
Ruthenium Complexes in the Fight against Pathogenic Microorganisms. An Extensive Review



Abstract
:1. Introduction
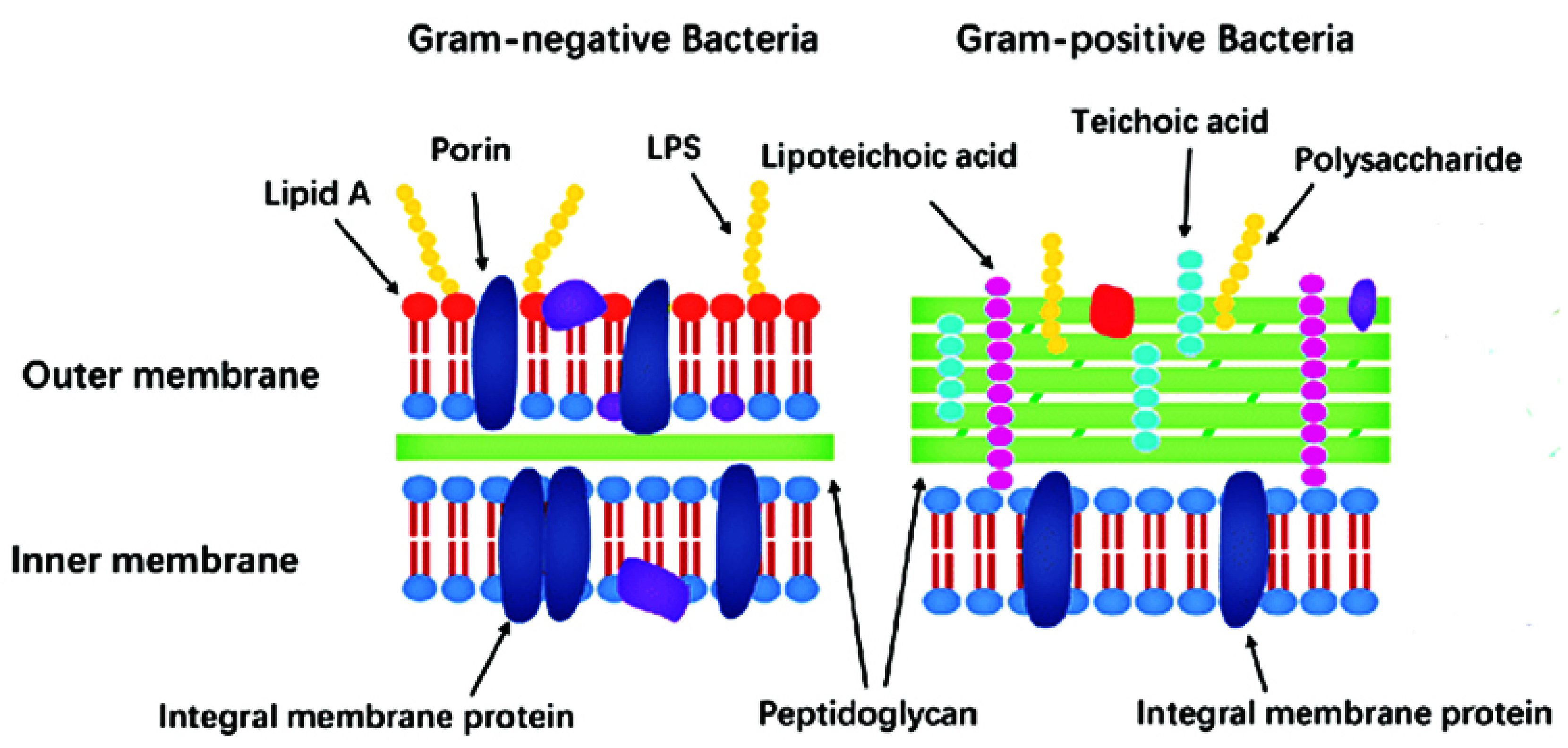
