
Combined Self-Nanoemulsifying and Solid Dispersion Systems Showed Enhanced Cinnarizine Release in Hypochlorhydria/Achlorhydria Dissolution Model



Abstract
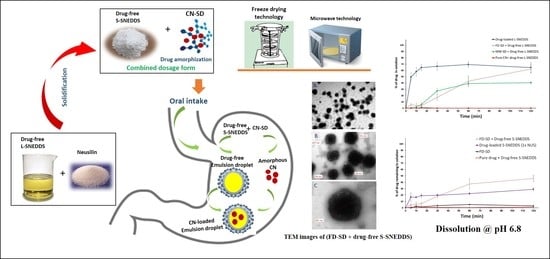
:
1. Introduction
- (1)
- self-emulsification of the drug-free S-SNEDDS leading to the formation of drug-free nanoemulsion;
- (2)
- simultaneously partitioning of the amorphized drug inside the formed nanoemulsion droplets, leading to enhanced drug dissolution: this system offers the advantage of avoiding the unfavorable drug interaction with the adsorbent (during storage) that could retard complete drug release from S-SNEDDS [13,14].
2. Materials and Methods
2.1. Materials
2.2. Preparation of Drug-Free and Drug-Loaded Liquid Self-Nanoemulsifying Drug Delivery Systems (L-SNEDDS)
2.3. Preparation of Drug-Free and Drug-Loaded Solid Self-Nanoemulsifying Drug Delivery Systems (S-SNEDDS)
2.4. Preparation of Drug Loaded Solid Dispersions (SD)
2.4.1. Freeze Dried Solid Dispersion (FD-SD)
2.4.2. Microwave Irradiation Solid Dispersion (MW-SD)
2.4.3. Preparation of Combined Formulation (SD + S-SNEDDS)
2.4.4. Determination of CN Encapsulation Efficiency
2.5. Optimization and Characterization of the Combined Formulation
2.5.1. Powder Properties
2.5.2. Differential Scanning Calorimetry (DSC)
2.5.3. X-ray Powder Diffraction (XRD)
2.5.4. Droplet Size Analysis and Zeta Potential of Liquid and Solid SNEDDS
2.5.5. Transmission Electron Microscopy
2.5.6. In Vitro Dissolution Tests
2.5.7. CN Quantification by UPLC Assay
2.6. Statistical Analysis
3. Results
3.1. Preparation and Optimization of Solid SNEDDS and SDs
3.2. Characterization of Solid SNEDDS and SDs
3.2.1. Powder Properties
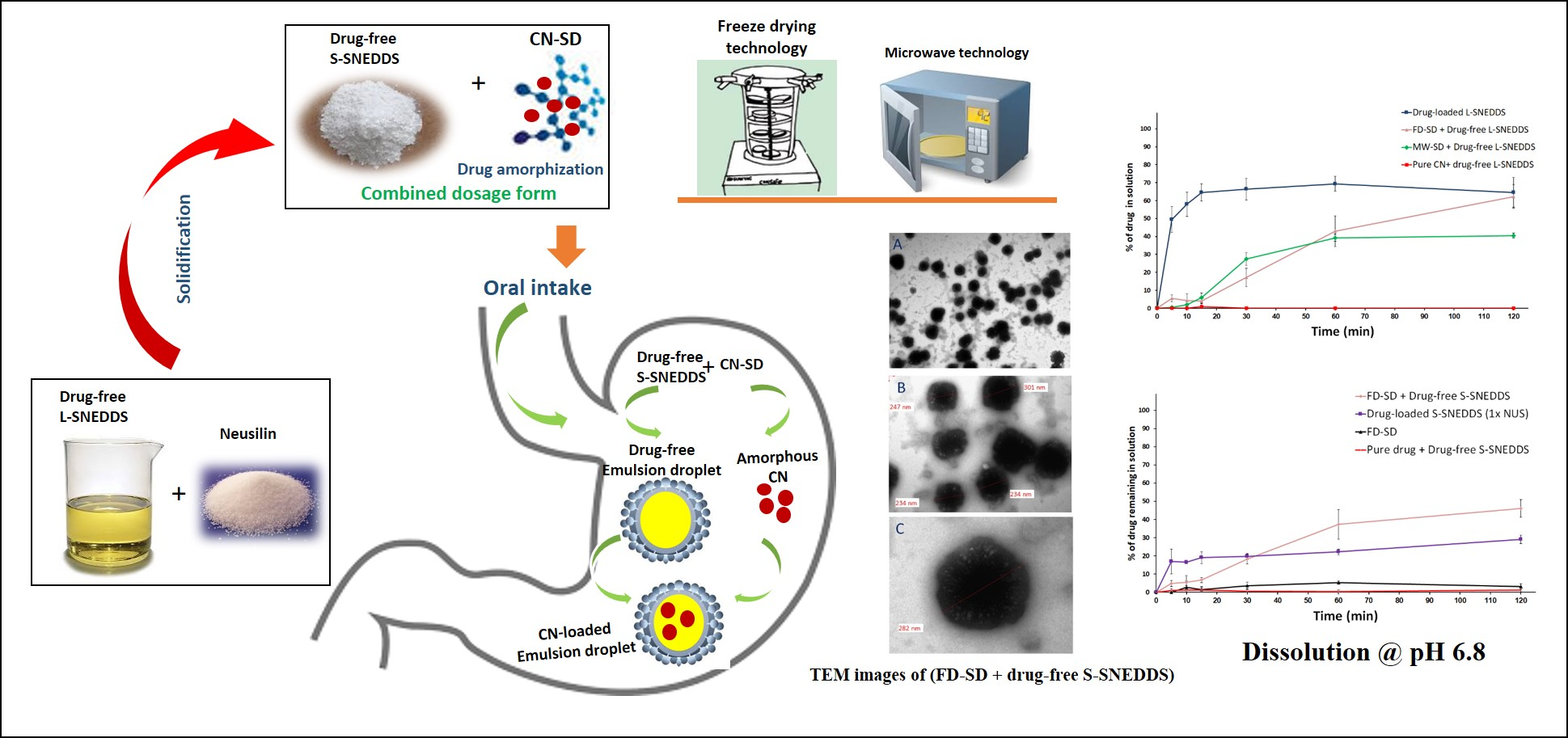
3.2.2. Differential Scanning Calorimetry (DSC)
3.2.3. X-ray Diffraction (XRD)
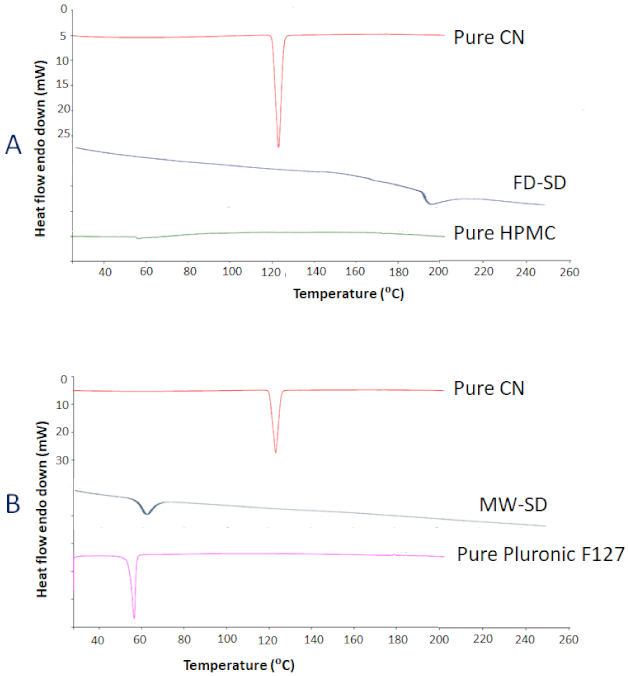
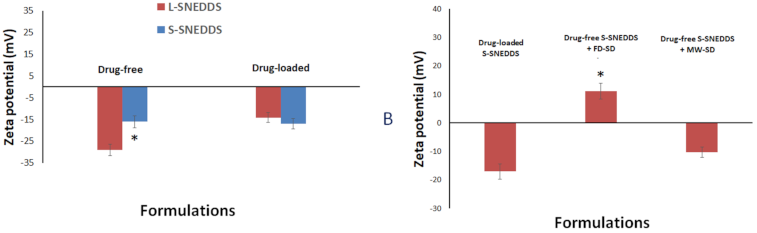
3.2.4. Droplet Size Analysis and Zeta Potential of Liquid/Solid SNEDDS
3.2.5. Transmission Electron Microscopy
3.2.6. In Vitro Dissolution
