
PK/PD Modeling of the PDE7 Inhibitor—GRMS-55 in a Mouse Model of Autoimmune Hepatitis

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Experimental Procedures
2.3.1. Pharmacokinetic Study
2.3.2. ConA-induced Hepatitis and Compound Administration
2.3.3. Assessment of Emetogenicity
2.3.4. Experimental Design and Sample Collection
2.4. Analytical Methods
2.5. Data and Statistical Analysis
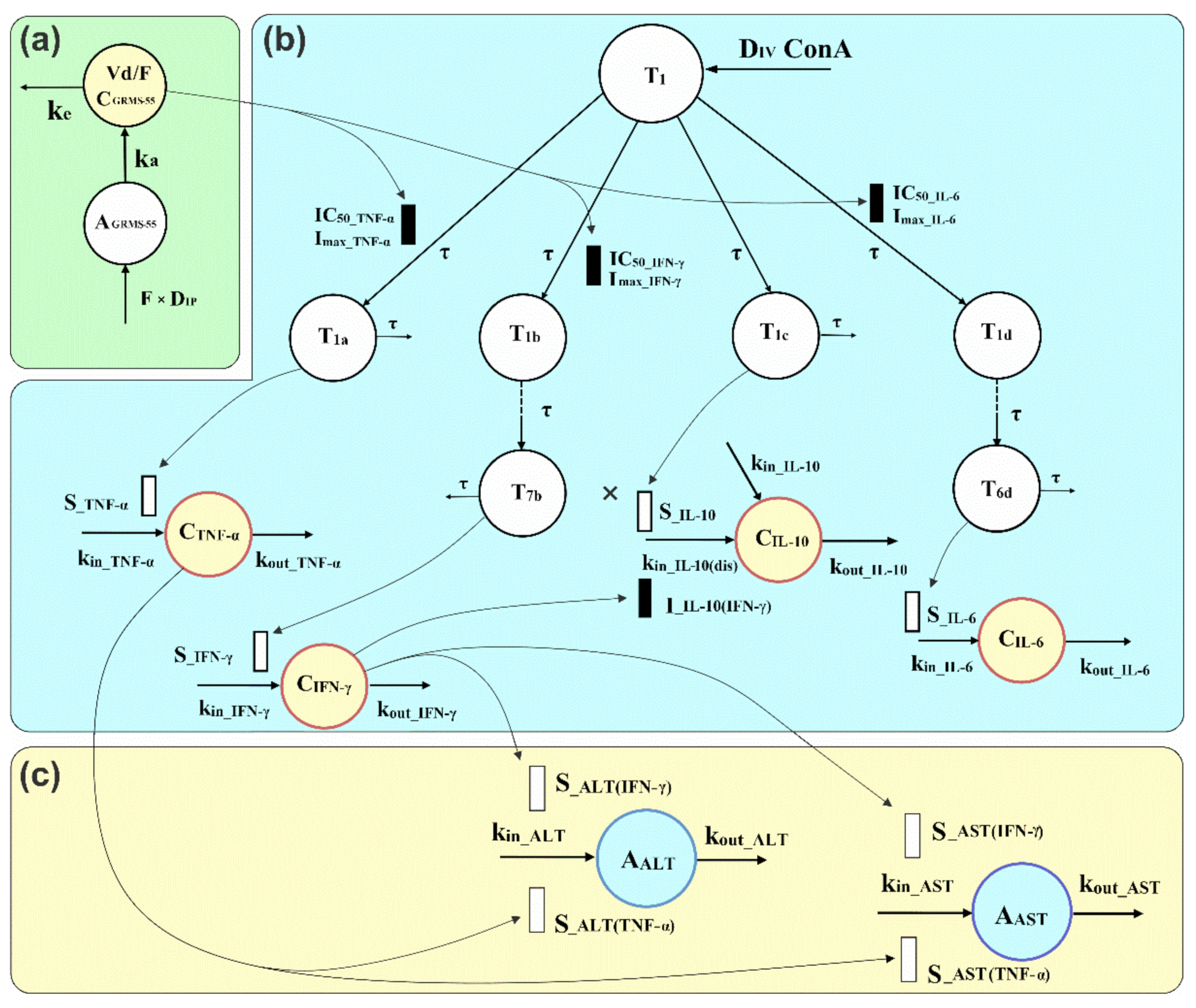
2.5.1. Pharmacokinetics and PK/PD/Disease Progression Modeling
2.5.2. Statistical Analysis
3. Results
3.1. Comparative Assessment of PDE Inhibitors in ConA-Induced Hepatitis
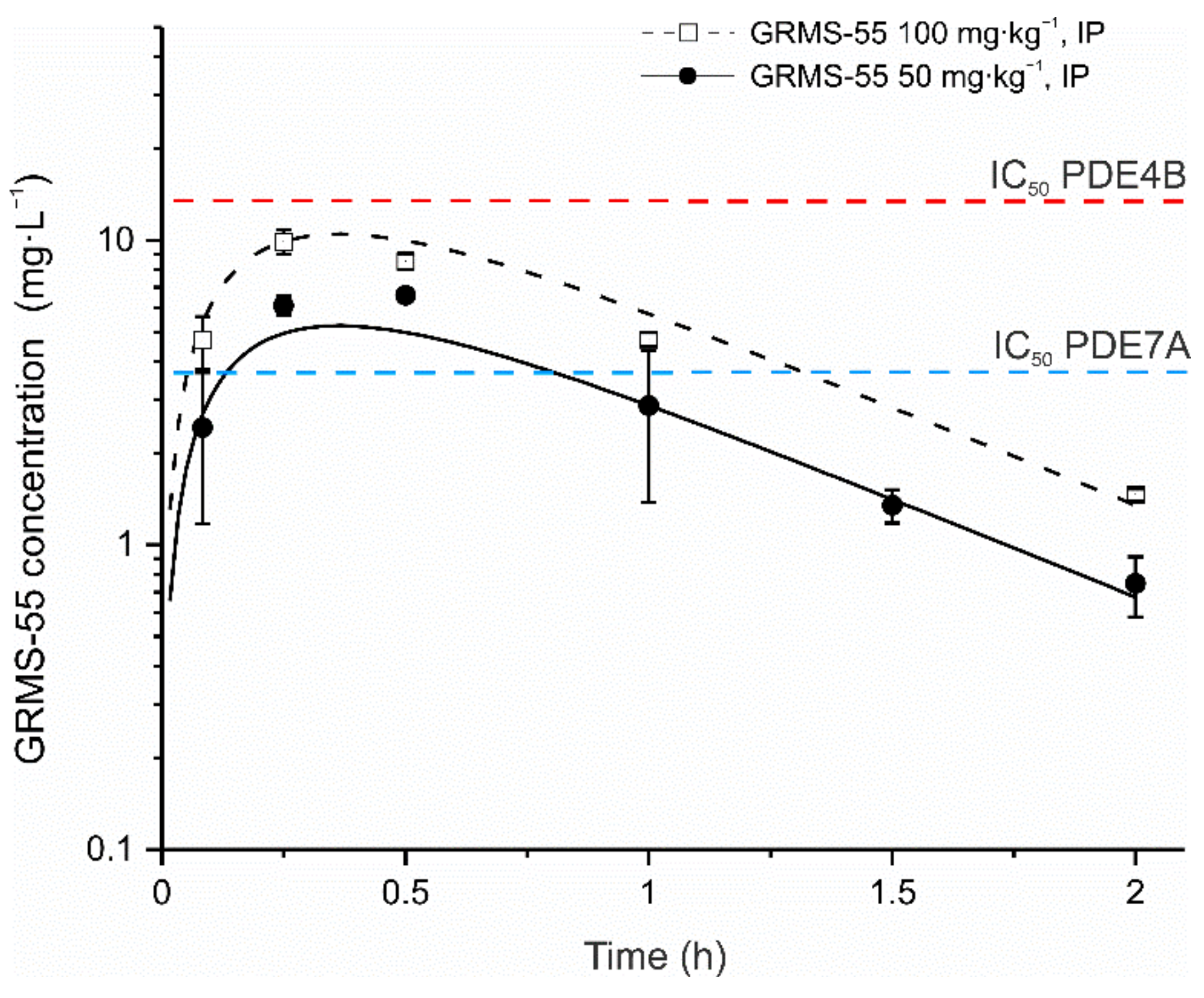
3.2. Pharmacokinetics of GRMS-55
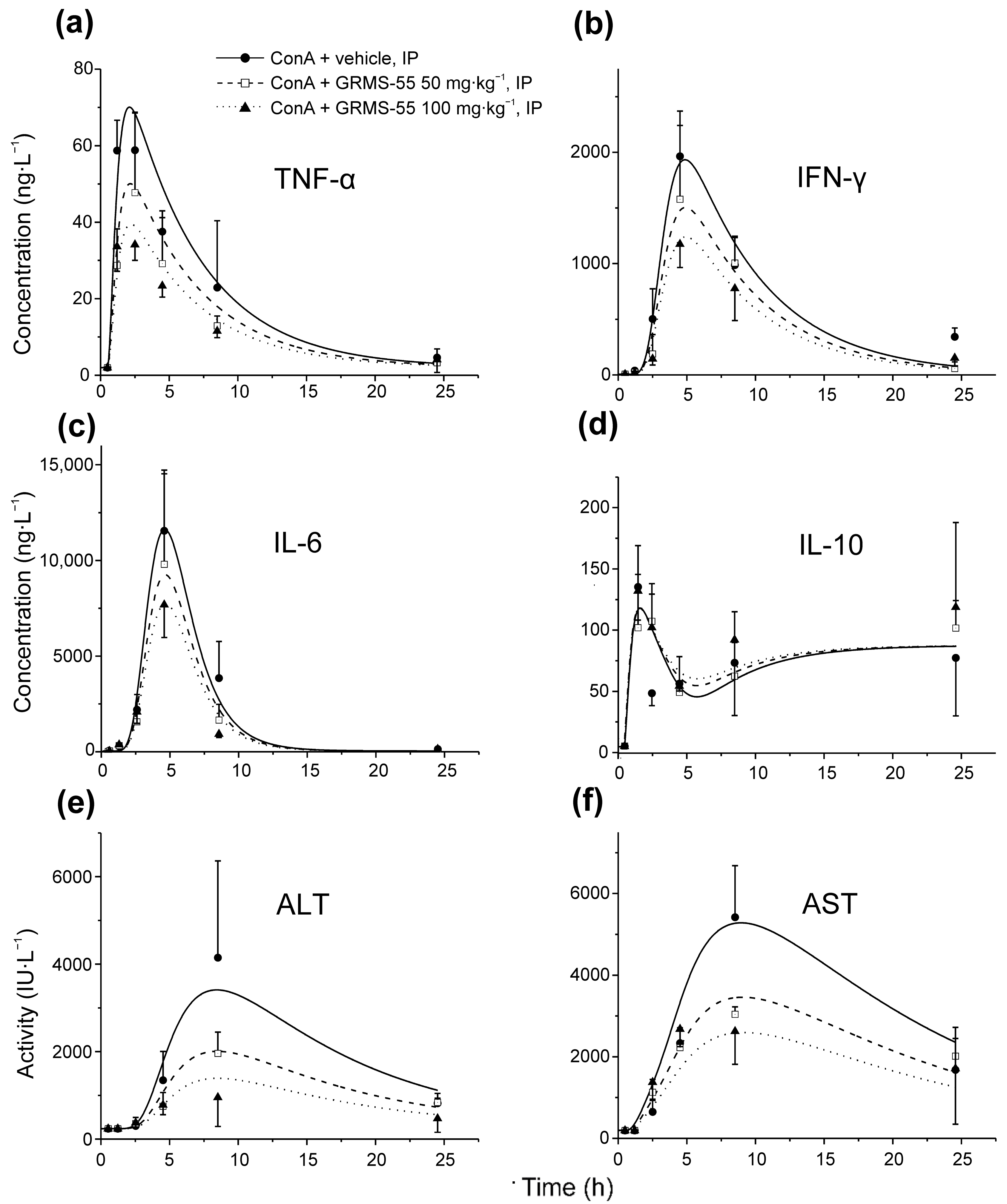
3.3. Pharmacodynamics of GRMS-55 and Hepatitis Progression
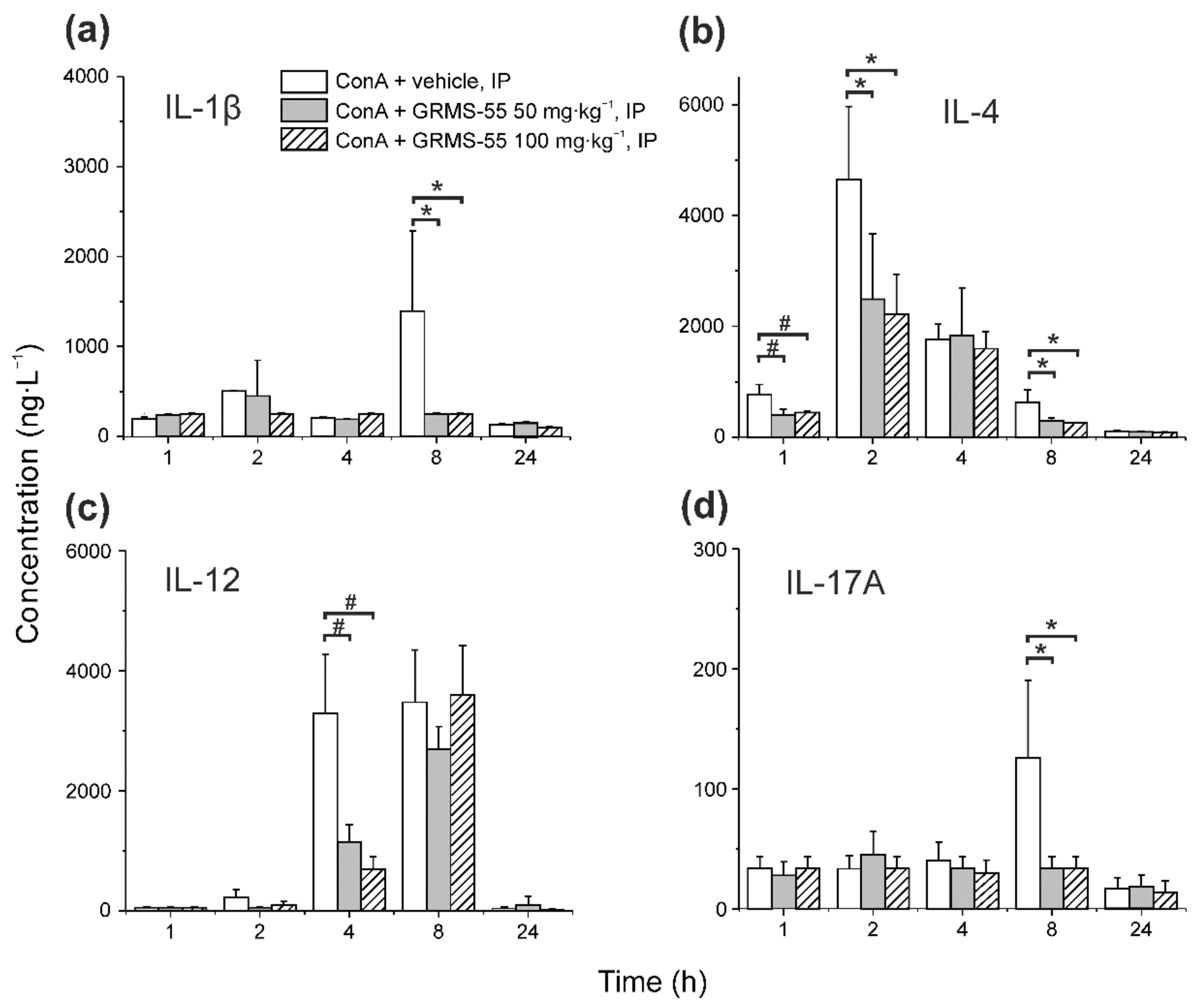
3.4. Influence of GRMS-55 Administration on the Release of Other Relevant Cytokines in ConA-Induced Hepatitis
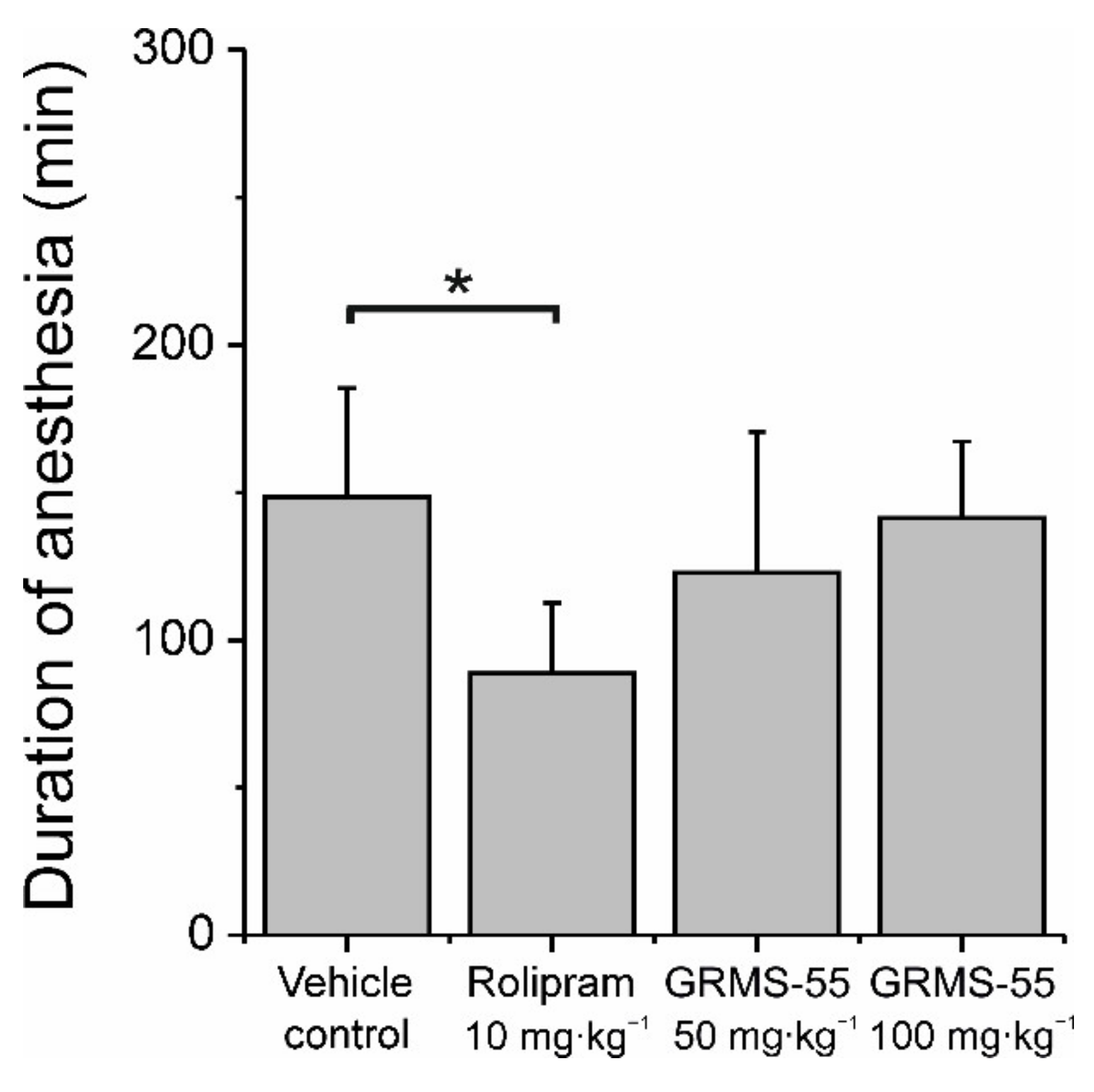
3.5. Assessment of Emetogenicity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gatselis, N.K.; Zachou, K.; Koukoulis, G.K.; Dalekos, G.N. Autoimmune Hepatitis, One Disease with Many Faces: Etiopathogenetic, Clinico-Laboratory and Histological Characteristics. World J. Gastroenterol. 2015, 21, 60–83. [Google Scholar] [CrossRef]
- Czaja, A.J. Diagnosis and Management of Autoimmune Hepatitis: Current Status and Future Directions. Gut Liver 2016, 10, 177–203. [Google Scholar] [CrossRef] [Green Version]
- Cropley, A.; Weltman, M. The Use of Immunosuppression in Autoimmune Hepatitis: A Current Literature Review. Clin. Mol. Hepatol. 2017, 23, 22–26. [Google Scholar] [CrossRef] [Green Version]
- Beretta-Piccoli, B.T.; Mieli-Vergani, G.; Vergani, D. Autoimmune Hepatitis: Standard Treatment and Systematic Review of Alternative Treatments. World J. Gastroenterol. 2017, 23, 6030–6048. [Google Scholar] [CrossRef] [PubMed]
- Houslay, M.D.; Schafer, P.; Zhang, K.Y. Keynote Review: Phosphodiesterase-4 as a Therapeutic Target. Drug Discov. Today 2005, 10, 1503–1519. [Google Scholar] [CrossRef]