2. General Remarks on Bacterial Cell Structure. Gram-Positive vs. Gram-Negative Strains
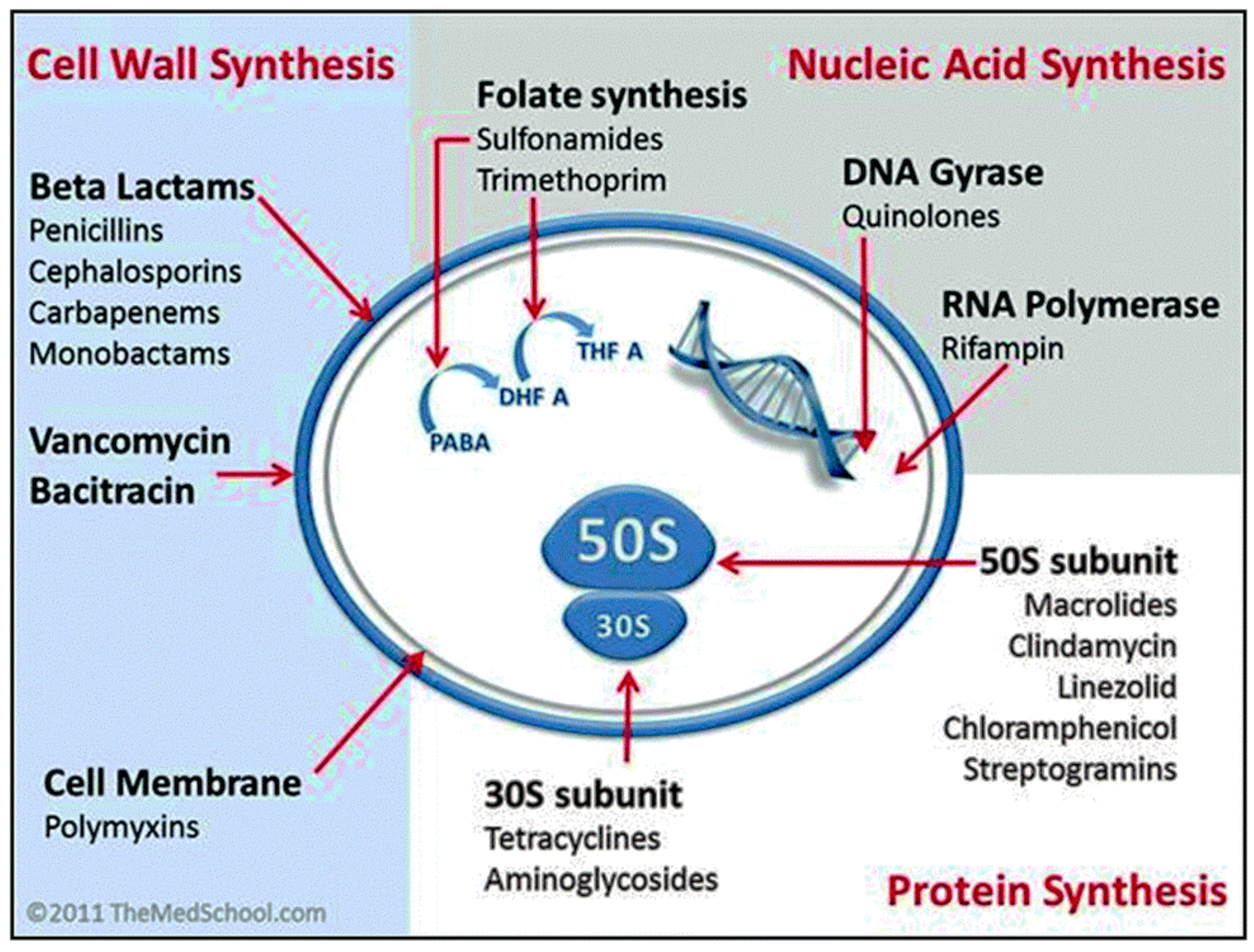
3. Mechanisms of Action of Current Drugs
