
Targeting KRAS in Solid Tumors: Current Challenges and Future Opportunities of Novel KRAS Inhibitors

 ,
, 
 , and
, and 
Abstract
:1. Introduction
2. KRAS Mutation in Solid Tumors
3. Non-Small Cell Lung Cancer
3.1. Role of KRAS
3.2. Therapeutic Approach
4. Pancreatic Cancer
4.1. Role of KRAS
4.2. Therapeutic Approach
5. Colorectal Cancer
5.1. Role of KRAS
5.2. Therapeutic Approach
6. Other Solid Tumors
6.1. Low-Grade Serous Ovarian Carcinoma
6.2. Endometrial Cancer
7. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO) 2018 Statistics. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 10 February 2021).
- Global Cancer Observatory, International Agency for Research on Cancer (IARC). Available online: https://gco.iarc.fr/today/ (accessed on 11 March 2021).
- Surveillance, Epidemiology, and End Results Program (SEER), Cancer Statistics. Available online: https://seer.cancer.gov/statfacts/ (accessed on 11 March 2021).
- Johnson, L.; Mercer, K.; Greenbaum, D. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 2001, 410, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- McCormick, F. KRAS as a Therapeutic Target. Clin. Cancer Res. 2015, 21, 1797–1801. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug. Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bournet, B.; Buscail, C.; Muscari, F.; Cordelier, P.; Buscail, L. Targeting KRAS for diagnosis, prognosis, and treatment of pancreatic cancer: Hopes and realities. Eur. J. Cancer. 2016, 54, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias Carvalho, P.; Guimarães, C.F.; Cardoso, A.P.; Mendonça, S.; Costa, Â.M.; Oliveira, M.J.; Velho, S. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res. 2018, 78, 7–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef]
- Mai, T.T.; Lito, P. A treatment strategy for KRAS-driven tumors. Nat. Med. 2018, 24, 902–904. [Google Scholar] [CrossRef] [PubMed]
- Riely, G.J.; Kris, M.G.; Rosenbaum, D.; Marks, J.; Li, A.; Chitale, D.A.; Nafa, K.; Riedel, E.R.; Hsu, M.; Pao, W.; et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin. Cancer Res. 2008, 14, 5731–5734. [Google Scholar] [CrossRef] [Green Version]
- El Osta, B.; Behera, M.; Kim, S.; Berry, L.D.; Sica, G.; Pillai, R.N.; Owonikoko, T.K.; Kris, M.G.; Johnson, B.E.; Kwiatkowski, D.J.; et al. Characteristics and Outcomes of Patients With Metastatic KRAS-Mutant Lung Adenocarcinomas: The Lung Cancer Mutation Consortium Experience. J. Thorac. Oncol. 2019, 14, 876–889. [Google Scholar] [CrossRef]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Scheffler, M.; Ihle, M.A.; Hein, R.; Merkelbach-Bruse, S.; Scheel, A.H.; Siemanowski, J.; Brägelmann, J.; Kron, A.; Abedpour, N. K-ras Mutation Subtypes in NSCLC and Associated Co-occuring Mutations in Other Oncogenic Pathways. J. Thorac. Oncol. 2019, 14, 606–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascaux, C.; Iannino, N.; Martin, B.; Paesmans, M.; Berghmans, T.; Dusart, M.; Haller, A.; Lothaire, P.; Meert, A.P.; Noel, S.; et al. The role of RAS oncogene in survival of patients with lung cancer: A systematic review of the literature with meta-analysis. Br. J. Cancer 2005, 92, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Goulding, R.E.; Chenoweth, M.; Carter, G.C.; Boye, M.E.; Sheffield, K.M.; John, W.J.; Leusch, M.S.; Muehlenbein, C.E.; Li, L.; Jen, M.H.; et al. KRAS mutation as a prognostic factor and predictive factor in advanced/metastatic non-small cell lung cancer: A systematic literature review and meta-analysis. Cancer Treat. Res. Commun. 2020, 24, 100200. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, H.S.; Kim, B.J. Prognostic value of KRAS mutation in advanced non-small-cell lung cancer treated with immune checkpoint inhibitors: A meta-analysis and review. Oncotarget 2017, 8, 48248–48252. [Google Scholar] [CrossRef] [Green Version]