- Jankowska, A.; Świerczek, A.; Chłoń-Rzepa, G.; Pawłowski, M.; Wyska, E. PDE7-Selective and Dual Inhibitors: Advances in Chemical and Biological Research. Curr. Med. Chem. 2017, 24, 673–700. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Sugahara, S.; Naito, R.; Ichikawa, A.; Ikeda, K.; Yamada, T.; Shimizu, Y. The Effects of a Novel Phosphodiesterase 7A and -4 Dual Inhibitor, YM-393059, on T-Cell-Related Cytokine Production in Vitro and in Vivo. Eur. J. Pharmacol. 2006, 541, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Świerczek, A.; Wyska, E.; Baś, S.; Woyciechowska, M.; Mlynarski, J. PK/PD Studies on Non-Selective PDE Inhibitors in Rats Using cAMP as a Marker of Pharmacological Response. Naunyn Schmiedeberg′s. Arch. Pharmacol. 2017, 390, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Giembycz, M.A.; Corrigan, C.J.; Seybold, J.; Newton, R.; Barnes, P.J. Identification of Cyclic AMP Phosphodiesterases 3, 4 and 7 in Human CD4+ and CD8+ T-Lymphocytes: Role in Regulating Proliferation and the Biosynthesis of Interleukin-2. Br. J. Pharmacol. 1996, 118, 1945–1958. [Google Scholar] [CrossRef] [Green Version]
- Świerczek, A.; Pociecha, K.; Ślusarczyk, M.; Chłoń-Rzepa, G.; Baś, S.; Mlynarski, J.; Więckowski, K.; Zadrożna, M.; Nowak, B.; Wyska, E. Comparative Assessment of the New PDE7 Inhibitor—GRMS-55 and Lisofylline in Animal Models of Immune-Related Disorders: A PK/PD Modeling Approach. Pharm. Res. 2020, 37. [Google Scholar] [CrossRef] [Green Version]
- Chłoń-Rzepa, G.; Jankowska, A.; Ślusarczyk, M.; Świerczek, A.; Pociecha, K.; Wyska, E.; Bucki, A.; Gawalska, A.; Kołaczkowski, M.; Pawłowski, M. Novel Butanehydrazide Derivatives of Purine-2,6-Dione as Dual PDE4/7 Inhibitors with Potential Anti-Inflammatory Activity: Design, Synthesis and Biological Evaluation. Eur. J. Med. Chem. 2018, 146, 381–394. [Google Scholar] [CrossRef]
- Heymann, F.; Hamesch, K.; Weiskirchen, R.; Tacke, F. The Concanavalin A Model of Acute Hepatitis in Mice. Lab. Anim. 2015, 49, 12–20. [Google Scholar] [CrossRef]