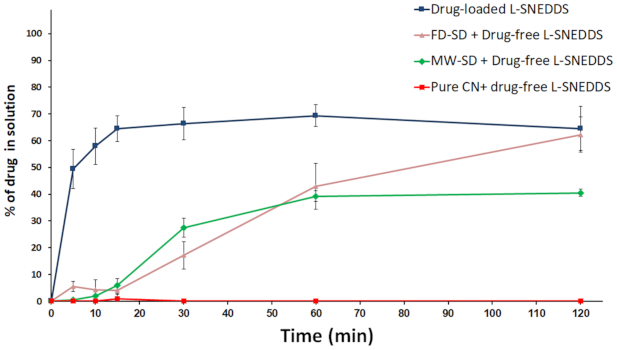
Effect of Neusilin® US2 on Drug Release from SNEDDS
Effect of SD + L-SNEDDS on Drug Release
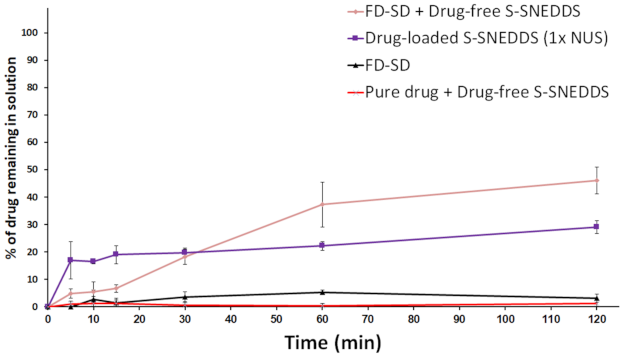
Effect of SD + S-SNEDDS on Drug Release
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schinke, T.; Amling, M. Gastrointestinal Tract and the Control of Bone Mass. In Translational Endocrinology of Bone; Karsenty, G., Ed.; Academic Press: San Diego, CA, USA, 2013; Chapter 6; pp. 63–71. ISBN 978-0-12-415784-2. [Google Scholar]
- Sharma, R.; Tandon, R.K. Nutrition, Dietary Fibers, and Cholelithiasis: Apple Pulp, Fibers, Clinical Trials. In Bioactive Food as Dietary Interventions for Liver and Gastrointestinal Disease; Elsevier Inc.: Amsterdam, The Netherlands, 2013; pp. 349–368. ISBN 9780123971548. [Google Scholar]
- Seaman, J.S.; Bowers, S.P.; Dixon, P.; Schindler, L. Dissolution of Common Psychiatric Medications in a Roux-en-Y Gastric Bypass Model. Psychosomatics 2005, 46, 250–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yska, J.P.; Punter, R.J.; Woerdenbag, H.J.; Emous, M.; Frijlink, H.W.; Wilffert, B.; Van Roon, E.N. A gastrointestinal simulation system for dissolution of oral solid dosage forms before and after Roux-en-Y gastric bypass. Eur. J. Hosp. Pharm. 2019, 26, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Kines, K.; Krupczak, T. Nutritional Interventions for Gastroesophageal Reflux, Irritable Bowel Syndrome, and Hypochlorhydria: A Case Report. Integr. Med. (Encinitas) 2016, 15, 49–53. [Google Scholar]
- Yago, M.R.; Frymoyer, A.R.; Smelick, G.S.; Frassetto, L.A.; Budha, N.R.; Dresser, M.J.; Ware, J.A.; Benet, L.Z. Gastric reacidification with betaine HCl in healthy volunteers with rabeprazole-induced hypochlorhydria. Mol. Pharm. 2013, 10, 4032–4037. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Kesarla, R.; Omri, A. Formulation Strategies to Improve the Bioavailability of Poorly Absorbed Drugs with Special Emphasis on Self-Emulsifying Systems. ISRN Pharm. 2013, 2013, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Lu, Y.; Qi, J.; Nie, S.; Hu, F.; Pan, W.; Wu, W. Solid self-nanoemulsifying cyclosporin a pellets prepared by fluid-bed coating: Preparation, characterization and in vitro redispersibility. Int. J. Nanomed. 2011, 6, 795–805. [Google Scholar] [CrossRef] [Green Version]