4. Mechanisms of Resistance to Antibiotics
5. Antibacterial and Antifungal Activities of Ruthenium Complexes
5.1. Mononuclear Ruthenium (II) Complexes
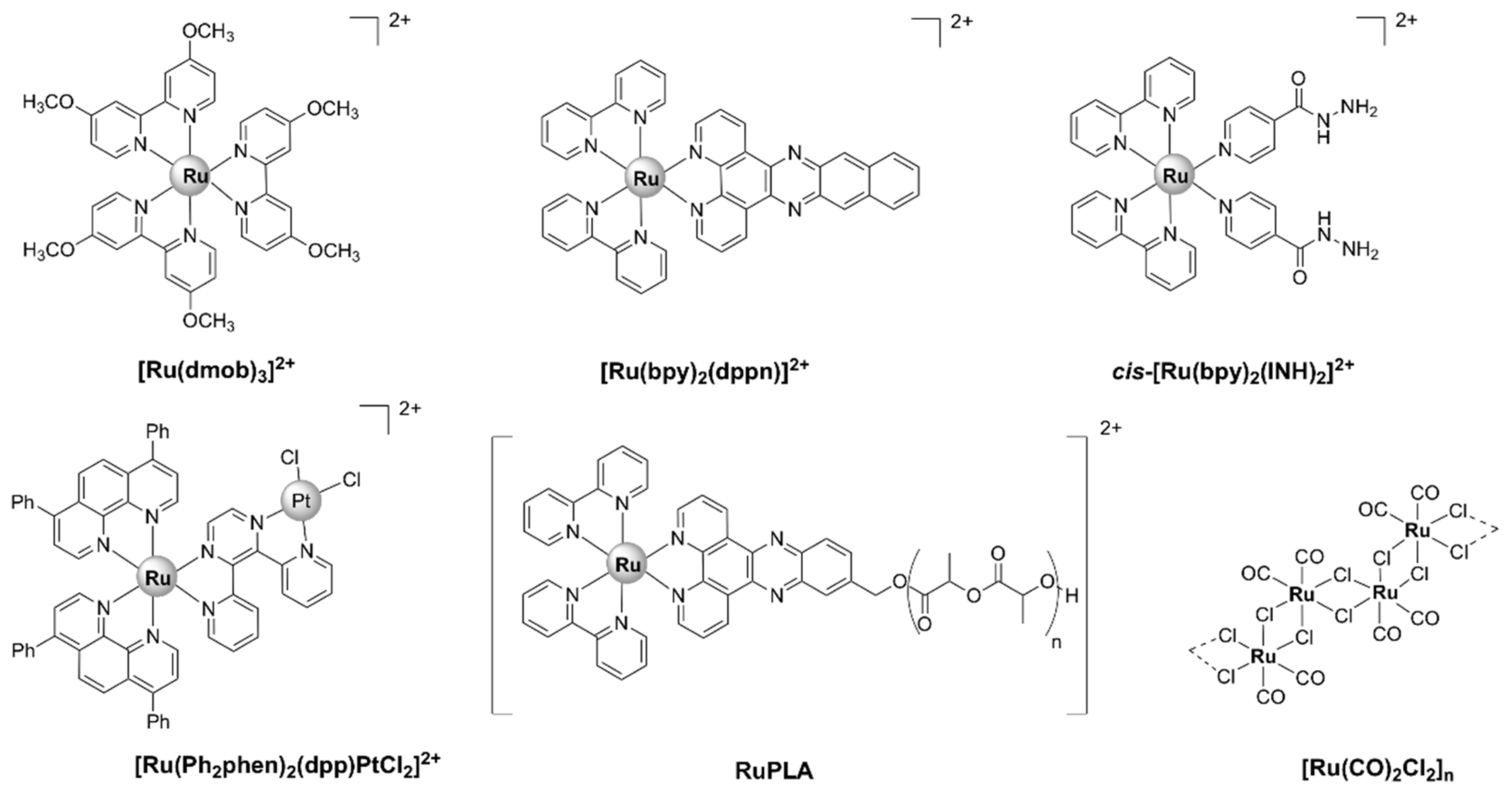
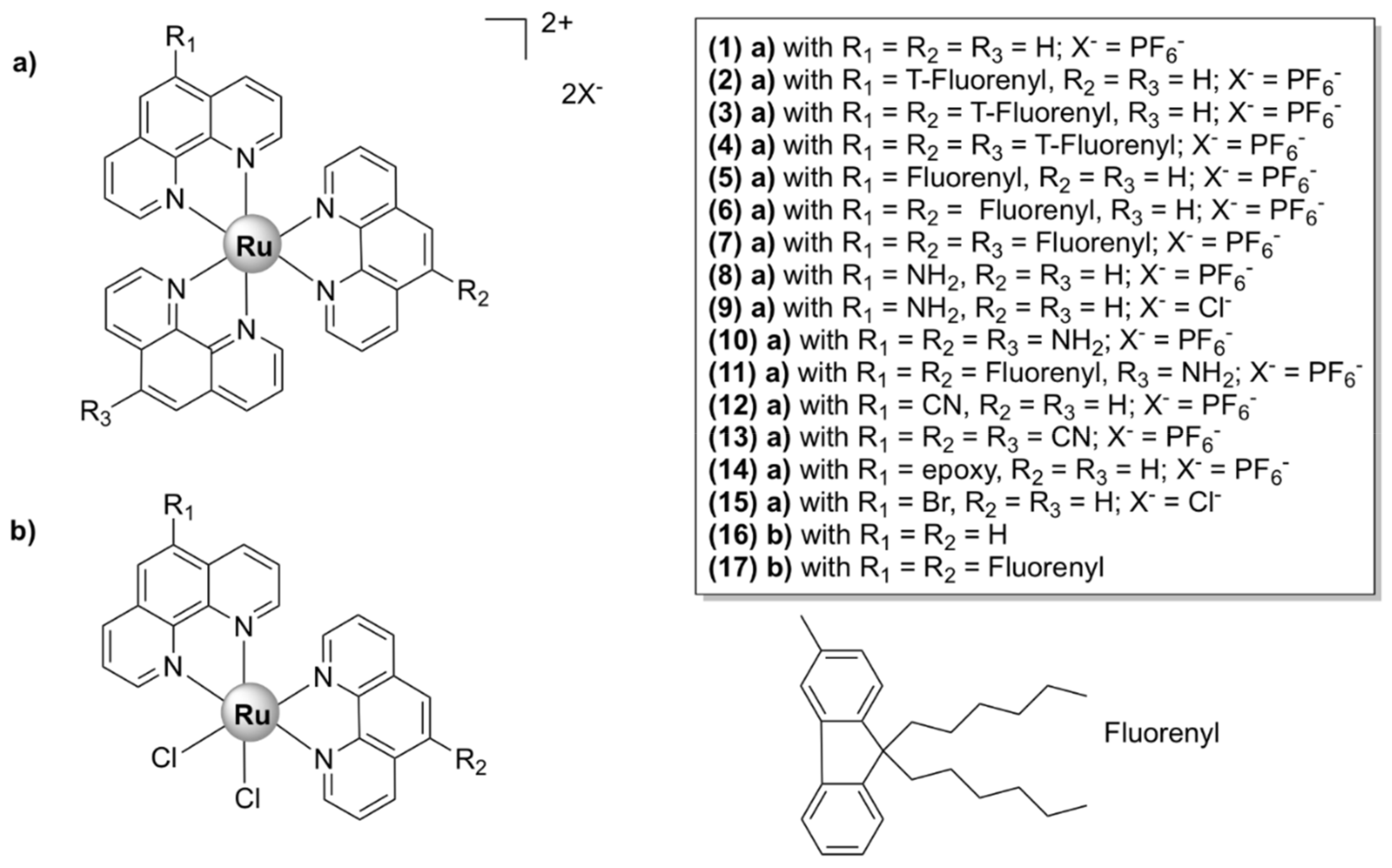
5.1.1. Mononuclear Polypyridyl Ru (II) Complexes
