
Mitochondrial Targeting Involving Cholesterol-Rich Lipid Rafts in the Mechanism of Action of the Antitumor Ether Lipid and Alkylphospholipid Analog Edelfosine


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
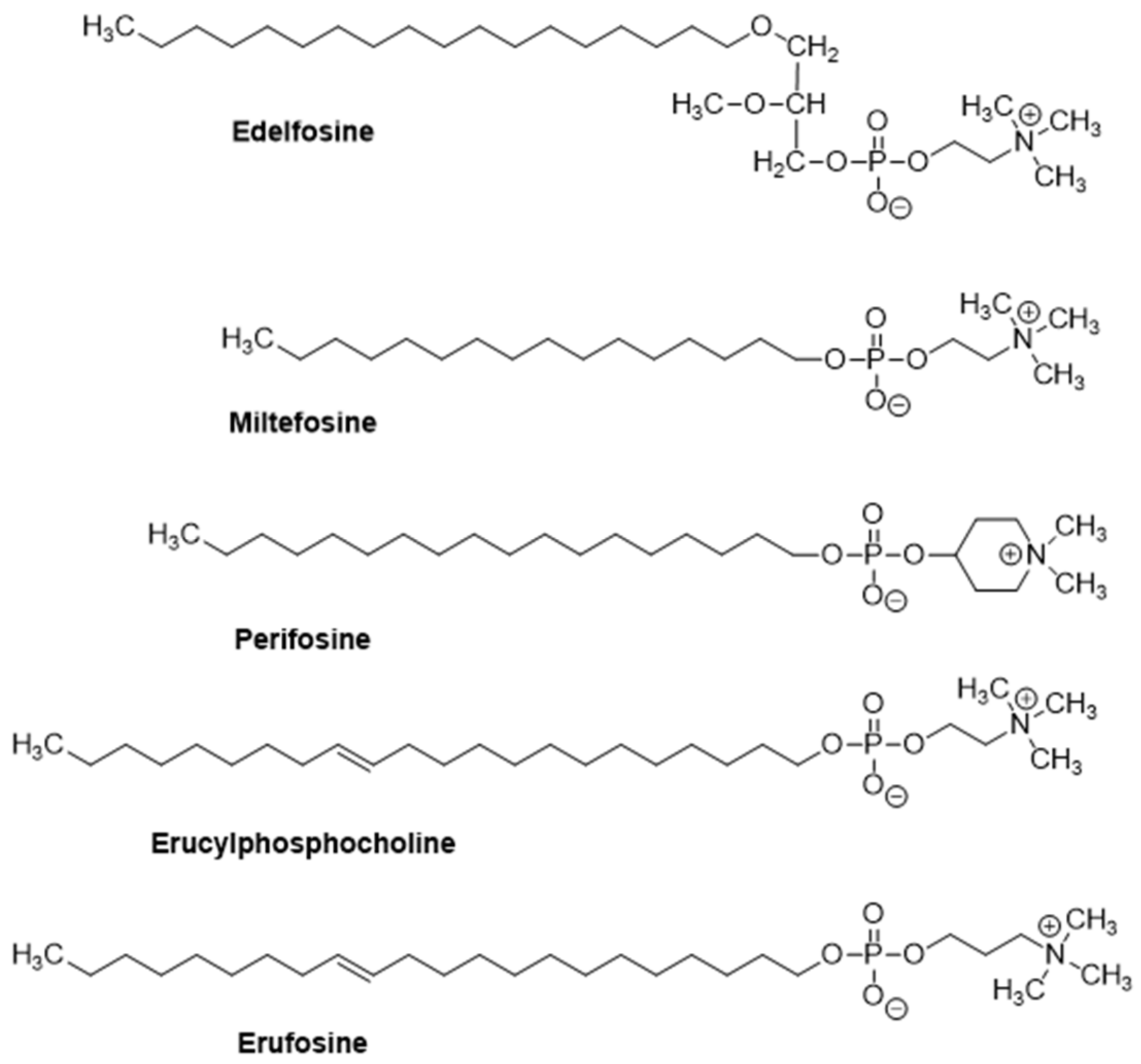
2. The Alkylphospholipid Analog Edelfosine Induces Apoptosis Selectively in Cancer Cells
3. Edelfosine Accumulates in Lipid Rafts and the Endoplasmic Reticulum of Cancer Cells, and the Generated Apoptotic Signals Converge on Mitochondria to Elicit Apoptosis
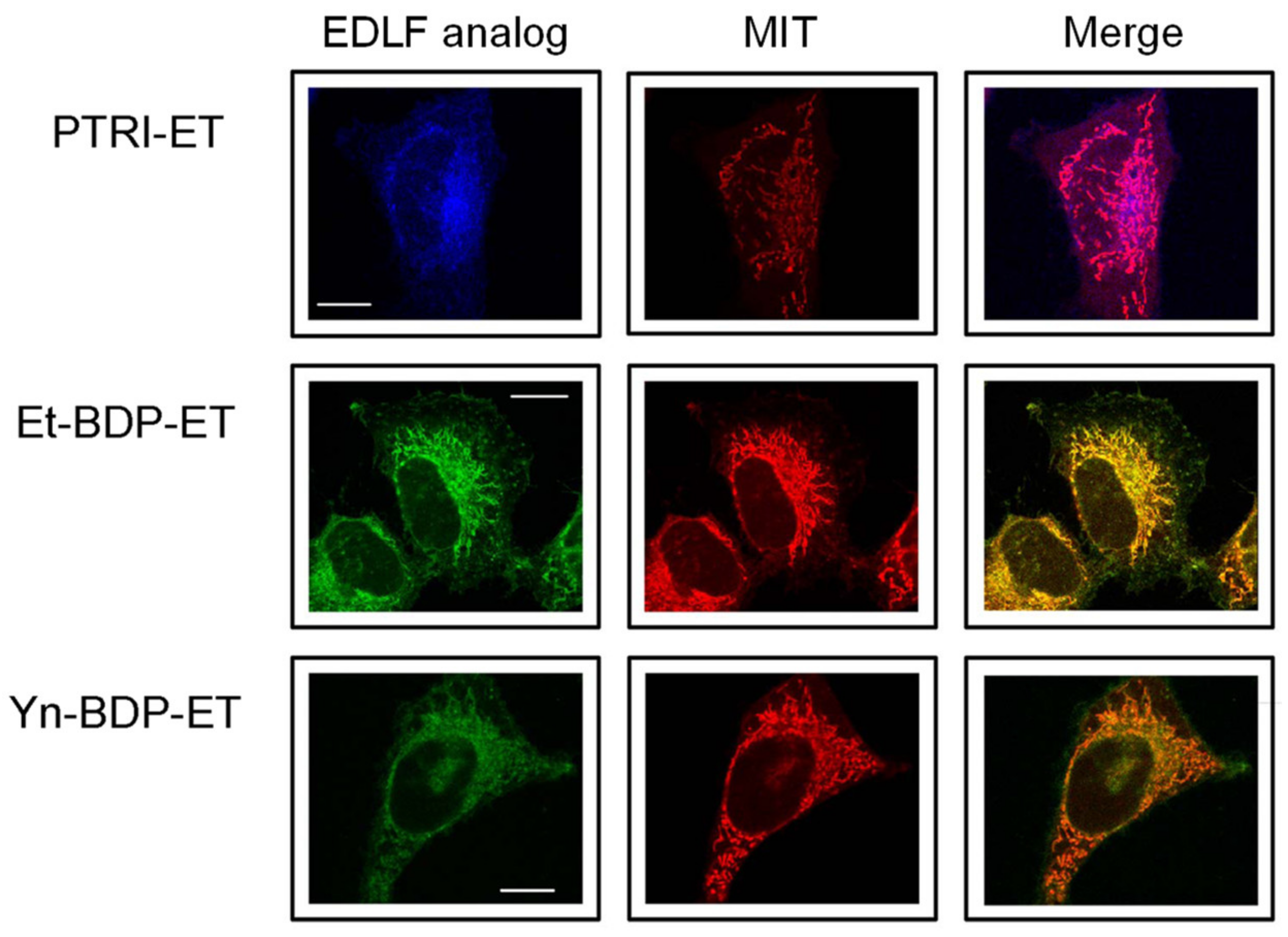
4. Localization of Edelfosine in the Mitochondria of Cancer Cells Using Fluorescent Analogs
5. Cholesterol in Mitochondria
6. Cholesterol Transport to Mitochondria
7. Mitochondrial Cholesterol in Cancer
8. Mitochondrial Permeability Transition Pore (mPTP) and Regulation of Cell Death
9. Edelfosine Induces Indirect and Direct Effects on Mitochondria
10. Edelfosine-Induced Apoptosis Involves F1FO–ATPase and Its Recruitment to Lipid Rafts
11. Edelfosine Promotes Raft Translocation to Mitochondria and Presence of Raft-Like Domains in Mitochondria
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AELs | antitumor ether lipids |
| ANT | adenine nucleotide translocator |
| APAF-1 | apoptosis protease-activating factor-1 |
| APLs | alkylphospholipid analogs |
| BODIPY | 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene; boron-dipyrromethene |
| CypD | cyclophilin D |
| ΔΨm | mitochondrial transmembrane potential |
| DiOC6(3) | 3,3′-dihexyloxacarbocyanine iodide |
| ER | endoplasmic reticulum |
| ET-18-OCH3 | 1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine (edelfosine) |
| ETC | electron transport chain |
| IMM | inner mitochondrial membrane |
| IMS | inter-membrane space |
| MAMs | mitochondria-associated membranes |
| MCD | methyl-β-cyclodextrin |
| MEFs | mouse embryonic fibroblasts |
| MLN64 | metastatic lymph node 64 protein |
| MOMP | mitochondrial outer membrane permeabilization |
| mPTP | mitochondrial permeability transition pore |
| NP-C | Niemann-Pick disease type C |
| NPC1 | Niemann-Pick type C1 protein |
| OMM | outer mitochondrial membrane |
| OSCP | oligomycin sensitivity-conferring protein |
| PiC | mitochondrial phosphate carrier (PiC) |
| ROS | reactive oxygen species |
| SLC25A3 | solute carrier family 25, member 3 |
| SNAP-23 | 23-kDa synaptosome associated protein |
| STARD1 | steroidogenic acute regulatory protein-related lipid transfer domain containing protein 1 |
| STARD3 | steroidogenic acute regulatory protein-related lipid transfer domain containing protein 3 |
| tBid | truncated Bid |
| TSPO | translocator protein |
| VDAC1 | voltage-dependent anion-selective channel 1 |
References
- Gajate, C.; Mollinedo, F. Biological activities, mechanisms of action and biomedical prospect of the antitumor ether phospholipid ET-18-OCH3 (Edelfosine), a proapoptotic agent in tumor cells. Curr. Drug Metab. 2002, 3, 491–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollinedo, F.; Gajate, C.; Martin-Santamaria, S.; Gago, F. ET-18-OCH3 (edelfosine): A selective antitumour lipid targeting apoptosis through intracellular activation of Fas/CD95 death receptor. Curr. Med. Chem. 2004, 11, 3163–3184. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F. Editorial: Antitumor alkylphospholipid analogs: A promising and growing family of synthetic cell membrane-targeting molecules for cancer treatment. Anticancer Agents Med. Chem. 2014, 14, 495–498. [Google Scholar] [CrossRef] [Green Version]
- Sundar, S.; Jha, T.K.; Thakur, C.P.; Bhattacharya, S.K.; Rai, M. Oral miltefosine for the treatment of Indian visceral leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 2006, 100 (Suppl. S1), S26–S33. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Jha, T.K.; Thakur, C.P.; Engel, J.; Sindermann, H.; Fischer, C.; Junge, K.; Bryceson, A.; Berman, J. Oral miltefosine for Indian visceral leishmaniasis. N. Engl. J. Med. 2002, 347, 1739–1746. [Google Scholar] [CrossRef] [Green Version]
- Dorlo, T.P.; Balasegaram, M.; Beijnen, J.H.; de Vries, P.J. Miltefosine: A review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J. Antimicrob. Chemother. 2012, 67, 2576–2597. [Google Scholar] [CrossRef]
- Smorenburg, C.H.; Seynaeve, C.; Bontenbal, M.; Planting, A.S.; Sindermann, H.; Verweij, J. Phase II study of miltefosine 6% solution as topical treatment of skin metastases in breast cancer patients. Anticancer Drugs 2000, 11, 825–828. [Google Scholar] [CrossRef]
- Dos Santos Nogueira, F.; Avino, V.C.; Galvis-Ovallos, F.; Pereira-Chioccola, V.L.; Moreira, M.A.B.; Romariz, A.; Molla, L.M.; Menz, I. Use of miltefosine to treat canine visceral leishmaniasis caused by Leishmania infantum in Brazil. Parasit Vectors 2019, 12, 79. [Google Scholar] [CrossRef] [PubMed]
- Gajate, C.; Mollinedo, F. Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts. Blood 2007, 109, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, P.G.; Eng, C.; Kolesar, J.; Hideshima, T.; Anderson, K.C. Perifosine, an oral, anti-cancer agent and inhibitor of the Akt pathway: Mechanistic actions, pharmacodynamics, pharmacokinetics, and clinical activity. Expert Opin. Drug Metab. Toxicol. 2012, 8, 623–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, K.; Kagabu, M.; Mizuno, M.; Oda, K.; Aoki, D.; Mabuchi, S.; Kamiura, S.; Yamaguchi, S.; Aoki, Y.; Saito, T.; et al. Phase II basket trial of perifosine monotherapy for recurrent gynecologic cancer with or without PIK3CA mutations. Invest. New Drugs 2017, 35, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Becher, O.J.; Millard, N.E.; Modak, S.; Kushner, B.H.; Haque, S.; Spasojevic, I.; Trippett, T.M.; Gilheeney, S.W.; Khakoo, Y.; Lyden, D.C.; et al. A phase I study of single-agent perifosine for recurrent or refractory pediatric CNS and solid tumors. PLoS ONE 2017, 12, e0178593. [Google Scholar] [CrossRef] [PubMed]
- Kaley, T.J.; Panageas, K.S.; Mellinghoff, I.K.; Nolan, C.; Gavrilovic, I.T.; DeAngelis, L.M.; Abrey, L.E.; Holland, E.C.; Lassman, A.B. Phase II trial of an AKT inhibitor (perifosine) for recurrent glioblastoma. J. Neurooncol. 2019, 144, 403–407. [Google Scholar] [CrossRef]
- Richardson, P.G.; Nagler, A.; Ben-Yehuda, D.; Badros, A.; Hari, P.N.; Hajek, R.; Spicka, I.; Kaya, H.; LeBlanc, R.; Yoon, S.-S.; et al. Randomized, placebo-controlled, phase 3 study of perifosine combined with bortezomib and dexamethasone in patients with relapsed, refractory multiple myeloma previously treated with bortezomib. eJHaem 2020, 1, 94–102. [Google Scholar] [CrossRef]
- Kaley, T.J.; Panageas, K.S.; Pentsova, E.I.; Mellinghoff, I.K.; Nolan, C.; Gavrilovic, I.; DeAngelis, L.M.; Abrey, L.E.; Holland, E.C.; Omuro, A.; et al. Phase I clinical trial of temsirolimus and perifosine for recurrent glioblastoma. Ann. Clin. Transl. Neurol. 2020, 7, 429–436. [Google Scholar] [CrossRef] [Green Version]
- Erdlenbruch, B.; Jendrossek, V.; Kugler, W.; Eibl, H.; Lakomek, M. Increased delivery of erucylphosphocholine to C6 gliomas by chemical opening of the blood-brain barrier using intracarotid pentylglycerol in rats. Cancer Chemother. Pharmacol. 2002, 50, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Erdlenbruch, B.; Jendrossek, V.; Marx, M.; Hunold, A.; Eibl, H.; Lakomek, M. Antitumor effects of erucylphosphocholine on brain tumor cells in vitro and in vivo. Anticancer Res. 1998, 18, 2551–2557. [Google Scholar]