- Passiglia, F.; Cappuzzo, F.; Alabiso, O.; Bettini, A.C.; Bidoli, P.; Chiari, R.; Defferrari, C.; Delmonte, A.; Finocchiaro, G.; Francini, G.; et al. Efficacy of nivolumab in pre-treated non-small-cell lung cancer patients harbouring KRAS mutations. Br. J. Cancer 2019, 120, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zheng, S.; Jin, R.; Wang, X.; Wang, F.; Zang, R.; Xu, H.; Lu, Z.; Huang, J.; Lei, Y.; et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020, 470, 95–105. [Google Scholar] [CrossRef]
- Herbst, S.T.; Lopes, G.; Kowalski, D.M.; Kasahara, M.; Wu, Y.-L.; De Castro Jr, G.; Cho, B.C.; Turna, H.Z.; Cristescu, R.; Aurora-Garg, D.; et al. LBA4–Association of KRAS mutational status with response to pembrolizumab monotherapy given as first-line therapy for PD-L1-positive advanced non-squamous NSCLC in Keynote-042. Ann. Oncol. 2019, 30, 63–64. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Biton, J.; Mansuet-Lupo, A.; Pécuchet, N.; Alifano, M.; Ouakrim, H.; Arrondeau, J.; Boudou-Rouquette, P.; Goldwasser, F.; Leroy, K.; Goc, J.; et al. TP53, STK11, and EGFR Mutations Predict Tumor Immune Profile and the Response to Anti-PD-1 in Lung Adenocarcinoma. Clin. Cancer Res. 2018, 24, 5710–5723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitai, H.; Ebi, H. Key roles of EMT for adaptive resistance to MEK inhibitor in KRAS mutant lung cancer. Small GTPases 2017, 8, 172–176. [Google Scholar] [CrossRef] [Green Version]
- Jänne, P.A.; Shaw, A.T.; Pereira, J.R.; Jeannin, G.; Vansteenkiste, J.; Barrios, C.; Franke, F.A.; Grinsted, L.; Zazulina, V.; Smith, P.; et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: A randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013, 14, 38–47. [Google Scholar] [CrossRef]
- Jänne, P.A.; van den Heuvel, M.M.; Barlesi, F.; Cobo, M.; Mazieres, J.; Crinò, L.; Orlov, S.; Blackhall, F.; Wolf, J.; Garrido, P.; et al. Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression-Free Survival in Patients With KRAS-Mutant Advanced Non-Small Cell Lung Cancer: The SELECT-1 Randomized Clinical Trial. JAMA 2017, 317, 1844–1853. [Google Scholar] [CrossRef] [Green Version]
- Blumenschein, G.R., Jr.; Smit, E.F.; Planchard, D.; Kim, D.W.; Cadranel, J.; De Pas, T.; Dunphy, F.; Udud, K.; Ahn, M.J.; Hanna, N.H.; et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC). Ann. Oncol. 2015, 26, 894–901. [Google Scholar] [CrossRef]
- Vansteenkiste, J.F.; Canon, J.L.; De Braud, F.; Grossi, F.; De Pas, T.; Gray, J.E.; Su, W.C.; Felip, E.; Yoshioka, H.; Gridelli, C.; et al. Safety and Efficacy of Buparlisib (BKM120) in Patients with PI3K Pathway-Activated Non-Small Cell Lung Cancer: Results from the Phase II BASALT-1 Study. J. Thorac. Oncol. 2015, 10, 1319–1327. [Google Scholar] [CrossRef] [Green Version]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Jänne, P.A.; Rybkin, I.I.; Spira, A.I.; Riely, G.J.; Papadopoulos, K.P.; Sabari, J.K.; Johnson, M.L.; Heist, R.S.; Bazhenova, L.; Barve, M.; et al. KRYSTAL-1, Activity and Safety of Adagrasib (MRTX849) in Advanced/Metastatic Non–Small-Cell Lung Cancer (NSCLC) Harboring KRAS G12C Mutation. Eur. J. Cancer 2020, 138, S1–S2, (Abstract Book). [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Spira, A.; Besse, B.; Wolf, J.; Skoulidis, F.; Borghaei, H.; Goto, K.; Park, K.; Griesinger, F.; Felip, E.; et al. CodeBreak 200, A phase III multicenter study of sotorasib (AMG 510), a KRAS(G12C) inhibitor, versus docetaxel in patients with previously treated advanced non-small cell lung cancer (NSCLC) harboring KRAS p.G12C mutation. Ann. Oncol. 2020, 31, S894. [Google Scholar] [CrossRef]