Mononuclear Ru(II) Heteroleptic Complexes Bearing 2,2’-Bipyridine (bpy) Ligands
Mononuclear Ru(II) Heteroleptic Complexes Bearing 1,10-phenanthroline (phen)
Mononuclear Ru (II) Heteroleptic Complexes Bearing Pyridophenazine Ligands
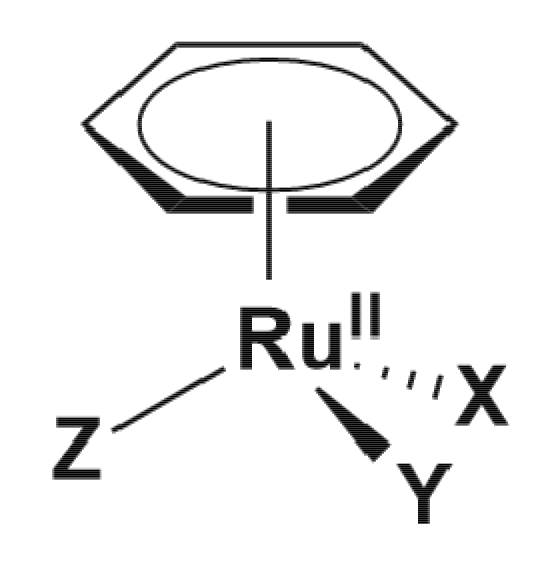
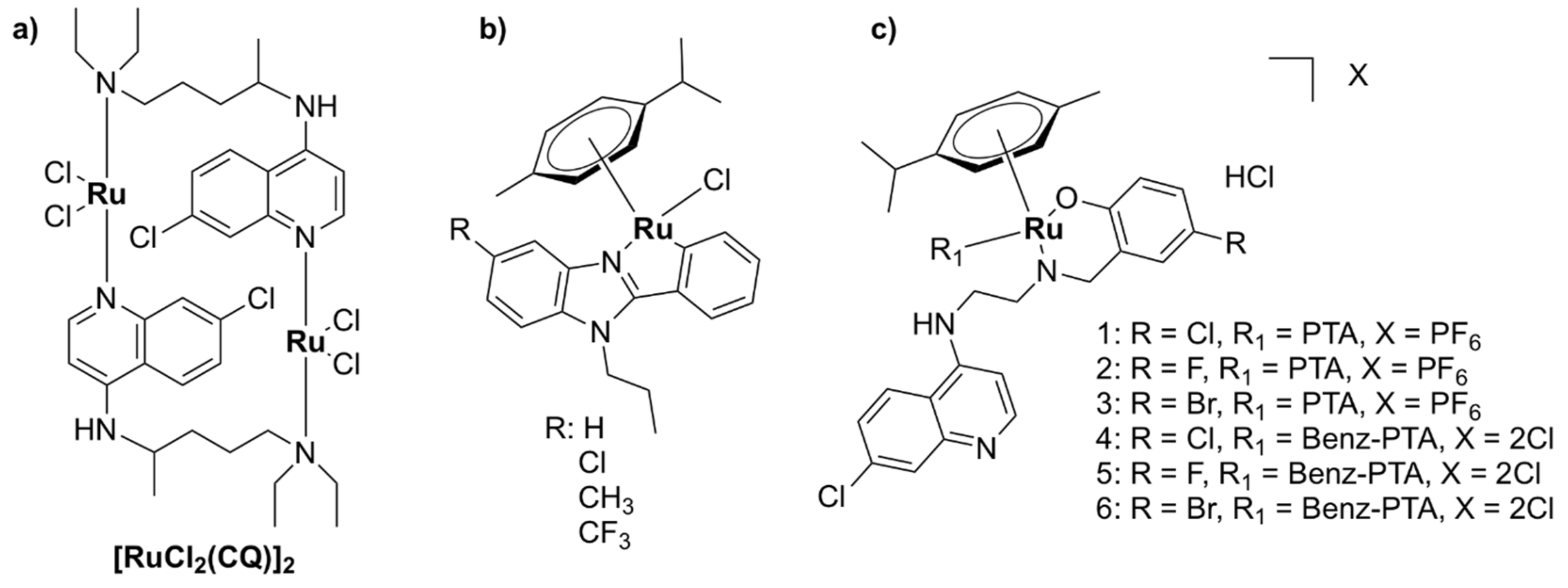