- Jendrossek, V.; Kugler, W.; Erdlenbruch, B.; Eibl, H.; Lang, F.; Lakomek, M. Erucylphosphocholine-induced apoptosis in chemoresistant glioblastoma cell lines: Involvement of caspase activation and mitochondrial alterations. Anticancer Res. 2001, 21, 3389–3396. [Google Scholar]
- Jendrossek, V.; Muller, I.; Eibl, H.; Belka, C. Intracellular mediators of erucylphosphocholine-induced apoptosis. Oncogene 2003, 22, 2621–2631. [Google Scholar] [CrossRef] [Green Version]
- Veenman, L.; Gavish, M.; Kugler, W. Apoptosis induction by erucylphosphohomocholine via the 18 kDa mitochondrial translocator protein: Implications for cancer treatment. Anticancer Agents Med. Chem. 2014, 14, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Chometon, G.; Cappuccini, F.; Raducanu, A.; Aumailley, M.; Jendrossek, V. The membrane-targeted alkylphosphocholine erufosine interferes with survival signals from the extracellular matrix. Anticancer Agents Med. Chem. 2014, 14, 578–591. [Google Scholar] [CrossRef]
- Henke, G.; Lindner, L.H.; Vogeser, M.; Eibl, H.J.; Worner, J.; Muller, A.C.; Bamberg, M.; Wachholz, K.; Belka, C.; Jendrossek, V. Pharmacokinetics and biodistribution of Erufosine in nude mice—Implications for combination with radiotherapy. Radiat. Oncol. 2009, 4, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henke, G.; Meier, V.; Lindner, L.H.; Eibl, H.; Bamberg, M.; Belka, C.; Budach, W.; Jendrossek, V. Effects of ionizing radiation in combination with Erufosine on T98G glioblastoma xenograft tumours: A study in NMRI nu/nu mice. Radiat. Oncol. 2012, 7, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, S.S.; Sharma, A.K.; Soni, H.; Ali, D.M.; Tews, B.; Konig, R.; Eibl, H.; Berger, M.R. Induction of ER and mitochondrial stress by the alkylphosphocholine erufosine in oral squamous cell carcinoma cells. Cell Death Dis. 2018, 9, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzoneva, R.; Stoyanova, T.; Petrich, A.; Popova, D.; Uzunova, V.; Momchilova, A.; Chiantia, S. Effect of erufosine on membrane lipid order in breast cancer cell models. Biomolecules 2020, 10, 802. [Google Scholar] [CrossRef] [PubMed]
- Varela-M, R.E.; Villa-Pulgarin, J.A.; Yepes, E.; Muller, I.; Modolell, M.; Munoz, D.L.; Robledo, S.M.; Muskus, C.E.; Lopez-Aban, J.; Muro, A.; et al. In vitro and in vivo efficacy of ether lipid edelfosine against Leishmania spp. and SbV-resistant parasites. PLoS Negl. Trop. Dis. 2012, 6, e1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajate, C.; Del Canto-Janez, E.; Acuna, A.U.; Amat-Guerri, F.; Geijo, E.; Santos-Beneit, A.M.; Veldman, R.J.; Mollinedo, F. Intracellular triggering of Fas aggregation and recruitment of apoptotic molecules into Fas-enriched rafts in selective tumor cell apoptosis. J. Exp. Med. 2004, 200, 353–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajate, C.; Mollinedo, F. Lipid rafts, endoplasmic reticulum and mitochondria in the antitumor action of the alkylphospholipid analog edelfosine. Anticancer Agents Med. Chem. 2014, 14, 509–527. [Google Scholar] [CrossRef] [Green Version]
- Jaffres, P.A.; Gajate, C.; Bouchet, A.M.; Couthon-Gourves, H.; Chantome, A.; Potier-Cartereau, M.; Besson, P.; Bougnoux, P.; Mollinedo, F.; Vandier, C. Alkyl ether lipids, ion channels and lipid raft reorganization in cancer therapy. Pharmacol. Ther. 2016, 165, 114–131. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid rafts as signaling hubs in cancer cell survival/death and invasion: Implications in tumor progression and therapy. J. Lipid Res. 2020, 61, 611–635. [Google Scholar] [CrossRef] [Green Version]
- Busto, J.V.; Sot, J.; Goni, F.M.; Mollinedo, F.; Alonso, A. Surface-active properties of the antitumour ether lipid 1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine (edelfosine). Biochim. Biophys. Acta 2007, 1768, 1855–1860. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Fernandez-Luna, J.L.; Gajate, C.; Martin-Martin, B.; Benito, A.; Martinez-Dalmau, R.; Modolell, M. Selective induction of apoptosis in cancer cells by the ether lipid ET-18-OCH3 (Edelfosine): Molecular structure requirements, cellular uptake, and protection by Bcl-2 and Bcl-XL. Cancer Res. 1997, 57, 1320–1328. [Google Scholar] [PubMed]
- Gajate, C.; Fonteriz, R.I.; Cabaner, C.; Alvarez-Noves, G.; Alvarez-Rodriguez, Y.; Modolell, M.; Mollinedo, F. Intracellular triggering of Fas, independently of FasL, as a new mechanism of antitumor ether lipid-induced apoptosis. Int. J. Cancer 2000, 85, 674–682. [Google Scholar] [CrossRef]
- Mollinedo, F.; de la Iglesia-Vicente, J.; Gajate, C.; Estella-Hermoso de Mendoza, A.; Villa-Pulgarin, J.A.; Campanero, M.A.; Blanco-Prieto, M.J. Lipid raft-targeted therapy in multiple myeloma. Oncogene 2010, 29, 3748–3757. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F.; de la Iglesia-Vicente, J.; Gajate, C.; Estella-Hermoso de Mendoza, A.; Villa-Pulgarin, J.A.; de Frias, M.; Roue, G.; Gil, J.; Colomer, D.; Campanero, M.A.; et al. In vitro and in vivo selective antitumor activity of Edelfosine against mantle cell lymphoma and chronic lymphocytic leukemia involving lipid rafts. Clin. Cancer Res. 2010, 16, 2046–2054. [Google Scholar] [CrossRef] [Green Version]
- Melo-Lima, S.; Gajate, C.; Mollinedo, F. Triggers and signaling cross-talk controlling cell death commitment. Cell Cycle 2015, 14, 465–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo-Lima, S.; Celeste Lopes, M.; Mollinedo, F. Necroptosis is associated with low procaspase-8 and active RIPK1 and -3 in human glioma cells. Oncoscience 2014, 1, 649–664. [Google Scholar] [CrossRef] [PubMed]
- Melo-Lima, S.; Lopes, M.C.; Mollinedo, F. ERK1/2 acts as a switch between necrotic and apoptotic cell death in ether phospholipid edelfosine-treated glioblastoma cells. Pharmacol. Res. 2015, 95, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Miguel, T.; Fonteriz, R.I.; Vay, L.; Gajate, C.; Lopez-Hernandez, S.; Mollinedo, F. Endoplasmic reticulum stress in the proapoptotic action of edelfosine in solid tumor cells. Cancer Res. 2007, 67, 10368–10378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajate, C.; Matos-da-Silva, M.; Dakir, E.L.; Fonteriz, R.I.; Alvarez, J.; Mollinedo, F. Antitumor alkyl-lysophospholipid analog edelfosine induces apoptosis in pancreatic cancer by targeting endoplasmic reticulum. Oncogene 2012, 31, 2627–2639. [Google Scholar] [CrossRef] [Green Version]
- Andreesen, R.; Modolell, M.; Weltzien, H.U.; Eibl, H.; Common, H.H.; Lohr, G.W.; Munder, P.G. Selective destruction of human leukemic cells by alkyl-lysophospholipids. Cancer Res. 1978, 38, 3894–3899. [Google Scholar] [PubMed]
- Modolell, M.; Andreesen, R.; Pahlke, W.; Brugger, U.; Munder, P.G. Disturbance of phospholipid metabolism during the selective destruction of tumor cells induced by alkyl-lysophospholipids. Cancer Res. 1979, 39, 4681–4686. [Google Scholar] [PubMed]
- Andreesen, R.; Modolell, M.; Oepke, G.H.; Common, H.; Lohr, G.W.; Munder, P.G. Studies on various parameters influencing leukemic cell destruction by alkyl-lysophospholipids. Anticancer Res. 1982, 2, 95–100. [Google Scholar] [PubMed]
- Scholar, E.M. Inhibition of the growth of human lung cancer cells by alkyl-lysophospholipid analogs. Cancer Lett. 1986, 33, 199–204. [Google Scholar] [CrossRef]