- The AACR Project GENIE Consortium. AACR Project GENIE: Powering precision medicine through an international consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Delpu, Y.; Hanoun, N.; Lulka, H.; Sicard, F.; Selves, J.; Buscail, L.; Torrisani, J.; Cordelier, P. Genetic and epigenetic alterations in pancreatic carcinogenesis. Curr. Genom. 2011, 12, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.J.; Jang, H.J.; Kim, J.H.; Kim, H.S.; Lee, J. KRAS mutation as a prognostic factor in ampullary adenocarcinoma: A meta-analysis and review. Oncotarget 2016, 7, 58001–58006. [Google Scholar] [CrossRef] [Green Version]
- Jonckheere, N.; Vasseur, R.; Van Seuningen, I. The cornerstone K-RAS mutation in pancreatic adenocarcinoma: From cell signaling network, target genes, biological processes to therapeutic targeting. Crit Rev. Oncol. Hematol. 2017, 111, 7–19. [Google Scholar] [CrossRef]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef] [Green Version]
- di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013, 144, 1220–1229. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.; Jiao, X.; Jiang, Y.; Sun, S. ERK1/2 activity contributes to gemcitabine resistance in pancreatic cancer cells. J. Int. Med. Res. 2013, 41, 300–306. [Google Scholar] [CrossRef]
- Baer, R.; Cintas, C.; Therville, N.; Guillermet-Guibert, J. Implication of PI3K/Akt pathway in pancreatic cancer: When PI3K isoforms matter? Adv. Biol. Regul. 2015, 59, 19–35. [Google Scholar] [CrossRef]
- Prabhu, L.; Mundade, R.; Korc, M.; Loehrer, P.J.; Lu, T. Critical role of NF-κB in pancreatic cancer. Oncotarget 2014, 5, 10969–10975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.; et al. An integrin β3-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014, 16, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Mann, K.M.; Ying, H.; Juan, J.; Jenkins, N.A.; Copeland, N.G. KRAS-related proteins in pancreatic cancer. Pharmacol. Ther. 2016, 168, 29–42. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Goldberg, J.; Varghese, A.M.; Yu, K.H.; Park, W.; O’Reilly, E.M. Genomic profiling in pancreatic ductal adenocarcinoma and a pathway towards therapy individualization: A scoping review. Cancer Treat. Rev. 2019, 75, 27–38. [Google Scholar] [CrossRef]
- Fleming, J.B.; Shen, G.L.; Holloway, S.E.; Davis, M.; Brekken, R.A. Molecular consequences of silencing mutant K-ras in pancreatic cancer cells: Justification for K-ras-directed therapy. Mol. Cancer Res. 2005, 3, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Yuan, T.L.; Fellmann, C.; Lee, C.S.; Ritchie, C.D.; Thapar, V.; Lee, L.C.; Hsu, D.J.; Grace, D.; Carver, J.O.; Zuber, J.; et al. Development of siRNA payloads to target KRAS-mutant cancer. Cancer Discov. 2014, 4, 1182–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; David, E.B.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://clinicaltrials.gov/ct2/show/NCT01676259 (accessed on 11 March 2021).
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef]