- Beg, S.; Katare, O.P.; Saini, S.; Garg, B.; Khurana, R.K.; Singh, B. Solid self-nanoemulsifying systems of olmesartan medoxomil: Formulation development, micromeritic characterization, in vitro and in vivo evaluation. Powder Technol. 2016, 294, 93–104. [Google Scholar] [CrossRef]
- Gumaste, S.G.; Freire, B.O.S.; Serajuddin, A.T.M. Development of solid SEDDS, VI: Effect of precoating of Neusilin® US2 with PVP on drug release from adsorbed self-emulsifying lipid-based formulations. Eur. J. Pharm. Sci. 2017, 110, 124–133. [Google Scholar] [CrossRef]
- Alwadei, M.; Kazi, M.; Alanazi, F.K. Novel oral dosage regimen based on self-nanoemulsifying drug delivery systems for codelivery of phytochemicals—Curcumin and thymoquinone. Saudi Pharm. J. 2019, 27, 866–876. [Google Scholar] [CrossRef]
- Gumaste, S.G.; Dalrymple, D.M.; Serajuddin, A.T.M. Development of solid SEDDS, V: Compaction and drug release properties of tablets prepared by adsorbing lipid-based formulations onto neusilin® US2. Pharm. Res. 2013, 30, 3186–3199. [Google Scholar] [CrossRef] [Green Version]
- Alhasani, K.F.; Kazi, M.; Ibrahim, M.A.; Shahba, A.A.; Alanazi, F.K. Self-nanoemulsifying ramipril tablets: A novel delivery system for the enhancement of drug dissolution and stability. Int. J. Nanomed. 2019, 14, 5435–5448. [Google Scholar] [CrossRef] [Green Version]
- Patki, M.; Patel, K. Development of a solid supersaturated self-nanoemulsifying preconcentrate (S-superSNEP) of fenofibrate using dimethylacetamide and a novel co-processed excipient. Drug Dev. Ind. Pharm. 2019, 45, 405–414. [Google Scholar] [CrossRef]
- Shahba, A.A.W.; Alanazi, F.K.; Abdel-Rahman, S.I. Stabilization benefits of single and multi-layer self-nanoemulsifying pellets: A poorly-water soluble model drug with hydrolytic susceptibility. PLoS ONE 2018, 13, e0198469. [Google Scholar] [CrossRef]