- Mollinedo, F.; Martinez-Dalmau, R.; Modolell, M. Early and selective induction of apoptosis in human leukemic cells by the alkyl-lysophospholipid ET-18-OCH3. Biochem. Biophys. Res. Commun. 1993, 192, 603–609. [Google Scholar] [CrossRef]
- Diomede, L.; Colotta, F.; Piovani, B.; Re, F.; Modest, E.J.; Salmona, M. Induction of apoptosis in human leukemic cells by the ether lipid 1-octadecyl-2-methyl-rac-glycero-3-phosphocholine. A possible basis for its selective action. Int. J. Cancer 1993, 53, 124–130. [Google Scholar] [CrossRef]
- Gajate, C.; Santos-Beneit, A.M.; Macho, A.; Lazaro, M.; Hernandez-De Rojas, A.; Modolell, M.; Munoz, E.; Mollinedo, F. Involvement of mitochondria and caspase-3 in ET-18-OCH3-induced apoptosis of human leukemic cells. Int. J. Cancer 2000, 86, 208–218. [Google Scholar] [CrossRef]
- Gajate, C.; Mollinedo, F. The antitumor ether lipid ET-18-OCH3 induces apoptosis through translocation and capping of Fas/CD95 into membrane rafts in human leukemic cells. Blood 2001, 98, 3860–3863. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, X.; Dakir el, H.; Mollinedo, F.; Gajate, C. Endoplasmic reticulum targeting in Ewing’s sarcoma by the alkylphospholipid analog edelfosine. Oncotarget 2015, 6, 14596–14613. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F.; Gajate, C.; Morales, A.I.; del Canto-Janez, E.; Justies, N.; Collia, F.; Rivas, J.V.; Modolell, M.; Iglesias, A. Novel anti-inflammatory action of edelfosine lacking toxicity with protective effect in experimental colitis. J. Pharmacol. Exp. Ther. 2009, 329, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.; Tait, S.W. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Luit, A.H.; Budde, M.; Ruurs, P.; Verheij, M.; van Blitterswijk, W.J. Alkyl-lysophospholipid accumulates in lipid rafts and induces apoptosis via raft-dependent endocytosis and inhibition of phosphatidylcholine synthesis. J. Biol. Chem. 2002, 277, 39541–39547. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F.; Gajate, C. Fas/CD95 death receptor and lipid rafts: New targets for apoptosis-directed cancer therapy. Drug Resist. Updat. 2006, 9, 51–73. [Google Scholar] [CrossRef]
- Gajate, C.; Gonzalez-Camacho, F.; Mollinedo, F. Involvement of raft aggregates enriched in Fas/CD95 death-inducing signaling complex in the antileukemic action of edelfosine in Jurkat cells. PLoS ONE 2009, 4, e5044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis-Sobreiro, M.; Gajate, C.; Mollinedo, F. Involvement of mitochondria and recruitment of Fas/CD95 signaling in lipid rafts in resveratrol-mediated antimyeloma and antileukemia actions. Oncogene 2009, 28, 3221–3234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollinedo, F.; Gajate, C. Lipid rafts and clusters of apoptotic signaling molecule-enriched rafts in cancer therapy. Future Oncol. 2010, 6, 811–821. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid rafts, death receptors and CASMERs: New insights for cancer therapy. Future Oncol. 2010, 6, 491–494. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F. Death receptors in multiple myeloma and therapeutic opportunities. In Myeloma Therapy. Pursuing the Plasma Cell; Lonial, S., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 393–419. [Google Scholar]
- Gajate, C.; Mollinedo, F. Lipid rafts and Fas/CD95 signaling in cancer chemotherapy. Recent Pat. Anticancer Drug Discov. 2011, 6, 274–283. [Google Scholar] [CrossRef] [Green Version]
- Gajate, C.; Mollinedo, F. Lipid rafts and raft-mediated supramolecular entities in the regulation of CD95 death receptor apoptotic signaling. Apoptosis 2015, 20, 584–606. [Google Scholar] [CrossRef]
- Gajate, C.; Mollinedo, F. Lipid raft-mediated Fas/CD95 apoptotic signaling in leukemic cells and normal leukocytes and therapeutic implications. J. Leukoc. Biol. 2015, 98, 739–759. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid rafts as major platforms for signaling regulation in cancer. Adv. Biol. Regul. 2015, 57, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Gajate, C. Fas/CD95, Lipid rafts, and cancer. In TRAIL, Fas Ligand, TNF and TLR3 in Cancer; Micheau, O., Ed.; Springer International Publishing AG: Cham, Switzerland, 2017; pp. 187–227. [Google Scholar]
- Mollinedo, F.; Gajate, C. FasL-independent activation of Fas. In Fas Signaling; Wajant, H., Ed.; Landes Bioscience and Springer Science: Georgetown, TX, USA, 2006; pp. 13–27. [Google Scholar]
- Reis-Sobreiro, M.; Roue, G.; Moros, A.; Gajate, C.; de la Iglesia-Vicente, J.; Colomer, D.; Mollinedo, F. Lipid raft-mediated Akt signaling as a therapeutic target in mantle cell lymphoma. Blood Cancer J. 2013, 3, e118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollinedo, F. Lipid raft involvement in yeast cell growth and death. Front. Oncol. 2012, 2, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, R.; Kielland-Brandt, M.C.; Fink, G.R. Yeast plasma membrane ATPase is essential for growth and has homology with (Na+ + K+), K+- and Ca2+-ATPases. Nature 1986, 319, 689–693. [Google Scholar] [CrossRef]
- Bagnat, M.; Chang, A.; Simons, K. Plasma membrane proton ATPase Pma1p requires raft association for surface delivery in yeast. Mol. Biol. Cell 2001, 12, 4129–4138. [Google Scholar] [CrossRef] [Green Version]
- Zaremberg, V.; Gajate, C.; Cacharro, L.M.; Mollinedo, F.; McMaster, C.R. Cytotoxicity of an anti-cancer lysophospholipid through selective modification of lipid raft composition. J. Biol. Chem. 2005, 280, 38047–38058. [Google Scholar] [CrossRef] [Green Version]
- Cuesta-Marban, A.; Botet, J.; Czyz, O.; Cacharro, L.M.; Gajate, C.; Hornillos, V.; Delgado, J.; Zhang, H.; Amat-Guerri, F.; Acuna, A.U.; et al. Drug uptake, lipid rafts, and vesicle trafficking modulate resistance to an anticancer lysophosphatidylcholine analogue in yeast. J. Biol. Chem. 2013, 288, 8405–8418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czyz, O.; Bitew, T.; Cuesta-Marban, A.; McMaster, C.R.; Mollinedo, F.; Zaremberg, V. Alteration of plasma membrane organization by an anticancer lysophosphatidylcholine analogue induces intracellular acidification and internalization of plasma membrane transporters in yeast. J. Biol. Chem. 2013, 288, 8419–8432. [Google Scholar] [CrossRef] [Green Version]
- Malinska, K.; Malinsky, J.; Opekarova, M.; Tanner, W. Visualization of protein compartmentation within the plasma membrane of living yeast cells. Mol. Biol. Cell 2003, 14, 4427–4436. [Google Scholar] [CrossRef]
- Hearn, J.D.; Lester, R.L.; Dickson, R.C. The uracil transporter Fur4p associates with lipid rafts. J. Biol. Chem. 2003, 278, 3679–3686. [Google Scholar] [CrossRef] [Green Version]
- Ausili, A.; Martinez-Valera, P.; Torrecillas, A.; Gomez-Murcia, V.; de Godos, A.M.; Corbalan-Garcia, S.; Teruel, J.A.; Gomez Fernandez, J.C. Anticancer agent edelfosine exhibits a high affinity for cholesterol and disorganizes liquid-ordered membrane structures. Langmuir 2018, 34, 8333–8346. [Google Scholar] [CrossRef]