- Wang, H.X.; Liu, M.; Weng, S.Y.; Li, J.J.; Xie, C.; He, H.L.; Guan, W.; Yuan, Y.S.; Gao, J. Immune Mechanisms of Concanavalin A Model of Autoimmune Hepatitis. World J. Gastroenterol. 2012, 18, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Christen, U. Animal Models of Autoimmune Hepatitis. Biochim. Biophys. Acta -Mol. Basis Dis. 2019, 1865, 970–981. [Google Scholar] [CrossRef]
- Cao, Q.; Batey, R.; Pang, G.; Russell, A.; Clancy, R. IL-6, IFN-Gamma and TNF-Alpha Production by Liver-Associated T Cells and Acute Liver Injury in Rats Administered Concanavalin A. Immunol. Cell Biol. 1998, 76, 542–549. [Google Scholar] [CrossRef]
- Seki, S.; Kitada, T.; Iwai, S.; Kadoya, H.; Yamada, T.; Kawada, N.; Sakaguchi, H.; Wakasa, K. Immunohistochemical Detection of Fas and Apoptosis in Type-1 Autoimmune Hepatitis. Hepatogastroenterology 2003, 50, 1274–1277. [Google Scholar]
- Butler, D.C.; Lewin, D.N.; Batalis, N.I. Differential Diagnosis of Hepatic Necrosis Encountered at Autopsy. Acad. Forensic Pathol. 2018, 8, 256–295. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Tang, M.H.; Chen, X.C.; Chen, L.J.; Wei, Y.Q.; Wang, Y.S. Inhibitory Effect of Liposomal Quercetin on Acute Hepatitis and Hepatic Fibrosis Induced by Concanavalin A. Braz. J. Med. Biol. Res. 2014, 47, 655–661. [Google Scholar] [CrossRef] [Green Version]
- Shirin, H.; Bruck, R.; Aeed, H.; Frenkel, D.; Kenet, G.; Zaidel, L.; Avni, Y.; Halpern, Z.; Hershkoviz, R. Pentoxifylline Prevents Concanavalin A-Induced Hepatitis by Reducing Tumor Necrosis Factor α Levels and Inhibiting Adhesion of T Lymphocytes to Extracellular Matrix. J. Hepatol. 1998, 29, 60–67. [Google Scholar] [CrossRef]
- Fukuda, T.; Mogami, A.; Hisadome, M.; Komatsu, H. Therapeutic Administration of Y-40138, a Multiple Cytokine Modulator, Inhibits Concanavalin A-Induced Hepatitis in Mice. Eur. J. Pharmacol. 2005, 523, 137–142. [Google Scholar] [CrossRef]