- Pecot, C.V.; Wu, S.Y.; Bellister, S.; Filant, J.; Rupaimoole, R.; Hisamatsu, T.; Bhattacharya, R.; Maharaj, A.; Azam, S.; Rodriguez-Aguayo, C.; et al. Therapeutic silencing of KRAS using systemically delivered siRNAs. Mol. Cancer Ther. 2014, 13, 2876–2885. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://clinicaltrials.gov/ct2/show/NCT03608631 (accessed on 11 March 2021).
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.Y.; Liu, Y.; Redhu, S.; Steplewski, K.; et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Van Laethem, J.L.; Riess, H.; Jassem, J.; Haas, M.; Martens, U.M.; Weekes, C.; Peeters, M.; Ross, P.; Bridgewater, J.; Melichar, B.; et al. Phase I/II Study of Refametinib (BAY 86-9766) in Combination with Gemcitabine in Advanced Pancreatic cancer. Target. Oncol. 2017, 12, 97–109. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Hidalgo, M.; Canon, J.L.; Macarulla, T.; Bazin, I.; Poddubskaya, E.; Manojlovic, N.; Radenkovic, D.; Verslype, C.; Raymond, E.; et al. Phase I/II trial of pimasertib plus gemcitabine in patients with metastatic pancreatic cancer. Int. J. Cancer 2018, 143, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.N.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Fountzilas, G.; Bobos, M.; Kalogera-Fountzila, A.; Xiros, N.; Murray, S.; Linardou, H.; Karayannopoulou, G.; Koutras, A.K.; Bafaloukos, D.; Samantas, E.; et al. Gemcitabine combined with gefitinib in patients with inoperable or metastatic pancreatic cancer: A phase II Study of the Hellenic Cooperative Oncology Group with biomarker evaluation. Cancer Investig. 2008, 26, 784–793. [Google Scholar] [CrossRef]
- Halfdanarson, T.R.; Foster, N.R.; Kim, G.P.; Meyers, J.P.; Smyrk, T.C.; McCullough, A.E.; Ames, M.M.; Jaffe, J.P.; Alberts, S.R. A Phase II Randomized Trial of Panitumumab, Erlotinib, and Gemcitabine Versus Erlotinib and Gemcitabine in Patients with Untreated, Metastatic Pancreatic Adenocarcinoma: North Central Cancer Treatment Group Trial N064B (Alliance). Oncologist 2019, 24, 589589-e160. [Google Scholar] [CrossRef] [Green Version]
- Belmont, P.J.; Jiang, P.; McKee, T.D.; Xie, T.; Isaacson, J.; Baryla, N.E.; Roper, J.; Sinnamon, M.J.; Lee, N.V.; Kan, J.L.; et al. Resistance to dual blockade of the kinases PI3K and mTOR in KRAS-mutant colorectal cancer models results in combined sensitivity to inhibition of the receptor tyrosine kinase EGFR. Sci. Signal. 2014, 7, ra107. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.N.; Zhao, L.; Yu, L.F.; Wei, M.J. BRAF and KRAS mutations in metastatic colorectal cancer: Future perspectives for personalized therapy. Gastroenterol. Rep. 2020, 8, 192–205. [Google Scholar] [CrossRef]
- Allegra, C.J.; Jessup, J.M.; Somerfield, M.R.; Hamilton, S.R.; Hammond, E.H.; Hayes, D.F.; McAllister, P.K. Morton RF, Schilsky RL. American Society of Clinical Oncology provisional clinical opinion: Testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J. Clin. Oncol. 2009, 27, 2091–2096. [Google Scholar]
- Giopanou, I.; Pintzas, A. RAS and BRAF in the foreground for non-small cell lung cancer and colorectal cancer: Similarities and main differences for prognosis and therapies. Crit. Rev. Oncol. Hematol. 2020, 146, 102859. [Google Scholar] [CrossRef]
- Fakih, M.; Desai, J.; Kuboki, Y.; Strickler, J.H.; Price, T.J.; Durm, G.A.; Falchook, G.S.; Denlinger, C.S.; Krauss, J.C.; Shapiro, G.; et al. CodeBreak 100, Activity of AMG 510, a novel small molecule inhibitor of KRASG12C, in patients with advanced colorectal cancer. J. Clin. Oncol. 2020, 38 (Suppl. 15), 4018. [Google Scholar] [CrossRef]