- Busto, J.V.; del Canto-Jañez, E.; Goñi, F.M.; Mollinedo, F.; Alonso, A. Combination of the anti-tumour cell ether lipid edelfosine with sterols abolishes haemolytic side effects of the drug. J. Chem. Biol. 2008, 1, 89–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto-Miguel, T.; Gajate, C.; Mollinedo, F. Differential targets and subcellular localization of antitumor alkyl-lysophospholipid in leukemic versus solid tumor Cells. J. Biol. Chem. 2006, 281, 14833–14840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Gajate, C.; Yu, L.P.; Fang, Y.X.; Mollinedo, F. Mitochondrial-derived ROS in edelfosine-induced apoptosis in yeasts and tumor cells. Acta Pharmacol. Sin. 2007, 28, 888–894. [Google Scholar] [CrossRef] [Green Version]
- Quesada, E.; Delgado, J.; Gajate, C.; Mollinedo, F.; Acuna, A.U.; Amat-Guerri, F. Fluorescent phenylpolyene analogues of the ether phospholipid edelfosine for the selective labeling of cancer cells. J. Med. Chem. 2004, 47, 5333–5335. [Google Scholar] [CrossRef] [PubMed]
- Mateo, C.R.; Souto, A.A.; Amat-Guerri, F.; Acuna, A.U. New fluorescent octadecapentaenoic acids as probes of lipid membranes and protein-lipid interactions. Biophys. J. 1996, 71, 2177–2191. [Google Scholar] [CrossRef] [Green Version]
- Quesada, E.; Acuna, A.U.; Amat-Guerri, F. New Transmembrane Polyene Bolaamphiphiles as Fluorescent Probes in Lipid Bilayers. Angew. Chem. Int. Ed. Engl. 2001, 40, 2095–2097. [Google Scholar] [CrossRef]
- Mollinedo, F.; Fernandez, M.; Hornillos, V.; Delgado, J.; Amat-Guerri, F.; Acuna, A.U.; Nieto-Miguel, T.; Villa-Pulgarin, J.A.; Gonzalez-Garcia, C.; Cena, V.; et al. Involvement of lipid rafts in the localization and dysfunction effect of the antitumor ether phospholipid edelfosine in mitochondria. Cell Death Dis. 2011, 2, e158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa-Pulgarin, J.A.; Gajate, C.; Botet, J.; Jimenez, A.; Justies, N.; Varela, M.R.; Cuesta-Marban, A.; Muller, I.; Modolell, M.; Revuelta, J.L.; et al. Mitochondria and lipid raft-located FOF1-ATP synthase as major therapeutic targets in the antileishmanial and anticancer activities of ether lipid edelfosine. PLoS Negl. Trop. Dis. 2017, 11, e0005805. [Google Scholar] [CrossRef]
- Kuerschner, L.; Richter, D.; Hannibal-Bach, H.K.; Gaebler, A.; Shevchenko, A.; Ejsing, C.S.; Thiele, C. Exogenous ether lipids predominantly target mitochondria. PLoS ONE 2012, 7, e31342. [Google Scholar] [CrossRef] [Green Version]
- Dowhan, W. Molecular basis for membrane phospholipid diversity: Why are there so many lipids? Annu. Rev. Biochem. 1997, 66, 199–232. [Google Scholar] [CrossRef] [Green Version]
- Olson, R.E. Discovery of the lipoproteins, their role in fat transport and their significance as risk factors. J. Nutr. 1998, 128, 439S–443S. [Google Scholar] [CrossRef]
- Kuijpers, P. History in medicine: The story of cholesterol, lipids and cardiology. J. Cradiol. Pract. 2021, 19, 1–5. [Google Scholar]
- Walusinski, O. Charcot and cholesterin. Eur. Neurol. 2019, 81, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Maxfield, F.R.; van Meer, G. Cholesterol, the central lipid of mammalian cells. Curr. Opin. Cell Biol. 2010, 22, 422–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, D. Phase transitions and fluidity characteristics of lipids and cell membranes. Q. Rev. Biophys. 1975, 8, 185–235. [Google Scholar] [CrossRef] [PubMed]
- Jaipuria, G.; Ukmar-Godec, T.; Zweckstetter, M. Challenges and approaches to understand cholesterol-binding impact on membrane protein function: An NMR view. Cell Mol. Life Sci. 2018, 75, 2137–2151. [Google Scholar] [CrossRef]
- Mondal, M.; Mesmin, B.; Mukherjee, S.; Maxfield, F.R. Sterols are mainly in the cytoplasmic leaflet of the plasma membrane and the endocytic recycling compartment in CHO cells. Mol. Biol. Cell 2009, 20, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.L.; Sheng, R.; Jung, J.H.; Wang, L.; Stec, E.; O’Connor, M.J.; Song, S.; Bikkavilli, R.K.; Winn, R.A.; Lee, D.; et al. Orthogonal lipid sensors identify transbilayer asymmetry of plasma membrane cholesterol. Nat. Chem. Biol. 2017, 13, 268–274. [Google Scholar] [CrossRef] [Green Version]
- Lange, Y.; Swaisgood, M.H.; Ramos, B.V.; Steck, T.L. Plasma membranes contain half the phospholipid and 90% of the cholesterol and sphingomyelin in cultured human fibroblasts. J. Biol. Chem. 1989, 264, 3786–3793. [Google Scholar] [CrossRef]
- Das, A.; Brown, M.S.; Anderson, D.D.; Goldstein, J.L.; Radhakrishnan, A. Three pools of plasma membrane cholesterol and their relation to cholesterol homeostasis. eLife 2014, 3, e02882. [Google Scholar] [CrossRef]
- Fantini, J.; Barrantes, F.J. How cholesterol interacts with membrane proteins: An exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 2013, 4, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daum, G.; Vance, J.E. Import of lipids into mitochondria. Prog. Lipid Res. 1997, 36, 103–130. [Google Scholar] [CrossRef]
- Horvath, S.E.; Daum, G. Lipids of mitochondria. Prog Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef]
- Simons, K.; Vaz, W.L. Model systems, lipid rafts, and cell membranes. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 269–295. [Google Scholar] [CrossRef]
- Subczynski, W.K.; Pasenkiewicz-Gierula, M.; Widomska, J.; Mainali, L.; Raguz, M. High Cholesterol/Low Cholesterol: Effects in biological membranes: A review. Cell Biochem. Biophys. 2017, 75, 369–385. [Google Scholar] [CrossRef]
- Zakany, F.; Kovacs, T.; Panyi, G.; Varga, Z. Direct and indirect cholesterol effects on membrane proteins with special focus on potassium channels. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158706. [Google Scholar] [CrossRef]
- Ohvo-Rekila, H.; Ramstedt, B.; Leppimaki, P.; Slotte, J.P. Cholesterol interactions with phospholipids in membranes. Prog. Lipid Res. 2002, 41, 66–97. [Google Scholar] [CrossRef]
- Desai, R.; Campanella, M. Exploring mitochondrial cholesterol signalling for therapeutic intervention in neurological conditions. Br. J. Pharmacol. 2019, 176, 4284–4292. [Google Scholar] [CrossRef]
- Miller, W.L.; Bose, H.S. Early steps in steroidogenesis: Intracellular cholesterol trafficking. J. Lipid Res. 2011, 52, 2111–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elustondo, P.; Martin, L.A.; Karten, B. Mitochondrial cholesterol import. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, J.; Liu, N.; Kuhn, L.A.; Garavito, R.M.; Ferguson-Miller, S. Translocator protein 18 kDa (TSPO): An old protein with new functions? Biochemistry 2016, 55, 2821–2831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, V.; Fan, J.; Zirkin, B. Translocator protein (18 kDa): An update on its function in steroidogenesis. J. Neuroendocrinol. 2018, 30, e12500. [Google Scholar] [CrossRef]
- Liu, J.; Rone, M.B.; Papadopoulos, V. Protein-protein interactions mediate mitochondrial cholesterol transport and steroid biosynthesis. J. Biol. Chem. 2006, 281, 38879–38893. [Google Scholar] [CrossRef] [Green Version]
- Shoshan-Barmatz, V.; Pittala, S.; Mizrachi, D. VDAC1 and the TSPO: Expression, interactions, and associated functions in health and disease states. Int. J. Mol. Sci. 2019, 20, 3348. [Google Scholar] [CrossRef] [Green Version]
- Delavoie, F.; Li, H.; Hardwick, M.; Robert, J.C.; Giatzakis, C.; Peranzi, G.; Yao, Z.X.; Maccario, J.; Lacapere, J.J.; Papadopoulos, V. In vivo and in vitro peripheral-type benzodiazepine receptor polymerization: Functional significance in drug ligand and cholesterol binding. Biochemistry 2003, 42, 4506–4519. [Google Scholar] [CrossRef]