- Robichaud, A.; Savoie, C.; Stamatiou, P.B.; Tattersall, F.D.; Chan, C.C. PDE4 Inhibitors Induce Emesis in Ferrets via a Noradrenergic Pathway. Neuropharmacology 2001, 40, 262–269. [Google Scholar] [CrossRef]
- FDA. Bioanalytical Method Validation Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry (accessed on 9 April 2021).
- Donnelly, R.P.; Freeman, S.L.; Hayes, M.P. Inhibition of IL-10 Expression by IFN-γ up-Regulates Transcription of TNF-α in Human Monocytes. J. Immunol. 1995, 155, 1420–1427. [Google Scholar]
- Nicoletti, F.; Zaccone, P.; Xiang, M.; Magro, G.; Di Mauro, M.; Di Marco, R.; Garotta, G.; Meroni, P. Essential Pathogenetic Role for Interferon (IFN-)γ in Concanavalin A-Induced T Cell-Dependent Hepatitis: Exacerbation by Exogenous IFN-γ and Prevention by IFN-γ Receptor-Immunoglobulin Fusion Protein. Cytokine 2000, 12, 315–323. [Google Scholar] [CrossRef]
- Gantner, F.; Leist, M.; Lohse, A.W.; Germann, P.G.; Tiegs, G. Concanavalin A-Induced T-Cell-Mediated Hepatic Injury in Mice: The Role of Tumor Necrosis Factor. Hepatology 1995, 21, 190–198. [Google Scholar] [CrossRef]
- Tagawa, Y.I.; Matthys, P.; Heremans, H.; Dillen, C.; Zaman, Z.; Iwakura, Y.; Billiau, A. Bimodal Role of Endogenous Interleukin-6 in Concanavalin A-Induced Hepatitis in Mice. J. Leukoc. Biol. 2000, 67, 90–96. [Google Scholar] [CrossRef]
- Kunz, M.; Ibrahim, S.M. Cytokines and Cytokine Profiles in Human Autoimmune Diseases and Animal Models of Autoimmunity. Mediat. Inflamm. 2009, 2009, 979258. [Google Scholar] [CrossRef]
- Robichaud, A.; Stamatiou, P.B.; Jin, S.-L.C.; Lachance, N.; MacDonald, D.; Laliberté, F.; Liu, S.; Huang, Z.; Conti, M.; Chan, C.-C. Deletion of Phosphodiesterase 4D in Mice Shortens Α2-Adrenoceptor-Mediated Anesthesia, a Behavioral Correlate of Emesis. J. Clin. Investig. 2002, 110, 1045–1052. [Google Scholar] [CrossRef]
- Ye, T.; Wang, T.; Yang, X.; Fan, X.; Wen, M.; Shen, Y.; Xi, X.; Men, R.; Yang, L. Cellular Physiology and Biochemistry Cellular Physiology and Biochemistry Comparison of Concanavalin A-Induced Murine Autoimmune Hepatitis Models. Cell Physiol. Biochem. 2018, 46, 1241–1251. [Google Scholar] [CrossRef]