- Bennouna, J.; Lang, I.; Valladares-Ayerbes, M.; Boer, K.; Adenis, A.; Escudero, P.; Kim, T.Y.; Pover, G.M.; Morris, C.D.; Douillard, J.Y. A Phase II, open-label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY-142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Investig. New Drugs 2011, 29, 1021–1028. [Google Scholar] [CrossRef]
- Jifu, E.; Xing, J.; Gong, H.; He, J.; Zhang, W. Combine MEK inhibition with PI3K/mTOR inhibition exert inhibitory tumor growth effect on KRAS and PIK3CA mutation CRC xenografts due to reduced expression of VEGF and matrix metallopeptidase-9. Tumor Biol. 2015, 36, 1091–1097. [Google Scholar]
- Porru, M.; Artuso, S.; Salvati, E.; Bianco, A.; Franceschin, M.; Diodoro, M.G.; Passeri, D.; Orlandi, A.; Savorani, F.; D’Incalci, M.; et al. Targeting G-Quadruplex DNA Structures by EMICORON Has a Strong Antitumor Efficacy against Advanced Models of Human Colon Cancer. Mol. Cancer Ther. 2015, 14, 2541–2551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, G.; Papke, B.; Ismail, S.; Vartak, N.; Chandra, A.; Hoffmann, M.; Hahn, S.A.; Triola, G.; Wittinghofer, A.; Bastiaens, P.I. Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature 2013, 497, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Kim, S.H.; Im, C.Y.; Hwang, H.J. Recent Development of Small Molecule Glutaminase Inhibitors. Curr. Top. Med. Chem. 2018, 18, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.Y.; Zhong, W.Z.; Zhang, X.C.; Su, J.; Xie, Z.; Liu, S.Y.; Tu, H.Y.; Chen, H.J.; Sun, Y.L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Cheng, K.A.; Hata, A.N.; Faber, A.C.; Ebi, H.; Coffee, E.M.; Greninger, P.; Brown, R.D.; Godfrey, J.T.; Cohoon, T.J.; et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013, 23, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Gourley, C.; Farley, J.; Provencher, D.M.; Pignata, S.; Mileshkin, L.; Harter, P.; Maenpaa, J.; Kim, J.W.; Pujaide-Lauraine, E.; Glasspool, R.M.; et al. Gynecologic Cancer InterGroup (GCIG) consensus review for ovarian and primary peritoneal low-grade serous carcinomas. Int. J. Gynecol. Cancer 2014, 24, S9–S13. [Google Scholar] [CrossRef]
- Van Nieuwenhuysen, E.; Busschaert, P.; Laenen, A.; Moerman, P.; Han, S.N.; Neven, P.; Lambrechts, D.; Vergote, I. Loss of 1p36.33 Frequent in Low-Grade Serous Ovarian Cancer. Neoplasia 2019, 21, 582–590. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. Low-grade serous ovarian carcinoma: An evolution toward targeted therapy. Int. J. Gynecol. Cancer 2020, 30, 1619–1626. [Google Scholar] [CrossRef]
- Farley, J.; Brady, W.E.; Vathipadiekal, V.; Lankes, H.A.; Coleman, R.; Morgan, M.A.; Mannel, R.; Yamada, S.D.; Mutch, D.; Rodgers, W.H.; et al. Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: An open-label, single-arm, phase 2 study. Lancet. Oncol. 2013, 14, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Bellone, S.; Zammataro, L.; Schwartz, P.E.; Santin, A.D. Binimetinib (MEK162) in recurrent low-grade serous ovarian cancer resistant to chemotherapy and hormonal treatment. Gynecol. Oncol. Rep. 2018, 25, 41–44. [Google Scholar] [CrossRef]
- Monk, B.J.; Grisham, R.N.; Banerjee, S.; Kalbacher, E.; Mirza, M.R.; Romero, I.; Vuylsteke, P.; Coleman, R.L.; Hilpert, F.; Oza, A.M.; et al. MILO/ENGOT-ov11, Binimetinib Versus Physician’s Choice Chemotherapy in Recurrent or Persistent Low-Grade Serous Carcinomas of the Ovary, Fallopian Tube, or Primary Peritoneum. J. Clin. Oncol. 2020, 38, 3753–3762. [Google Scholar] [CrossRef] [PubMed]