- Fantini, J.; Di Scala, C.; Evans, L.S.; Williamson, P.T.; Barrantes, F.J. A mirror code for protein-cholesterol interactions in the two leaflets of biological membranes. Sci. Rep. 2016, 6, 21907. [Google Scholar] [CrossRef] [Green Version]
- Vance, J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar] [CrossRef]
- Flis, V.V.; Daum, G. Lipid transport between the endoplasmic reticulum and mitochondria. Cold Spring Harb. Perspect. Biol. 2013, 5, a013235. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Csordas, G.; Renken, C.; Varnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnoczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helle, S.C.; Kanfer, G.; Kolar, K.; Lang, A.; Michel, A.H.; Kornmann, B. Organization and function of membrane contact sites. Biochim. Biophys. Acta 2013, 1833, 2526–2541. [Google Scholar] [CrossRef] [Green Version]
- Van Vliet, A.R.; Verfaillie, T.; Agostinis, P. New functions of mitochondria associated membranes in cellular signaling. Biochim. Biophys. Acta 2014, 1843, 2253–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annunziata, I.; Sano, R.; d’Azzo, A. Mitochondria-associated ER membranes (MAMs) and lysosomal storage diseases. Cell Death Dis. 2018, 9, 328. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Min, K.T. The interface between ER and mitochondria: Molecular compositions and functions. Mol. Cells 2018, 41, 1000–1007. [Google Scholar] [PubMed]
- Barbosa, A.D.; Siniossoglou, S. Function of lipid droplet-organelle interactions in lipid homeostasis. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1459–1468. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Murphy, S.; Martin, S.; Parton, R.G. Lipid droplet-organelle interactions; sharing the fats. Biochim. Biophys. Acta 2009, 1791, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Hou, X.; Shen, W.J.; Hanssen, R.; Khor, V.K.; Cortez, Y.; Roseman, A.N.; Azhar, S.; Kraemer, F.B. SNARE-mediated cholesterol movement to mitochondria supports steroidogenesis in rodent cells. Mol. Endocrinol. 2016, 30, 234–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagerstrom, S.; Polesie, S.; Wickstrom, Y.; Johansson, B.R.; Schroder, H.D.; Hojlund, K.; Bostrom, P. Lipid droplets interact with mitochondria using SNAP23. Cell Biol. Int. 2009, 33, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, V.; Chawla, A.; Roche, P.A. Identification of a novel syntaxin- and synaptobrevin/VAMP-binding protein, SNAP-23, expressed in non-neuronal tissues. J. Biol. Chem. 1996, 271, 13300–13303. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F.; Lazo, P.A. Identification of two isoforms of the vesicle-membrane fusion protein SNAP-23 in human neutrophils and HL-60 cells. Biochem. Biophys. Res. Commun. 1997, 231, 808–812. [Google Scholar] [CrossRef]
- Martin-Martin, B.; Nabokina, S.M.; Blasi, J.; Lazo, P.A.; Mollinedo, F. Involvement of SNAP-23 and syntaxin 6 in human neutrophil exocytosis. Blood 2000, 96, 2574–2583. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Calafat, J.; Janssen, H.; Martin-Martin, B.; Canchado, J.; Nabokina, S.M.; Gajate, C. Combinatorial SNARE complexes modulate the secretion of cytoplasmic granules in human neutrophils. J. Immunol. 2006, 177, 2831–2841. [Google Scholar] [CrossRef] [Green Version]
- Bissig, C.; Gruenberg, J. Lipid sorting and multivesicular endosome biogenesis. Cold Spring Harb. Perspect. Biol. 2013, 5, a016816. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Jiang, L.; Yang, H.; Song, B.L. Routes and mechanisms of post-endosomal cholesterol trafficking: A story that never ends. Traffic 2017, 18, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Ouweneel, A.B.; Thomas, M.J.; Sorci-Thomas, M.G. The ins and outs of lipid rafts: Functions in intracellular cholesterol homeostasis, microparticles, and cell membranes. J. Lipid Res. 2020, 61, 676–686. [Google Scholar] [CrossRef] [Green Version]
- Vanier, M.T.; Millat, G. Niemann-Pick disease type C. Clin. Genet. 2003, 64, 269–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.Y.; Reid, P.C.; Sugii, S.; Ohgami, N.; Cruz, J.C.; Chang, C.C. Niemann-Pick type C disease and intracellular cholesterol trafficking. J. Biol. Chem. 2005, 280, 20917–20920. [Google Scholar] [CrossRef] [Green Version]
- Sevin, M.; Lesca, G.; Baumann, N.; Millat, G.; Lyon-Caen, O.; Vanier, M.T.; Sedel, F. The adult form of Niemann-Pick disease type C. Brain 2007, 130, 120–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piroth, T.; Boelmans, K.; Amtage, F.; Rijntjes, M.; Wierciochin, A.; Musacchio, T.; Weiller, C.; Volkmann, J.; Klebe, S. Adult-onset niemann-pick disease type C: Rapid treatment initiation advised but early diagnosis remains difficult. Front. Neurol. 2017, 8, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, R.E.; Abi-Mosleh, L.; Radhakrishnan, A.; Dale, J.D.; Brown, M.S.; Goldstein, J.L. Purified NPC1 protein. I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J. Biol. Chem. 2008, 283, 1052–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naureckiene, S.; Sleat, D.E.; Lackland, H.; Fensom, A.; Vanier, M.T.; Wattiaux, R.; Jadot, M.; Lobel, P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 2000, 290, 2298–2301. [Google Scholar] [CrossRef] [PubMed]
- Storch, J.; Xu, Z. Niemann-Pick C2 (NPC2) and intracellular cholesterol trafficking. Biochim. Biophys. Acta 2009, 1791, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Cheruku, S.R.; Xu, Z.; Dutia, R.; Lobel, P.; Storch, J. Mechanism of cholesterol transfer from the Niemann-Pick type C2 protein to model membranes supports a role in lysosomal cholesterol transport. J. Biol. Chem. 2006, 281, 31594–31604. [Google Scholar] [CrossRef]
- Berger, A.C.; Hanson, P.K.; Wylie Nichols, J.; Corbett, A.H. A yeast model system for functional analysis of the Niemann-Pick type C protein 1 homolog, Ncr1p. Traffic 2005, 6, 907–917. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, P.; Dwyer, N.K.; Christenson, L.K.; Fujimoto, T.; Martinez, F.; Comly, M.; Hanover, J.A.; Blanchette-Mackie, E.J.; Strauss, J.F., 3rd. MLN64 mediates mobilization of lysosomal cholesterol to steroidogenic mitochondria. J. Biol. Chem. 2002, 277, 33300–33310. [Google Scholar] [CrossRef] [Green Version]
- Bose, H.S.; Whittal, R.M.; Ran, Y.; Bose, M.; Baker, B.Y.; Miller, W.L. StAR-like activity and molten globule behavior of StARD6, a male germ-line protein. Biochemistry 2008, 47, 2277–2288. [Google Scholar] [CrossRef]
- Soccio, R.E.; Breslow, J.L. Intracellular cholesterol transport. Arterioscler. Thromb Vasc. Biol. 2004, 24, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.A.; Kennedy, B.E.; Karten, B. Mitochondrial cholesterol: Mechanisms of import and effects on mitochondrial function. J. Bioenerg. Biomembr. 2016, 48, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Arenas, F.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Intracellular Cholesterol Trafficking and Impact in Neurodegeneration. Front. Mol. Neurosci. 2017, 10, 382. [Google Scholar] [CrossRef] [Green Version]