- Shahba, A.A.; Alanazi, F.K.; Mohsin, K.; Abdel-Hamid, M. Stability assessment of cinnarizine in self-emulsifying drug delivery systems. Lat. Am. J. Pharm. 2012, 31, 549–554. [Google Scholar]
- Shahba, A.A.-W.; Mohsin, K.; Alanazi, F.K. Novel self-nanoemulsifying drug delivery systems (SNEDDS) for oral delivery of cinnarizine: Design, optimization, and in-vitro assessment. AAPS PharmSciTech 2012, 13. [Google Scholar] [CrossRef] [Green Version]
- Shahba, A.A.W.; Ahmed, A.R.; Alanazi, F.K.; Mohsin, K.; Abdel-Rahman, S.I. Multi-Layer Self-Nanoemulsifying Pellets: An Innovative Drug Delivery System for the Poorly Water-Soluble Drug Cinnarizine. AAPS PharmSciTech 2018, 19, 2087–2102. [Google Scholar] [CrossRef]
- Shahba, A.A.W.; Mohsin, K.; Alanazi, F.K.; Abdel-Rahman, S.I. Optimization of self-nanoemulsifying formulations for weakly basic lipophilic drugs: Role of acidification and experimental design. Braz. J. Pharm. Sci. 2016, 52, 653–668. [Google Scholar] [CrossRef] [Green Version]
- Shahba, A.A.W.; Ahmed, A.R.; Mohsin, K.; Abdel-Rahman, S.I.; Alanazi, F.K. Solidification of cinnarizine self-nanoemulsifying drug delivery systems by fluid bed coating: Optimization of the process and formulation variables. Pharmazie 2017, 72, 143–151. [Google Scholar]
- Kazi, M.; Shahba, A.A.; Alrashoud, S.; Alwadei, M.; Sherif, A.Y.; Alanazi, F.K. Bioactive self-nanoemulsifying drug delivery systems (Bio-SNEDDS) for combined oral delivery of curcumin and piperine. Molecules 2020, 25, 1703. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.A.; Shazly, G.A.; Elossaily, G.M.; Ezzeldin, E.; Aleanizy, F.S. Physicochemical, pharmacokinetics, and histological evaluation of new naproxen-quercetin co-lyophilizate to diminish drug-induced gastric irritations in rats. Saudi Pharm. J. 2019, 27, 413–421. [Google Scholar] [CrossRef]