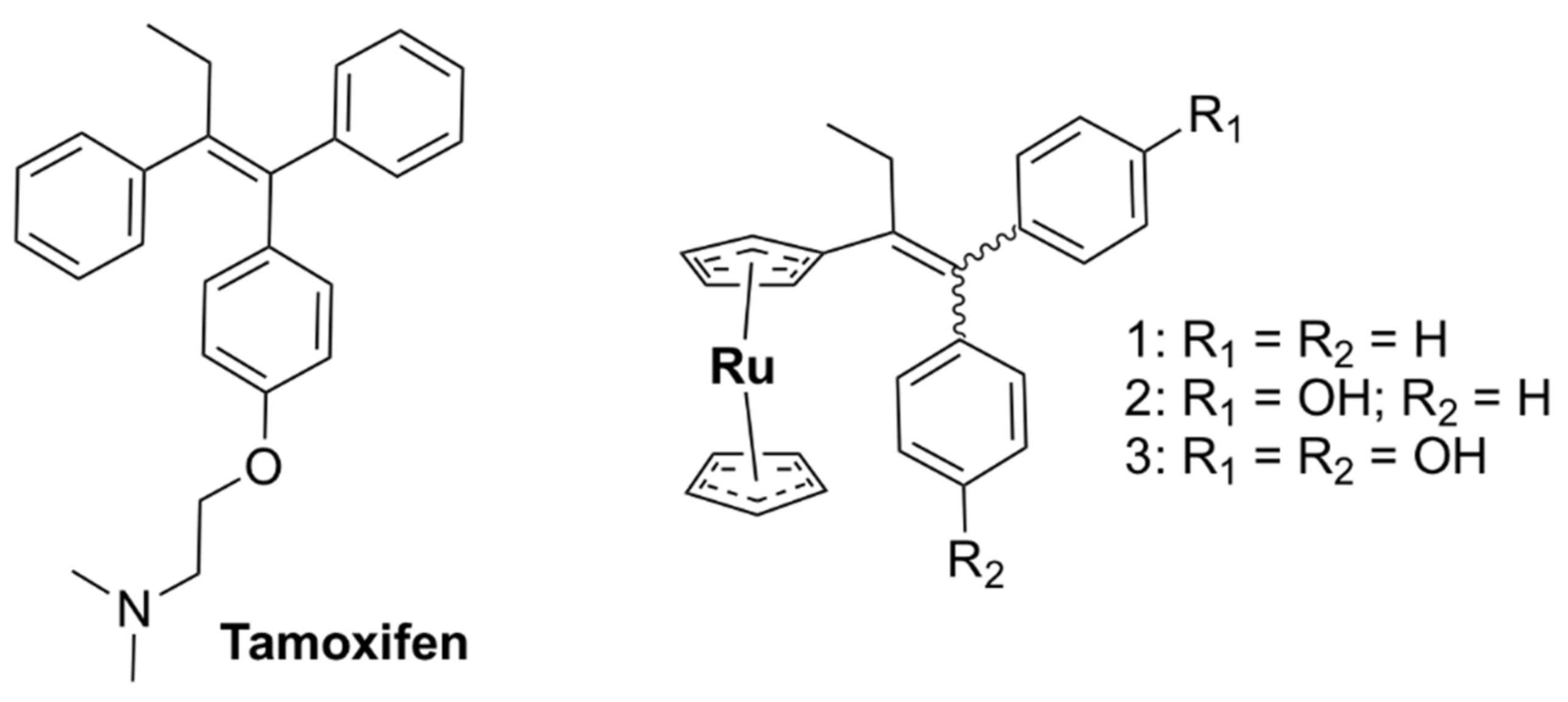
5.1.2. Mononuclear Ru (II)–arene Complexes
5.1.3. Other Mononuclear Ru Complexes
5.2. Polynuclear Ruthenium (II) Complexes