- Roca-Agujetas, V.; Barbero-Camps, E.; de Dios, C.; Podlesniy, P.; Abadin, X.; Morales, A.; Mari, M.; Trullas, R.; Colell, A. Cholesterol alters mitophagy by impairing optineurin recruitment and lysosomal clearance in Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Ribas, V.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondria, cholesterol and cancer cell metabolism. Clin. Transl. Med. 2016, 5, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feo, F.; Canuto, R.A.; Garcea, R.; Gabriel, L. Effect of cholesterol content on some physical and functional properties of mitochondria isolated from adult rat liver, fetal liver, cholesterol-enriched liver and hepatomas AH-130, 3924A and 5123. Biochim. Biophys. Acta 1975, 413, 116–134. [Google Scholar] [CrossRef]
- Crain, R.C.; Clark, R.W.; Harvey, B.E. Role of lipid transfer proteins in the abnormal lipid content of Morris hepatoma mitochondria and microsomes. Cancer Res. 1983, 43, 3197–3202. [Google Scholar]
- Montero, J.; Morales, A.; Llacuna, L.; Lluis, J.M.; Terrones, O.; Basanez, G.; Antonsson, B.; Prieto, J.; Garcia-Ruiz, C.; Colell, A.; et al. Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res. 2008, 68, 5246–5256. [Google Scholar] [CrossRef] [Green Version]
- Krycer, J.R.; Brown, A.J. Cholesterol accumulation in prostate cancer: A classic observation from a modern perspective. Biochim. Biophys. Acta 2013, 1835, 219–229. [Google Scholar] [CrossRef]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The role of cholesterol in cancer. Cancer Res. 2016, 76, 2063–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parlo, R.A.; Coleman, P.S. Enhanced rate of citrate export from cholesterol-rich hepatoma mitochondria. The truncated Krebs cycle and other metabolic ramifications of mitochondrial membrane cholesterol. J. Biol. Chem. 1984, 259, 9997–10003. [Google Scholar] [CrossRef]
- Baggetto, L.G.; Clottes, E.; Vial, C. Low mitochondrial proton leak due to high membrane cholesterol content and cytosolic creatine kinase as two features of the deviant bioenergetics of Ehrlich and AS30-D tumor cells. Cancer Res. 1992, 52, 4935–4941. [Google Scholar]
- Lucken-Ardjomande, S.; Montessuit, S.; Martinou, J.C. Bax activation and stress-induced apoptosis delayed by the accumulation of cholesterol in mitochondrial membranes. Cell Death Differ. 2008, 15, 484–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoratti, M.; Szabo, I. The mitochondrial permeability transition. Biochim. Biophys. Acta 1995, 1241, 139–176. [Google Scholar] [CrossRef]
- Halestrap, A.P.; McStay, G.P.; Clarke, S.J. The permeability transition pore complex: Another view. Biochimie 2002, 84, 153–166. [Google Scholar] [CrossRef]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef]
- Rao, V.K.; Carlson, E.A.; Yan, S.S. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim. Biophys. Acta 2014, 1842, 1267–1272. [Google Scholar] [CrossRef] [Green Version]
- Kwong, J.Q.; Molkentin, J.D. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P.; Brenner, C. The adenine nucleotide translocase: A central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 2003, 10, 1507–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camara, A.K.S.; Zhou, Y.; Wen, P.C.; Tajkhorshid, E.; Kwok, W.M. Mitochondrial VDAC1: A key gatekeeper as potential therapeutic target. Front. Physiol. 2017, 8, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, V.; Baraldi, M.; Guilarte, T.R.; Knudsen, T.B.; Lacapere, J.J.; Lindemann, P.; Norenberg, M.D.; Nutt, D.; Weizman, A.; Zhang, M.R.; et al. Translocator protein (18kDa): New nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006, 27, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Si Chaib, Z.; Marchetto, A.; Dishnica, K.; Carloni, P.; Giorgetti, A.; Rossetti, G. Impact of cholesterol on the stability of monomeric and dimeric forms of the translocator protein TSPO: A molecular simulation study. Molecules 2020, 25, 4299. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, V.; Stocco, D.M.; Tu, L.N. Minireview: Translocator protein (TSPO) and steroidogenesis: A reappraisal. Mol. Endocrinol. 2015, 29, 490–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochette, L.; Meloux, A.; Zeller, M.; Malka, G.; Cottin, Y.; Vergely, C. Mitochondrial SLC25 carriers: Novel targets for cancer therapy. Molecules 2020, 25, 2417. [Google Scholar] [CrossRef]
- Giorgio, V.; Bisetto, E.; Soriano, M.E.; Dabbeni-Sala, F.; Basso, E.; Petronilli, V.; Forte, M.A.; Bernardi, P.; Lippe, G. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J. Biol. Chem. 2009, 284, 33982–33988. [Google Scholar] [CrossRef] [Green Version]
- Beutner, G.; Alanzalon, R.E.; Porter, G.A., Jr. Cyclophilin D regulates the dynamic assembly of mitochondrial ATP synthase into synthasomes. Sci. Rep. 2017, 7, 14488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokoszka, J.E.; Waymire, K.G.; Levy, S.E.; Sligh, J.E.; Cai, J.; Jones, D.P.; MacGregor, G.R.; Wallace, D.C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004, 427, 461–465. [Google Scholar] [CrossRef]
- Krauskopf, A.; Eriksson, O.; Craigen, W.J.; Forte, M.A.; Bernardi, P. Properties of the permeability transition in VDAC1(-/-) mitochondria. Biochim. Biophys. Acta 2006, 1757, 590–595. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Sheiko, T.; Craigen, W.J.; Molkentin, J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 2007, 9, 550–555. [Google Scholar] [CrossRef]
- Sileikyte, J.; Blachly-Dyson, E.; Sewell, R.; Carpi, A.; Menabo, R.; Di Lisa, F.; Ricchelli, F.; Bernardi, P.; Forte, M. Regulation of the mitochondrial permeability transition pore by the outer membrane does not involve the peripheral benzodiazepine receptor (Translocator Protein of 18 kDa (TSPO)). J. Biol. Chem. 2014, 289, 13769–13781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez-Aguilar, M.; Douglas, D.L.; Gibson, A.K.; Domeier, T.L.; Molkentin, J.D.; Baines, C.P. Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. J. Mol. Cell Cardiol. 2014, 72, 316–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, S. A novel conceptual model for the dual role of FOF1-ATP synthase in cell life and cell death. Biomol. Concepts 2020, 11, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; Burchell, V.; Schiavone, M.; Bassot, C.; Minervini, G.; Petronilli, V.; Argenton, F.; Forte, M.; Tosatto, S.; Lippe, G.; et al. Ca2+ binding to F-ATP synthase beta subunit triggers the mitochondrial permeability transition. EMBO Rep. 2017, 18, 1065–1076. [Google Scholar] [CrossRef]
- Watt, I.N.; Montgomery, M.G.; Runswick, M.J.; Leslie, A.G.; Walker, J.E. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc. Natl. Acad. Sci. USA 2010, 107, 16823–16827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Ford, H.C.; Carroll, J.; Douglas, C.; Gonzales, E.; Ding, S.; Fearnley, I.M.; Walker, J.E. Assembly of the membrane domain of ATP synthase in human mitochondria. Proc. Natl. Acad. Sci. USA 2018, 115, 2988–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Pfanner, N.; Becker, T. Assembling the mitochondrial ATP synthase. Proc. Natl. Acad. Sci. USA 2018, 115, 2850–2852. [Google Scholar] [CrossRef] [Green Version]
- Okuno, D.; Iino, R.; Noji, H. Rotation and structure of FoF1-ATP synthase. J. Biochem. 2011, 149, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Davies, K.M.; Strauss, M.; Daum, B.; Kief, J.H.; Osiewacz, H.D.; Rycovska, A.; Zickermann, V.; Kuhlbrandt, W. Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 14121–14126. [Google Scholar] [CrossRef] [Green Version]