- Champer, M.; Miller, D.; Kuo, D.Y. Response to trametinib in recurrent low-grade serous ovarian cancer with NRAS mutation: A case report. Gynecol. Oncol. Rep. 2019, 28, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Tabernero, J.; Janku, F.; Wainberg, Z.A.; Paz-Ares, L.; Vansteenkiste, J.; Van Cutsem, E.; Pérez-García, J.; Stathis, A.; Britten, C.D.; et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin. Cancer Res. 2015, 21, 730–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piulats, J.M.; Guerra, E.; Gil-Martín, M.; Roman-Canal, B.; Gatius, S.; Sanz-Pamplona, R.; Velasco, A.; Vidal, A.; Matias-Guiu, X. Molecular approaches for classifying endometrial carcinoma. Gynecol. Oncol. 2017, 145, 200–207. [Google Scholar] [CrossRef]
- Sideris, M.; Emin, E.I.; Abdullah, Z.; Hanrahan, J.; Stefatou, K.M.; Sevas, V.; Emin, E.; Hollingworth, T.; Odejinmi, F.; Papagrigoriadis, S.; et al. The Role of KRAS in Endometrial Cancer: A Mini-Review. Anticancer Res. 2019, 39, 533–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraki, Y.; Banno, K.; Yanokura, M.; Kobayashi, Y.; Kawaguchi, M.; Nomura, H.; Hirasawa, A.; Susumu, N.; Aoki, D. Epigenetic DNA hypermethylation: Clinical applications in endometrial cancer (Review). Oncol. Rep. 2009, 22, 967–972. [Google Scholar]
- Bogani, G.; Ricci, M.T.; Vitellaro, M.; Ditto, A.; Chiappa, V.; Raspagliesi, F. Impact of gene-specific germline pathogenic variants on presentation of endometrial cancer in Lynch syndrome. Int. J. Gynecol. Cancer 2019, 29, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Ring, K.L.; Yates, M.S.; Schmandt, R.; Onstad, M.; Zhang, Q.; Celestino, J.; Kwan, S.Y.; Lu, K.H. Endometrial Cancers With Activating KRas Mutations Have Activated Estrogen Signaling and Paradoxical Response to MEK Inhibition. Int. J. Gynecol. Cancer 2017, 27, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Sill, M.W.; Thaker, P.H.; Bender, D.P.; Street, D.; McGuire, W.P.; Johnston, C.M.; Rotmensch, J. A phase II evaluation of selumetinib (AZD6244, ARRY-142886), a selective MEK-1/2 inhibitor in the treatment of recurrent or persistent endometrial cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2015, 138, 30–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; de Stanchina, E.; Mazutis, L.; et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 2020, 577, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.B.; Fece de la Cruz, F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRASG12C Inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrogio, C. Mechanisms of Resistance to KRAS G12C Inhibitors. In Proceedings of the 2020 World Conference of Lung Cancer (WCLC), Singapore, 28 January 2021. [Google Scholar]
- Molina-Arcas, M. Combination Therapies to Maximize the Impact of KRAS-G12C Inhibitors. In Proceedings of the 2020 World Conference of Lung Cancer (WCLC), Singapore, 30 January 2021. [Google Scholar]
- Nagasaka, M. Emerging Mechanisms To Target KRAS Directly. In Proceedings of the 2020 World Conference of Lung Cancer (WCLC), Singapore, 29 January 2021. [Google Scholar]
- Burslem, G.M.; Smith, B.E.; Lai, A.C.; Jaime-Figueroa, S.; McQuaid, D.C.; Bondeson, D.P.; Toure, M.; Dong, H.; Qian, Y.; Wang, J.; et al. The advantages of targeted protein degradation over inhibition: An RTK case study. Cell Chem. Biol. 2018, 25, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Xiong, Y.; Safaee, N.; Nowak, R.P.; Donovan, K.A.; Yuan, C.J.; Nabet, B.; Gero, T.W.; Feru, F.; Li, L.; et al. Exploring Targeted Degradation Strategy for Oncogenic KRASG12C. Cell Chem. Biol. 2020, 27, 19–31. [Google Scholar] [CrossRef]