- Gantner, F.; Kusters, S.; Wendel, A.; Hatzelmann, A.; Schudt, C.; Tiegs, G. Protection from T Cell-Mediated Murine Liver Failure by Phosphodiesterase Inhibitors. J. Pharmacol. Exp. Ther. 1997, 280, 53–60. [Google Scholar]
- Goto, M.; Tanaka, Y.; Murakawa, M.; Kadoshima-Yamaoka, K.; Inoue, H.; Murafuji, H.; Nagahira, A.; Kanki, S.; Hayashi, Y.; Nagahira, K.; et al. Inhibition of Phosphodiesterase 7A Ameliorates Concanavalin A-Induced Hepatitis in Mice. Int. Immunopharmacol. 2009, 9, 1347–1351. [Google Scholar] [CrossRef]
- Li, X.; Dubois, D.C.; Song, D.; Almon, R.R.; Jusko, W.J.; Chen, X. Modeling Combined Immunosuppressive and Anti-Inflammatory Effects of Dexamethasone and Naproxen in Rats Predicts the Steroid-Sparing Potential of Naproxen. Drug Metab. Dispos. 2017, 45, 834–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lon, H.K.; DuBois, D.C.; Earp, J.C.; Almon, R.R.; Jusko, W.J. Modeling Effects of Dexamethasone on Disease Progression of Bone Mineral Density in Collagen-Induced Arthritic Rats. Pharmacol. Res. Perspect. 2015, 3, e00169. [Google Scholar] [CrossRef]
- Wehbi, V.L.; Taskén, K. Molecular Mechanisms for cAMP-Mediated Immunoregulation in T Cells—Role of Anchored Protein Kinase a Signaling Units. Front. Immunol. 2016, 7, 00222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staats, D.A.; King, G.A.; Gearhart, J.M.; Conolly, R.I. Clearance of Alanine Aminotransferase from Blood in Mice and Rats. In 1989 Toxic Hazards Research Unit Annual Report; Kutzman, R.S., Wall, H.G., Vinegar, A., Eds.; Armstrong Aerospace Medical Research Laboratory: Wright-Patterson AFB, OH, USA, 1990; pp. 133–139. [Google Scholar]
- Hofstra, C.L.; van Ingrid, A.; Hofman, G.; Nijkamp, F.P.; Jardieu, P.M.; van Oosterhout, A.J.M. Differential Effects of Endogenous and Exogenous Interferon-γ on Immunoglobulin E, Cellular Infiltration, and Airway Responsiveness in a Murine Model of Allergic Asthma. Am. J. Respir. Cell Mol. Biol. 1998, 19, 826–835. [Google Scholar] [CrossRef]