- Strauss, M.; Hofhaus, G.; Schroder, R.R.; Kuhlbrandt, W. Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 2008, 27, 1154–1160. [Google Scholar] [CrossRef] [Green Version]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brethes, D.; di Rago, J.P.; Velours, J. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 2002, 21, 221–230. [Google Scholar] [CrossRef]
- Bonora, M.; Bononi, A.; De Marchi, E.; Giorgi, C.; Lebiedzinska, M.; Marchi, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; Wojtala, A.; et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 2013, 12, 674–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azarashvili, T.; Odinokova, I.; Bakunts, A.; Ternovsky, V.; Krestinina, O.; Tyynela, J.; Saris, N.E. Potential role of subunit c of F0F1-ATPase and subunit c of storage body in the mitochondrial permeability transition. Effect of the phosphorylation status of subunit c on pore opening. Cell Calcium 2014, 55, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E.V. ATP synthase C-subunit-deficient mitochondria have a small cyclosporine A-sensitive channel, but lack the permeability transition pore. Cell Rep. 2019, 26, 11–17.e12. [Google Scholar] [CrossRef] [Green Version]
- Mnatsakanyan, N.; Llaguno, M.C.; Yang, Y.; Yan, Y.; Weber, J.; Sigworth, F.J.; Jonas, E.A. A mitochondrial megachannel resides in monomeric F1FO ATP synthase. Nat. Commun. 2019, 10, 5823. [Google Scholar] [CrossRef] [PubMed]
- Mnatsakanyan, N.; Jonas, E.A. ATP synthase c-subunit ring as the channel of mitochondrial permeability transition: Regulator of metabolism in development and degeneration. J. Mol. Cell Cardiol. 2020, 144, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Karch, J.; Kwong, J.Q.; Burr, A.R.; Sargent, M.A.; Elrod, J.W.; Peixoto, P.M.; Martinez-Caballero, S.; Osinska, H.; Cheng, E.H.; Robbins, J.; et al. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. eLife 2013, 2, e00772. [Google Scholar] [CrossRef]
- Naumova, N.; Sachl, R. Regulation of cell death by mitochondrial transport systems of calcium and Bcl-2 proteins. Membranes 2020, 10, 299. [Google Scholar] [CrossRef]
- Lamb, H.M. Double agents of cell death: Novel emerging functions of apoptotic regulators. FEBS J. 2020, 287, 2647–2663. [Google Scholar] [CrossRef] [Green Version]
- Alavian, K.N.; Li, H.; Collis, L.; Bonanni, L.; Zeng, L.; Sacchetti, S.; Lazrove, E.; Nabili, P.; Flaherty, B.; Graham, M.; et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat. Cell Biol. 2011, 13, 1224–1233. [Google Scholar] [CrossRef]
- Chen, Y.B.; Aon, M.A.; Hsu, Y.T.; Soane, L.; Teng, X.; McCaffery, J.M.; Cheng, W.C.; Qi, B.; Li, H.; Alavian, K.N.; et al. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J. Cell Biol. 2011, 195, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Lardy, H.A.; Johnson, D.; Mc, M.W. Antibiotics as tools for metabolic studies. I. A survey of toxic antibiotics in respiratory, phosphorylative and glycolytic systems. Arch. Biochem. Biophys. 1958, 78, 587–597. [Google Scholar] [CrossRef]
- Racker, E. A mitochondrial factor conferring oligomycin sensitivity on soluble mitochondrial ATPase. Biochem. Biophys. Res. Commun. 1963, 10, 435–439. [Google Scholar] [CrossRef]
- Kagawa, Y.; Racker, E. Partial resolution of the enzymes catalyzing oxidative phosphorylation. 8. Properties of a factor conferring oligomycin sensitivity on mitochondrial adenosine triphosphatase. J. Biol. Chem. 1966, 241, 2461–2466. [Google Scholar] [CrossRef]
- Symersky, J.; Osowski, D.; Walters, D.E.; Mueller, D.M. Oligomycin frames a common drug-binding site in the ATP synthase. Proc. Natl. Acad. Sci. USA 2012, 109, 13961–13965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniel, M.; Giorgio, V.; Fogolari, F.; Glick, G.D.; Bernardi, P.; Lippe, G. The oligomycin-sensitivity conferring protein of mitochondrial ATP synthase: Emerging new roles in mitochondrial pathophysiology. Int. J. Mol. Sci. 2014, 15, 7513–7536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpe, J.C.; Arnoult, D.; Youle, R.J. Control of mitochondrial permeability by Bcl-2 family members. Biochim. Biophys. Acta 2004, 1644, 107–113. [Google Scholar] [CrossRef]
- Di Lisa, F.; Bernardi, P. Mitochondrial function as a determinant of recovery or death in cell response to injury. Mol. Cell Biochem. 1998, 184, 379–391. [Google Scholar] [CrossRef]
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 2005, 17, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Breckenridge, D.G.; Stojanovic, M.; Marcellus, R.C.; Shore, G.C. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 2003, 160, 1115–1127. [Google Scholar] [CrossRef]
- Burgeiro, A.; Pereira, C.V.; Carvalho, F.S.; Pereira, G.C.; Mollinedo, F.; Oliveira, P.J. Edelfosine and perifosine disrupt hepatic mitochondrial oxidative phosphorylation and induce the permeability transition. Mitochondrion 2013, 13, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Christian, A.E.; Haynes, M.P.; Phillips, M.C.; Rothblat, G.H. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 1997, 38, 2264–2272. [Google Scholar] [CrossRef]
- Pagliarani, A.; Nesci, S.; Ventrella, V. Novel Drugs Targeting the c-Ring of the F1FO-ATP Synthase. Mini Rev. Med. Chem. 2016, 16, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.P.; Luo, M.; Zhou, W.; Symersky, J.; Bai, D.; Chambers, M.G.; Faraldo-Gomez, J.D.; Liao, M.; Mueller, D.M. High-resolution cryo-EM analysis of the yeast ATP synthase in a lipid membrane. Science 2018, 360, 6389. [Google Scholar] [CrossRef] [Green Version]
- Veenman, L.; Alten, J.; Linnemannstons, K.; Shandalov, Y.; Zeno, S.; Lakomek, M.; Gavish, M.; Kugler, W. Potential involvement of F0F1-ATP(synth)ase and reactive oxygen species in apoptosis induction by the antineoplastic agent erucylphosphohomocholine in glioblastoma cell lines: A mechanism for induction of apoptosis via the 18 kDa mitochondrial translocator protein. Apoptosis 2010, 15, 753–768. [Google Scholar] [PubMed] [Green Version]
- Norais, N.; Prome, D.; Velours, J. ATP synthase of yeast mitochondria. Characterization of subunit d and sequence analysis of the structural gene ATP7. J. Biol. Chem. 1991, 266, 16541–16549. [Google Scholar] [CrossRef]
- Ausili, A.; Torrecillas, A.; Aranda, F.J.; Mollinedo, F.; Gajate, C.; Corbalan-Garcia, S.; de Godos, A.; Gomez-Fernandez, J.C. Edelfosine is incorporated into rafts and alters their organization. J. Phys. Chem. B 2008, 112, 11643–11654. [Google Scholar] [CrossRef] [PubMed]
- Castro, B.M.; Fedorov, A.; Hornillos, V.; Delgado, J.; Acuna, A.U.; Mollinedo, F.; Prieto, M. Edelfosine and miltefosine effects on lipid raft properties: Membrane biophysics in cell death by antitumor lipids. J. Phys. Chem. B 2013, 117, 7929–7940. [Google Scholar] [CrossRef]
- Bernardi, P.; Vassanelli, S.; Veronese, P.; Colonna, R.; Szabo, I.; Zoratti, M. Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J. Biol. Chem. 1992, 267, 2934–2939. [Google Scholar] [CrossRef]
- Matsuyama, S.; Xu, Q.; Velours, J.; Reed, J.C. The Mitochondrial F0F1-ATPase proton pump is required for function of the proapoptotic protein Bax in yeast and mammalian cells. Mol. Cell 1998, 1, 327–336. [Google Scholar] [CrossRef]
- Gajate, C.; Mollinedo, F. Isolation of lipid rafts through discontinuous sucrose gradient centrifugation and Fas/CD95 death receptor localization in raft fractions. Methods Mol. Biol. 2017, 1557, 125–138. [Google Scholar] [PubMed]
