pH 6.5 | / |
| Initial Volume | 50 mL SGF + 250 mL of water | 50 mL | / |
| Secretions | 1 mL/min of SGF | 1 mL/min of Phosphate Buffer 100 mM pH 6.5 | / |
| Parameter | ETO NoBE | ETO BE | Reference Product | Reference |
|---|---|---|---|---|
| Dose (mg) | 120 | 120 | 120 | |
| ksec_s (mL/min) | 1 | 1 | 1 | [20] |
| ksec_d (mL/min) | 1 | 1 | 1 | [20] |
| t1/2,G (min) | 13 | 13 | 13 | [20] |
| Vs (mL) | 300 to 5 | 300 to 5 | 300 to 5 | [20] |
| Vd (mL) | 50 | 50 | 50 | [20] |
| Vj (mL) | 0 to 390 | 0 to 390 | 0 to 390 | [20] |
| Z (mL/mg/min) | 3.51 × 10−5 | 3.10 × 10−5 | 1.45 × 10−5 | Optimized by fitting |
| kpre (min−1) | 3.18 × 10−2 | 8.30 × 10−2 | 7.84 × 10−2 | Optimized by fitting |
| Cs mg/mL pH 2.0 | 13.21 | 13.21 | 13.21 | [6,7] |
| Cs mg/mL pH 4.5 | 0.44 | 0.44 | 0.44 | [7] |
| Cs mg/mL pH 6.8 | 0.14 | 0.14 | 0.14 | [7] |
| Pharmacokinetic (PK) Parameters | Value |
|---|---|
| Central compartment volume—Vc (L) Average value from [6,9,21] | 27.40 |
| Elimination rate constant from central compartment—ke (h−1) [6] | 0.0899 |
| Distribution rate constant from central to peripheral compartment—k12 (h−1) [6] | 0.6180 |
| Distribution rate constant from peripheral to central compartment—k21 (h−1) [6] | 0.2820 |
| Effective Permeability Small Intestine—Peff (cm/h) [6] | 1.71 |
| Parameter | Reference | BE | NoBE |
|---|---|---|---|
| Cmax experimental ng/mL | 1657 | 1762 | 2060 |
| Cmax predicted ng/mL | 1583 | 1866 | 2017 |
| PE % | 4.4 | −5.9 | 2.1 |
| Cmax Ratio | Ratio Predicted | Ratio Experimental |
|---|---|---|
| Test BE | 1.18 | 1.12 |
| Test NoBE | 1.27 | 1.18 |
| AUC(0–72) ng/mL × h | Reference | BE | NoBE |
|---|---|---|---|
| Experimental | 38,101 | 38,693 | 38,749 |
| Predicted | 28,009 | 32,303 | 32,854 |
| Ratio exp. | 1.02 | 1.02 | |
| Ratio pred. | 1.15 | 1.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez-Alvarez, I.; Bermejo, M.; Tsume, Y.; Ruiz-Picazo, A.; Gonzalez-Alvarez, M.; Hens, B.; Garcia-Arieta, A.; Amidon, G.E.; Amidon, G.L. An In Vivo Predictive Dissolution Methodology (iPD Methodology) with a BCS Class IIb Drug Can Predict the In Vivo Bioequivalence Results: Etoricoxib Products. Pharmaceutics 2021, 13, 507. https://doi.org/10.3390/pharmaceutics13040507
Gonzalez-Alvarez I, Bermejo M, Tsume Y, Ruiz-Picazo A, Gonzalez-Alvarez M, Hens B, Garcia-Arieta A, Amidon GE, Amidon GL. An In Vivo Predictive Dissolution Methodology (iPD Methodology) with a BCS Class IIb Drug Can Predict the In Vivo Bioequivalence Results: Etoricoxib Products. Pharmaceutics. 2021; 13(4):507. https://doi.org/10.3390/pharmaceutics13040507
Chicago/Turabian StyleGonzalez-Alvarez, Isabel, Marival Bermejo, Yasuhiro Tsume, Alejandro Ruiz-Picazo, Marta Gonzalez-Alvarez, Bart Hens, Alfredo Garcia-Arieta, Greg E. Amidon, and Gordon L. Amidon. 2021. "An In Vivo Predictive Dissolution Methodology (iPD Methodology) with a BCS Class IIb Drug Can Predict the In Vivo Bioequivalence Results: Etoricoxib Products" Pharmaceutics 13, no. 4: 507. https://doi.org/10.3390/pharmaceutics13040507
APA StyleGonzalez-Alvarez, I., Bermejo, M., Tsume, Y., Ruiz-Picazo, A., Gonzalez-Alvarez, M., Hens, B., Garcia-Arieta, A., Amidon, G. E., & Amidon, G. L. (2021). An In Vivo Predictive Dissolution Methodology (iPD Methodology) with a BCS Class IIb Drug Can Predict the In Vivo Bioequivalence Results: Etoricoxib Products. Pharmaceutics, 13(4), 507. https://doi.org/10.3390/pharmaceutics13040507