- Waage, A.; Brandtzaeg, P.; Halstensen, A.; Kierulf, P.; Espevik, T. The Complex Pattern of Cytokines in Serum from Patients with Meningococcal Septic Shock. Association between Interleukin 6, Interleukin 1, and Fatal Outcome. J. Exp. Med. 1989, 169, 333–338. [Google Scholar] [CrossRef]
- Ma, Y.; Zhao, S.; Shen, S.; Fang, S.; Ye, Z.; Shi, Z.; Hong, A. A Novel Recombinant Slow-Release TNF α-Derived Peptide Effectively Inhibits Tumor Growth and Angiogensis. Sci. Rep. 2015, 5, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kadoshima-Yamaoka, K.; Murakawa, M.; Goto, M.; Tanaka, Y.; Inoue, H.; Murafuji, H.; Hayashi, Y.; Nagahira, K.; Miura, K.; Nakatsuka, T.; et al. Effect of Phosphodiesterase 7 Inhibitor ASB16165 on Development and Function of Cytotoxic T Lymphocyte. Int. Immunopharmacol. 2009, 9, 97–102. [Google Scholar] [CrossRef]
- Jones, N.A.; Leport, M.; Holand, T.; Vos, T.; Morgan, M.; Fink, M.; Pruniaux, M.P.; Berthelier, C.; O’Connor, B.J.; Bertrand, C.; et al. Phosphodiesterase (PDE) 7 in Inflammatory Cells from Patients with Asthma and COPD. Pulm. Pharmacol. Ther. 2007, 20, 60–68. [Google Scholar] [CrossRef]
- Sang, X.X.; Wang, R.L.; Zhang, C.E.; Liu, S.J.; Shen, H.H.; Guo, Y.M.; Zhang, Y.M.; Niu, M.; Wang, J.B.; Bai, Z.F.; et al. Sophocarpine Protects Mice from ConA-Induced Hepatitis via Inhibition of the IFN-Gamma/STAT1 Pathway. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Lohse, A.W.; Manns, M.; Dienes, H.-P.; Zum Büschenfelde, K.M.; Cohen, I.R. Experimental Autoimmune Hepatitis: Disease Induction, Time Course and T-cell Reactivity. Hepatology 1990, 11, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, A.; Yoshizawa, K.; Joshita, S.; Yoneda, S.; Umemura, T.; Ichijo, T.; Matsumoto, A.; Ota, M.; Tanaka, E. Cytokine Profiles Affecting the Pathogenesis of Autoimmune Hepatitis in Japanese Patients. Hepatol. Res. 2011, 41, 350–357. [Google Scholar] [CrossRef]