- Gajate, C.; Mollinedo, F. Lipid raft isolation by sucrose gradient centrifugation and visualization of raft-located proteins by fluorescence microscopy: The use of combined techniques to assess Fas/CD95 location in rafts during apoptosis triggering. Methods Mol. Biol. 2021, 2187, 147–186. [Google Scholar] [PubMed]
- Das, B.; Mondragon, M.O.; Sadeghian, M.; Hatcher, V.B.; Norin, A.J. A novel ligand in lymphocyte-mediated cytotoxicity: Expression of the beta subunit of H+ transporting ATP synthase on the surface of tumor cell lines. J. Exp. Med. 1994, 180, 273–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, T.J.; Kim, M.S.; Kim, J.W.; Kim, B.W.; Choo, H.J.; Lee, J.W.; Kim, K.B.; Lee, C.S.; Kim, J.H.; Chang, S.Y.; et al. Lipid raft proteome reveals ATP synthase complex in the cell surface. Proteomics 2004, 4, 3536–3548. [Google Scholar] [CrossRef]
- Ma, Z.; Cao, M.; Liu, Y.; He, Y.; Wang, Y.; Yang, C.; Wang, W.; Du, Y.; Zhou, M.; Gao, F. Mitochondrial F1Fo-ATP synthase translocates to cell surface in hepatocytes and has high activity in tumor-like acidic and hypoxic environment. Acta Biochim. Biophys. Sin. 2010, 42, 530–537. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.W.; Lee, C.S.; Yi, J.S.; Lee, J.H.; Lee, J.W.; Choo, H.J.; Jung, S.Y.; Kim, M.S.; Lee, S.W.; Lee, M.S.; et al. Lipid raft proteome reveals that oxidative phosphorylation system is associated with the plasma membrane. Expert Rev. Proteom. 2010, 7, 849–866. [Google Scholar] [CrossRef]
- Kawai, Y.; Kaidoh, M.; Yokoyama, Y.; Ohhashi, T. Cell surface F1/FO ATP synthase contributes to interstitial flow-mediated development of the acidic microenvironment in tumor tissues. Am. J. Physiol. Cell Physiol. 2013, 305, C1139–C1150. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.J.; Shi, X.X.; Liu, Y.W.; He, Y.Q.; Wang, Y.Z.; Yang, C.X.; Gao, F. The mechanism underlying the effects of the cell surface ATP synthase on the regulation of intracellular acidification during acidosis. J. Cell Biochem. 2013, 114, 1695–1703. [Google Scholar] [CrossRef]
- Allen-Worthington, K.; Xie, J.; Brown, J.L.; Edmunson, A.M.; Dowling, A.; Navratil, A.M.; Scavelli, K.; Yoon, H.; Kim, D.G.; Bynoe, M.S.; et al. The F0F1 ATP synthase complex localizes to membrane rafts in gonadotrope cells. Mol. Endocrinol. 2016, 30, 996–1011. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Feng, Z.; Guo, Y.; Zhang, T.; Mai, A.; Kang, Z.; Weijen, T.; Wang, D.; Yin, D.; Zhu, D.; et al. F0F1 ATP synthase regulates extracellular calcium influx in human neutrophils by interacting with Cav2.3 and modulates neutrophil accumulation in the lipopolysaccharide-challenged lung. Cell Commun. Signal. 2020, 18, 19. [Google Scholar] [CrossRef] [PubMed]
- Gajate, C.; Gonzalez-Camacho, F.; Mollinedo, F. Lipid raft connection between extrinsic and intrinsic apoptotic pathways. Biochem. Biophys. Res. Commun. 2009, 380, 780–784. [Google Scholar] [CrossRef] [PubMed]
- Schon, A.; Freire, E. Thermodynamics of intersubunit interactions in cholera toxin upon binding to the oligosaccharide portion of its cell surface receptor, ganglioside GM1. Biochemistry 1989, 28, 5019–5024. [Google Scholar] [CrossRef] [PubMed]
- Harder, T.; Scheiffele, P.; Verkade, P.; Simons, K. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 1998, 141, 929–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorice, M.; Matarrese, P.; Manganelli, V.; Tinari, A.; Giammarioli, A.M.; Mattei, V.; Misasi, R.; Garofalo, T.; Malorni, W. Role of GD3-CLIPR-59 association in lymphoblastoid T cell apoptosis triggered by CD95/Fas. PLoS ONE 2010, 5, e8567. [Google Scholar] [CrossRef]
- Sorice, M.; Garofalo, T.; Misasi, R.; Manganelli, V.; Vona, R.; Malorni, W. Ganglioside GD3 as a raft component in cell death regulation. Anticancer Agents Med. Chem. 2012, 12, 376–382. [Google Scholar] [CrossRef]
- Sorice, M.; Mattei, V.; Matarrese, P.; Garofalo, T.; Tinari, A.; Gambardella, L.; Ciarlo, L.; Manganelli, V.; Tasciotti, V.; Misasi, R.; et al. Dynamics of mitochondrial raft-like microdomains in cell life and death. Commun. Integr. Biol. 2012, 5, 217–219. [Google Scholar] [CrossRef] [Green Version]
- Ciarlo, L.; Manganelli, V.; Garofalo, T.; Matarrese, P.; Tinari, A.; Misasi, R.; Malorni, W.; Sorice, M. Association of fission proteins with mitochondrial raft-like domains. Cell Death Differ. 2010, 17, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, T.; Giammarioli, A.M.; Misasi, R.; Tinari, A.; Manganelli, V.; Gambardella, L.; Pavan, A.; Malorni, W.; Sorice, M. Lipid microdomains contribute to apoptosis-associated modifications of mitochondria in T cells. Cell Death Differ. 2005, 12, 1378–1389. [Google Scholar] [CrossRef]
- Sorice, M.; Manganelli, V.; Matarrese, P.; Tinari, A.; Misasi, R.; Malorni, W.; Garofalo, T. Cardiolipin-enriched raft-like microdomains are essential activating platforms for apoptotic signals on mitochondria. FEBS Lett. 2009, 583, 2447–2450. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.Z.; Berg, K.B.; Foster, L.J. Mitochondria do not contain lipid rafts, and lipid rafts do not contain mitochondrial proteins. J. Lipid Res. 2009, 50, 988–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, T.; Manganelli, V.; Grasso, M.; Mattei, V.; Ferri, A.; Misasi, R.; Sorice, M. Role of mitochondrial raft-like microdomains in the regulation of cell apoptosis. Apoptosis 2015, 20, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, M.; Hayashi, T.; Su, T.P. The role of cholesterol in the association of endoplasmic reticulum membranes with mitochondria. Biochem. Biophys. Res. Commun. 2012, 417, 635–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Area-Gomez, E.; Del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; de Groof, A.J.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [Green Version]
- Sano, R.; Annunziata, I.; Patterson, A.; Moshiach, S.; Gomero, E.; Opferman, J.; Forte, M.; d’Azzo, A. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol. Cell 2009, 36, 500–511. [Google Scholar] [CrossRef] [Green Version]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mollinedo, F.; Gajate, C. Mitochondrial Targeting Involving Cholesterol-Rich Lipid Rafts in the Mechanism of Action of the Antitumor Ether Lipid and Alkylphospholipid Analog Edelfosine. Pharmaceutics 2021, 13, 763. https://doi.org/10.3390/pharmaceutics13050763
Mollinedo F, Gajate C. Mitochondrial Targeting Involving Cholesterol-Rich Lipid Rafts in the Mechanism of Action of the Antitumor Ether Lipid and Alkylphospholipid Analog Edelfosine. Pharmaceutics. 2021; 13(5):763. https://doi.org/10.3390/pharmaceutics13050763
Chicago/Turabian StyleMollinedo, Faustino, and Consuelo Gajate. 2021. "Mitochondrial Targeting Involving Cholesterol-Rich Lipid Rafts in the Mechanism of Action of the Antitumor Ether Lipid and Alkylphospholipid Analog Edelfosine" Pharmaceutics 13, no. 5: 763. https://doi.org/10.3390/pharmaceutics13050763
APA StyleMollinedo, F., & Gajate, C. (2021). Mitochondrial Targeting Involving Cholesterol-Rich Lipid Rafts in the Mechanism of Action of the Antitumor Ether Lipid and Alkylphospholipid Analog Edelfosine. Pharmaceutics, 13(5), 763. https://doi.org/10.3390/pharmaceutics13050763