- Nabet, B.; Roberts, J.M.; Buckley, D.L.; Paulk, J.; Dastjerdi, S.; Yang, A.; Leggett, A.L.; Erb, M.A.; Lawlor, M.A.; Souza, A.; et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018, 14, 431–441. [Google Scholar] [CrossRef]
- Lee, W.; Lee, J.H.; Jun, S.; Lee, J.H.; Bang, D. Selective targeting of KRAS oncogenic alleles by CRISPR/Cas9 inhibits proliferation of cancer cells. Sci. Rep. 2018, 8, 11879. [Google Scholar] [CrossRef] [Green Version]


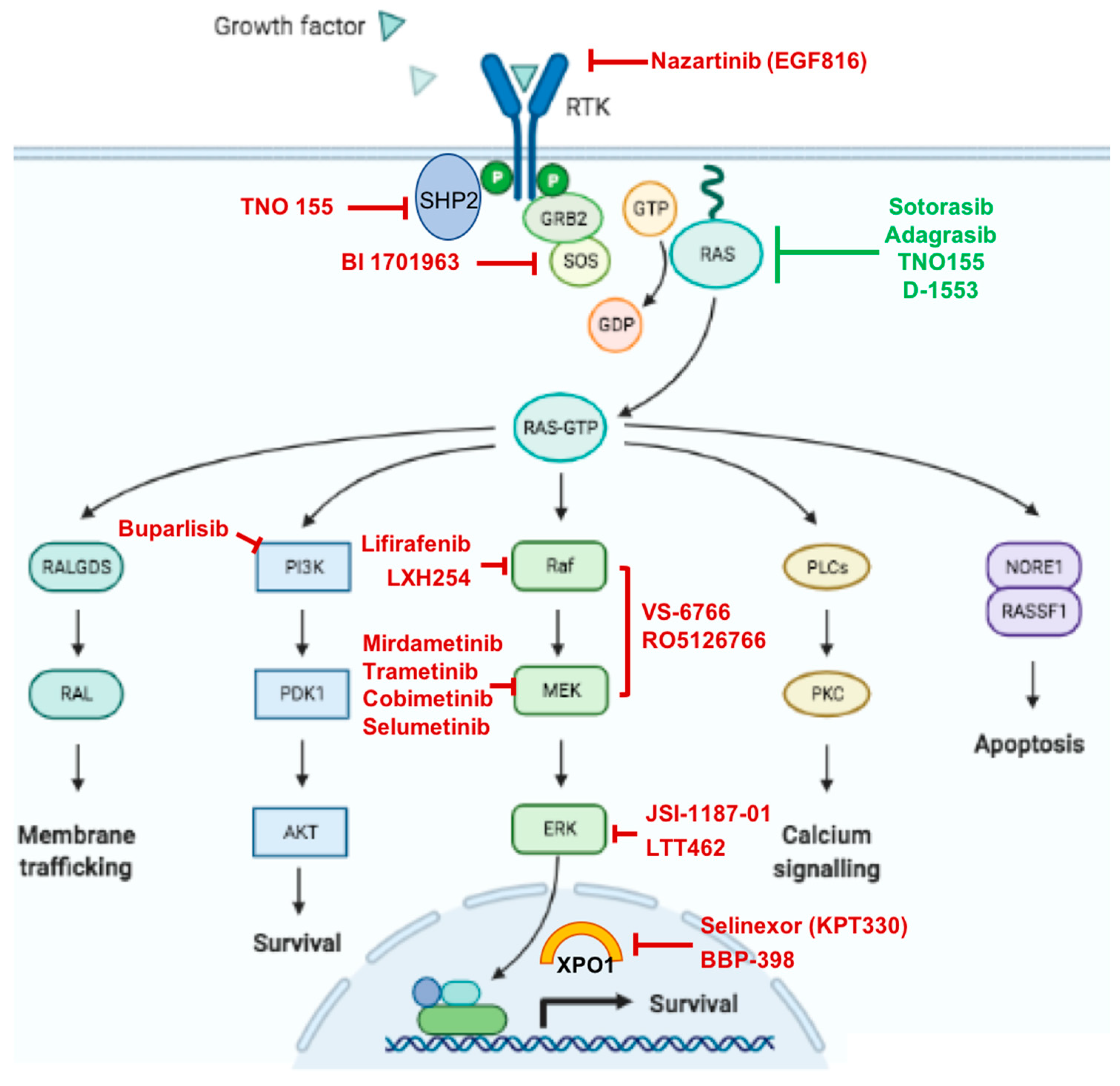{kind=link}
| Trial Name, NCT Number | Phase | Condition(s) | Drug(s) | Sample Size | Primary Endpoint(s) |
|---|---|---|---|---|---|
| CodeBreak 101, NCT04185883 | Ib | KRASG12C mutated advanced solid tumors | Sotorasib (AMG510) monotherapy or in combination with miscellaneous agents * | n = 1003 | DLTs incidence TEAEs |
| CodeBreak 200, NCT04303780 | III randomized | KRASG12C mutated NSCLC | Sotorasib vs. Docetaxel | n = 650 | PFS |
| KRYSTAL 1, NCT03785249 | I/II | KRASG12C mutated advanced solid tumors | MRTX849 monotherapy or in combination with pembrolizumab, or cetuximab, or Afatinib | n = 391 | Safety PK ORR |
| KRYSTAL 2, NCT04330664 | I/II | KRASG12C mutated advanced solid tumors | MRTX849 + TNO155 | n = 148 | Safety PK |
| KRYSTAL 7, NCT04613596 | II | KRASG12C mutated NSCLC | MRTX849 + Pembrolizumab | n = 120 | ORR |
| NCT04585035 | I/II | KRASG12C mutated advanced solid tumors | D-1553 | n = 200 | DLTs and AEs incidence, PK |
| NCT04627142 | I | KRAS mutated CRC | BI 1701963 + Irinotecan | n = 95 | MTD, DLTs incidence, ORR |
| NCT04449874 | Ia/Ib | KRASG12C mutated advanced solid tumors | GDC-6036 monotherapy or in combination with miscellaneous agents ** | n = 236 | AEs incidence, DLTs incidence |
| Trial Name, NCT Number | Phase | Condition(s) | Drug(s) | Sample Size | Primary Endpoint(s) |
|---|---|---|---|---|---|
| NCT03170206 | I/II | NSCLC | Palbociclib + Binimetinib | n = 72 | MTD, safety PFS |
| MEKiAUTO, NCT04214418 | I/II | Gastrointestinal tumors | Cobimetinib + Atezolizumab +Hydroxychloroquine | n = 175 | MTD |
| NCT03065387 | I | Advanced solid tumors | Neratinib + Palbociclib, or Everolimus, or Trametinib | n = 120 | MTD |
| NCT04620330 | II, randomized | NSCLC | VS-6766 +/− Defactinib | n =100 | ORR |
| NCT03981614 | III, randomized | CRC | Binimetinib + Palbociclib +/− TAS102 | n = 112 | PFS |
| NCT03095612 | I/II | NSCLC | Selinexor (KPT-330) + Docetaxel | n = 59 | MTD |
| NCT02079740 | Ib/II | Advanced solid tumors | Trametinib + Navitoclax | n = 130 | AEs incidence RR, PFS |
| FRAME, NCT03875820 | I | NSCLC, CRC, LGSOC | Defactinib + RO5126766 | n = 80 | RP2D AEs |
| NCT03299088 | Ib | NSCLC | Trametinib + Pembrolizumab | n = 42 | DLTs incidence |
| NCT04092673 | I/II | Advanced solid tumors | Zotatifin | n = 45 | TEAEs, AEs MTD, RP2D, PK |
| NCT03965845 | Ib/II | Advanced solid tumors | Telaglenestat (CB-839) + Palbociclib | n = 85 | Safety, MTD, RP2D |
| NCT03114319 | I | Advanced solid tumors | TNO155 +/− Nazartinib (EGF816) | n = 255 | AEs incidence, DLTs incidence |
| NCT02407509 | I | Advanced solid tumors | RO5126766 + Everolimus | n = 94 | Safety |
| NCT03756818 | I | Advanced solid tumors | TAK-659 + Paclitaxel | n = 64 | AEs incidence, MTD |
| NCT04528836 | I/Ib | KRASG12C mutated advanced solid tumors | BBP-398 | n = 60 | MTD |