- Than, N.N.; Jeffery, H.C.; Oo, Y.H. Autoimmune Hepatitis: Progress from Global Immunosuppression to Personalised Regulatory T Cell Therapy. Can. J. Gastroenterol. Hepatol. 2016, 2016, 7181685. [Google Scholar] [CrossRef]
- Wang, K. Molecular Mechanisms of Hepatic Apoptosis. Cell Death Dis. 2014, 5, e996. [Google Scholar] [CrossRef]
- Mori, F.; Pérez-Torres, S.; de Caro, R.; Porzionato, A.; Macchi, V.; Beleta, J.; Gavaldà, A.; Palacios, J.M.; Mengod, G. The Human Area Postrema and Other Nuclei Related to the Emetic Reflex Express cAMP Phosphodiesterases 4B and 4D. J. Chem. Neuroanat. 2010, 40, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Guo, Y.; Li, S.; Wang, L.; Tang, Y.; Li, T.; Chen, W.; Zhong, G.; Song, G. Structure-Based Design and Structure-Activity Relationships of 1,2,3,4-Tetrahydroisoquinoline Derivatives as Potential PDE4 Inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 1188–1193. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analysis | Parameter | Brief Description | Final Estimate | CV (%) |
|---|---|---|---|---|
| (a) Pharmacokinetic | Vd/F (L·kg−1) | Apparent volume of distribution of GRMS-55 | 1.81 | 32 |
| ka (h−1) | Absorption rate constant of GRMS-55 | 1.49 | 21 | |
| ke (h−1) | Elimination rate constant of GRMS-55 | 4.59 | 30 | |
| (b) Pharmacodynamic | τ (h) | Mean transit time | 0.405 | 9 |
| kout_IFN-γ (h−1) | Elimination rate constant of IFN-γ | 0.234 | 24 | |
| kout_IL-6 (h−1) | Elimination rate constant of IL-6 | 0.531 | 17 | |
| kout_TNF-α (h−1) | Elimination rate constant of TNF-α | 0.209 | 17 | |
| kout_IL-10 (h−1) | Elimination rate constant of IL-10 | 1.841 | 57 | |
| IC50_IFN-γ (mg·L−1) | GRMS-55 concentration resulting in 50% of Imax of IFN-γ | 12.27 | 24 | |
| IC50_ IL-6 (mg·L−1) | GRMS-55 concentration resulting in 50% of Imax of IL-6 | 13.40 | 28 | |
| IC50_TNF-α (mg·L−1) | GRMS-55 concentration resulting in 50% of Imax of TNF-α | 7.97 | 21 | |
| S_IFN-γ | IFN-γ synthesis stimulation coefficient | 5856 | 17 | |
| S_TNF- α | TNF-α synthesis stimulation coefficient | 453.9 | 16 | |