- Anwer, M.K.; Agarwal, S.P.; Ali, A.; Sultana, Y. Molecular complexes of aspirin with humic acid extracted from shilajit and their characterization. J. Incl. Phenom. Macrocycl. Chem. 2010, 67, 209–215. [Google Scholar] [CrossRef]
- Alshehri, S.; Shakeel, F.; Ibrahim, M.; Elzayat, E.; Altamimi, M.; Shazly, G.; Mohsin, K.; Alkholief, M.; Alsulays, B.; Alshetaili, A.; et al. Influence of the microwave technology on solid dispersions of mefenamic acid and flufenamic acid. PLoS ONE 2017, 12, e0182011. [Google Scholar] [CrossRef] [Green Version]
- Moneghini, M.; Zingone, G.; De Zordi, N. Influence of the microwave technology on the physical-chemical properties of solid dispersion with Nimesulide. Powder Technol. 2009, 195, 259–263. [Google Scholar] [CrossRef]
- Abdel-Hamid, M.; Shahba, A.; Mohsin, K.; Alanazi, F. Ultra performance liquid chromatography assay for cinnarizine in lipid-based formulations. Asian J. Chem. 2012, 24, 595–600. [Google Scholar]
- Shah, R.B.; Tawakkul, M.A.; Khan, M.A. Comparative evaluation of flow for pharmaceutical powders and granules. AAPS PharmSciTech 2008. [Google Scholar] [CrossRef] [Green Version]
- Geldart, D.; Abdullah, E.C.; Hassanpour, A.; Nwoke, L.C.; Wouters, I. Characterization of powder flowability using measurement of angle of repose. China Particuology 2006. [Google Scholar] [CrossRef]
- Beakawi Al-Hashemi, H.M.; Baghabra Al-Amoudi, O.S. A review on the angle of repose of granular materials. Powder Technol. 2018, 330, 397–417. [Google Scholar] [CrossRef]
- Badran, M.M.; Mady, M.M.; Ghannam, M.M.; Shakeel, F. Preparation and characterization of polymeric nanoparticles surface modified with chitosan for target treatment of colorectal cancer. Int. J. Biol. Macromol. 2017, 95, 643–649. [Google Scholar] [CrossRef]
- El Maghraby, G.M.; Elzayat, E.M.; Alanazi, F.K. Development of modified in situ gelling oral liquid sustained release formulation of dextromethorphan. Drug Dev. Ind. Pharm. 2012, 38, 971–978. [Google Scholar] [CrossRef]
- The United States Pharmacopeial Convention <1174> Powder Flow. US Pharm 2012, 35, 801–804.