| NCT03087071 | II | Anti-EGFR refractory CRC | Panitumumab + trametinib | n = 84 | RR |
| NCT04418167 | I | Advanced solid tumors with MAPK pathway mutations | JSI-1187-01 +/− dabrafenib | n = 124 | TEAEs |
| NCT03905148 | Ib | Advanced solid tumors | Lifirafenib (BGB-283) + Mirdametinib (PD-0325901) | n = 75 | AEs, DLTs, TEAEs ORR |
| NCT04263090 | I/IIa | NSCLC | Rigosertib + Nivolumab | n = 30 | MTD ORR |
| MAZEPPA, NCT04348045 | II, randomized | Metastatic PDAC patients with disease control after I line CT | Selumetinib + Nivolumab, or FOLFOXIRI | n = 307 | PFS |
| NCT02974725 | Ib | NSCLC | LXH254 + trametinib, or Ribociclib, or LTT462 | n = 331 | AEs incidence, DLTs incidence |
| NCT03829410 | Ib/II | CRC | Onvansertib (PCM-075) + FOLFIRI + bevacizumab | n = 44 | MTD, AEs incidence, ORR |
| NCT04132505 | I | PDAC | Binimetinib + Hydroxychloroquine | n = 39 | MTD |
| STOPTRAFFIC-1, NCT04599140 | Ib/II | RAS mutated MSS CRC | SX-682 +/− nivolumab | n = 53 | MTD, DLTs |
| NCT03520842 | II | NSCLC | Regorafenib + methotrexate | n = 18 | PFS |
| NCT03948763 | I | KRAS mutant NSCLC, CRC, PDAC | mRNA-5671/V941 +/− pembrolizumab | n = 100 | DLTs, AEs, discontinuation rate |
| NCT04146298 | I/II | RAS G12V mutant PDAC (HLA-A*11-01) | Mutant KRAS G12V-specific TCR transduced autologous T cells + antiPD1 | n = 30 | AEs incidence, ORR |
| Compound(s) Name(s) | Molecular Target and Mechanism of Action |
|---|---|
| Sotorasib (AMG510) Adagrasib (MRTX849) D-1553 GDC-6036 | KRAS G12C inhibitors |
| TNO155 | Src homology Phosphatase 2 (SHP2) inhibitor |
| BI 1701963 | SOS1 (panRAS inhibitor) |
| VS-6766 RO5126766 | RAF/MEK inhibitors |
| Selinexor (KPT330) BBP-398 | Exportin 1 (XPO1) inhibitor |
| Defactinib (VS-6063) | Focal adhesion kinase (FAK) and proline-rich tyrosine kinase-2 (Pyk2) inhibitor |
| Zotatifin (eFT226) | Eukaryotic translation initiation factor (eIF) 4A1-mediated translation inhibitor |
| Telaglenestat (CB-839) | Glutaminase inhibitor |
| Nazartinib (EGF816) | Epidermal growth factor receptor (EGFR) inhibitor |
| TAK-659 | Dual inhibitor of spleen tyrosine kinase (SYK) and FMS-like tyrosine kinase 3 (FLT3) |
| JSI-1187-01 LTT462 | ERK1/2 inhibitor |
| Lifirafenib (BGB-283) LXH254 | RAF inhibitor |
| Mirdametinib (PD-0325901) | MEK inhibitor |
| Onvansertib (PCM-075) | Polo-like kinase 1 (PLK1) inhibitor |
| SX-682 | CXC chemokine receptors 1 and 2 (CXCR1/2) inhibitors |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Indini, A.; Rijavec, E.; Ghidini, M.; Cortellini, A.; Grossi, F. Targeting KRAS in Solid Tumors: Current Challenges and Future Opportunities of Novel KRAS Inhibitors. Pharmaceutics 2021, 13, 653. https://doi.org/10.3390/pharmaceutics13050653
Indini A, Rijavec E, Ghidini M, Cortellini A, Grossi F. Targeting KRAS in Solid Tumors: Current Challenges and Future Opportunities of Novel KRAS Inhibitors. Pharmaceutics. 2021; 13(5):653. https://doi.org/10.3390/pharmaceutics13050653
Chicago/Turabian StyleIndini, Alice, Erika Rijavec, Michele Ghidini, Alessio Cortellini, and Francesco Grossi. 2021. "Targeting KRAS in Solid Tumors: Current Challenges and Future Opportunities of Novel KRAS Inhibitors" Pharmaceutics 13, no. 5: 653. https://doi.org/10.3390/pharmaceutics13050653
APA StyleIndini, A., Rijavec, E., Ghidini, M., Cortellini, A., & Grossi, F. (2021). Targeting KRAS in Solid Tumors: Current Challenges and Future Opportunities of Novel KRAS Inhibitors. Pharmaceutics, 13(5), 653. https://doi.org/10.3390/pharmaceutics13050653