| S_IL-6 | IL-6 synthesis stimulation coefficient | 10,320 | 13 | |
| S_IL-10 | IL-10 synthesis stimulation coefficient | 2.035 | 64 | |
| kin_IL-10(dis) (ng·L−1·h−1) | Production rate constant of IL-10 in diseased animals | 160.8 | 61 | |
| I_ IL-10(IFN-γ) (L·ng−1) | IL-10 synthesis inhibitory coefficient | 0.0002 | 36 | |
| (c) Disease progression | S_ALT(TNF-α) | Stimulation coeffcient of ALT production by TNF-α | 0.0072 | 84 |
| S_ALT(IFN-γ) | Stimulation coeffcient of ALT production by IFN-γ | 0.0002 | 48 | |
| kout_ALT (h−1) | Elimination rate constant of ALT | 0.0997 | 28 | |
| S_AST(TNF-α) (L·ng−1) | Stimulation coeffcient of AST production by TNF-α | 0.7727 | 37 | |
| S_AST(IFN-γ) (L·ng−1) | Stimulation coeffcient of AST production by IFN-γ | 0.0007 | 83 | |
| kout_AST (h−1) | Elimination rate constant of AST | 0.0817 | 20 | |
| α | Power coeffcient for stimlation of ALT production by TNF-α | 1.5 a | – | |
| β | Power coeffcient for stimlation of AST production by IFN-γ | 1.5 a | – |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Świerczek, A.; Plutecka, H.; Ślusarczyk, M.; Chłoń-Rzepa, G.; Wyska, E. PK/PD Modeling of the PDE7 Inhibitor—GRMS-55 in a Mouse Model of Autoimmune Hepatitis. Pharmaceutics 2021, 13, 597. https://doi.org/10.3390/pharmaceutics13050597
Świerczek A, Plutecka H, Ślusarczyk M, Chłoń-Rzepa G, Wyska E. PK/PD Modeling of the PDE7 Inhibitor—GRMS-55 in a Mouse Model of Autoimmune Hepatitis. Pharmaceutics. 2021; 13(5):597. https://doi.org/10.3390/pharmaceutics13050597
Chicago/Turabian StyleŚwierczek, Artur, Hanna Plutecka, Marietta Ślusarczyk, Grażyna Chłoń-Rzepa, and Elżbieta Wyska. 2021. "PK/PD Modeling of the PDE7 Inhibitor—GRMS-55 in a Mouse Model of Autoimmune Hepatitis" Pharmaceutics 13, no. 5: 597. https://doi.org/10.3390/pharmaceutics13050597
APA StyleŚwierczek, A., Plutecka, H., Ślusarczyk, M., Chłoń-Rzepa, G., & Wyska, E. (2021). PK/PD Modeling of the PDE7 Inhibitor—GRMS-55 in a Mouse Model of Autoimmune Hepatitis. Pharmaceutics, 13(5), 597. https://doi.org/10.3390/pharmaceutics13050597