- Abouelatta, S.M.; Aboelwafa, A.A.; Khalil, R.M.; Elgazayerly, O.N. Floating lipid beads for the improvement of bioavailability of poorly soluble basic drugs: In-vitro optimization and in-vivo performance in humans. Eur. J. Pharm. Biopharm. 2015, 89, 82–92. [Google Scholar] [CrossRef]
- Kumar, P.; Mohan, C.; Kanamsrinivasan Uma Shankar, M.; Gulati, M. Physiochemical Characterization and Release Rate Studies of SolidDispersions of Ketoconazole with Pluronic F127 and PVP K-30. Iran. J. Pharm. Res. IJPR 2011, 10, 685–694. [Google Scholar]
- Griesser, J.; Burtscher, S.; Köllner, S.; Nardin, I.; Prüfert, F.; Bernkop-Schnürch, A. Zeta potential changing self-emulsifying drug delivery systems containing phosphorylated polysaccharides. Eur. J. Pharm. Biopharm. 2017, 119, 264–270. [Google Scholar] [CrossRef]
- Venema, K.; Verhoeven, J.; Beckman, C.; Keller, D. Survival of a probiotic-containing product using capsule-within-capsule technology in an in vitro model of the stomach and small intestine (TIM-1). Benef. Microbes 2020, 11, 403–409. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Excipients | Formulations * | ||||||
|---|---|---|---|---|---|---|---|
| Drug-Free L-SNEDDS | Drug-Loaded L-SNEDDS | Drug-Free S-SNEDDS | Drug-Loaded S-SNEDDS (1× NUS) | Drug-Loaded S-SNEDDS (2× NUS) | MW-SD | FD-SD | |
| CN | - | 8 | - | 4 | 2.7 | 10 | 20 |
| Oleic acid | 25 | 23 | 12.5 | 11.5 | 7.7 | - | - |
| Imwitor I308 | 25 | 23 | 12.5 | 11.5 | 7.7 | - | - |
| Cremophor El | 50 | 46 | 25 | 23 | 15.3 | - | - |
| Neusilin US2 | - | - | 50 | 50 | 66.7 | - | - |
| HPMC E3 | - | - | - | - | - | - | 80 |
| Pluronic F127 | - | - | - | - | - | 90 | - |
| SUM | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Test | Pure NUS | Drug-Free S-SNEDDS | ||
|---|---|---|---|---|
| Result | Flow Property * | Result | Flow Property * | |
| Angle of repose | 24.7 ± 0.3 | Excellent | 33.6 ± 0.1 | Good |
| Bulk density | 0.19 ± 0.0 | 0.29 ± 0.0 | ||
| Tapped density | 0.20 ± 0.0 | 0.35 ± 0.0 | ||
| Compressibility index | 6.49 ± 0.2 | Excellent | 17.07 ± 0.37 | Fair |
| Hausner ratio | 1.07 ± 0.0 | Excellent | 1.21 ± 0.0 | Fair |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahba, A.A.; Tashish, A.Y.; Alanazi, F.K.; Kazi, M. Combined Self-Nanoemulsifying and Solid Dispersion Systems Showed Enhanced Cinnarizine Release in Hypochlorhydria/Achlorhydria Dissolution Model. Pharmaceutics 2021, 13, 627. https://doi.org/10.3390/pharmaceutics13050627
Shahba AA, Tashish AY, Alanazi FK, Kazi M. Combined Self-Nanoemulsifying and Solid Dispersion Systems Showed Enhanced Cinnarizine Release in Hypochlorhydria/Achlorhydria Dissolution Model. Pharmaceutics. 2021; 13(5):627. https://doi.org/10.3390/pharmaceutics13050627
Chicago/Turabian StyleShahba, Ahmad A., Ahmad Y. Tashish, Fars K. Alanazi, and Mohsin Kazi. 2021. "Combined Self-Nanoemulsifying and Solid Dispersion Systems Showed Enhanced Cinnarizine Release in Hypochlorhydria/Achlorhydria Dissolution Model" Pharmaceutics 13, no. 5: 627. https://doi.org/10.3390/pharmaceutics13050627
APA StyleShahba, A. A., Tashish, A. Y., Alanazi, F. K., & Kazi, M. (2021). Combined Self-Nanoemulsifying and Solid Dispersion Systems Showed Enhanced Cinnarizine Release in Hypochlorhydria/Achlorhydria Dissolution Model. Pharmaceutics, 13(5), 627. https://doi.org/10.3390/pharmaceutics13050627