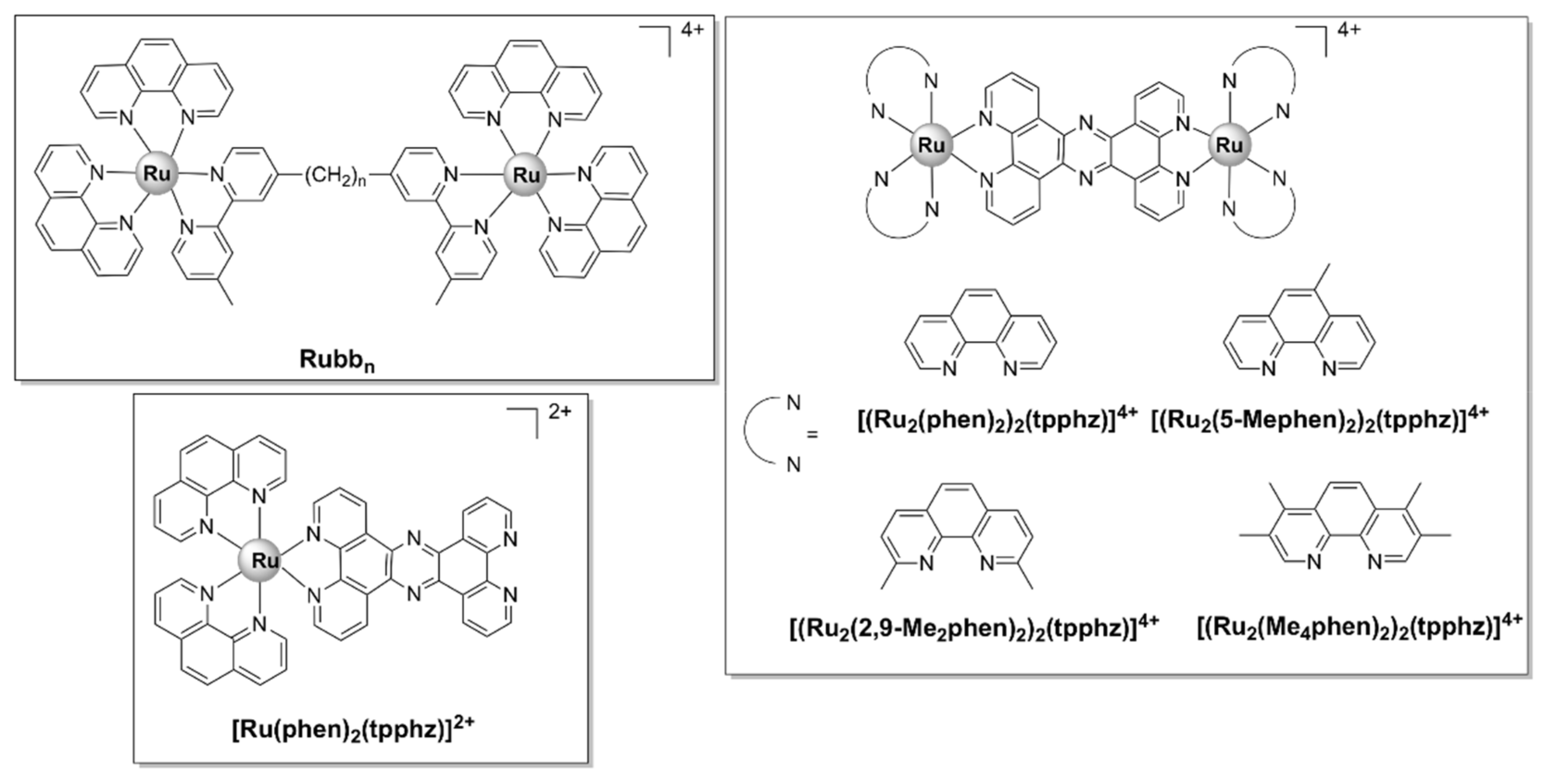
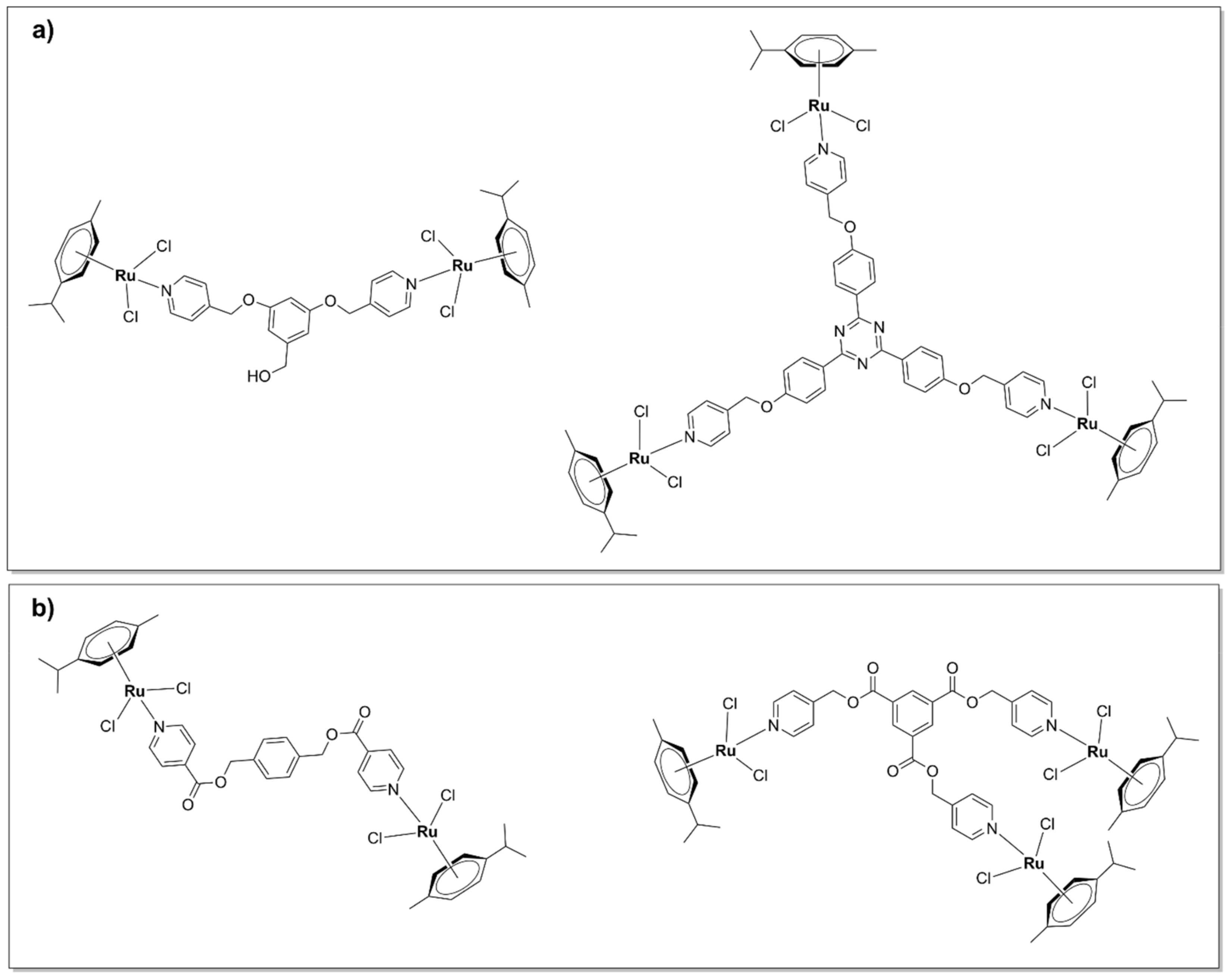
5.2.1. Kinetically Inert Dinuclear Polypyridylruthenium (II) Complexes
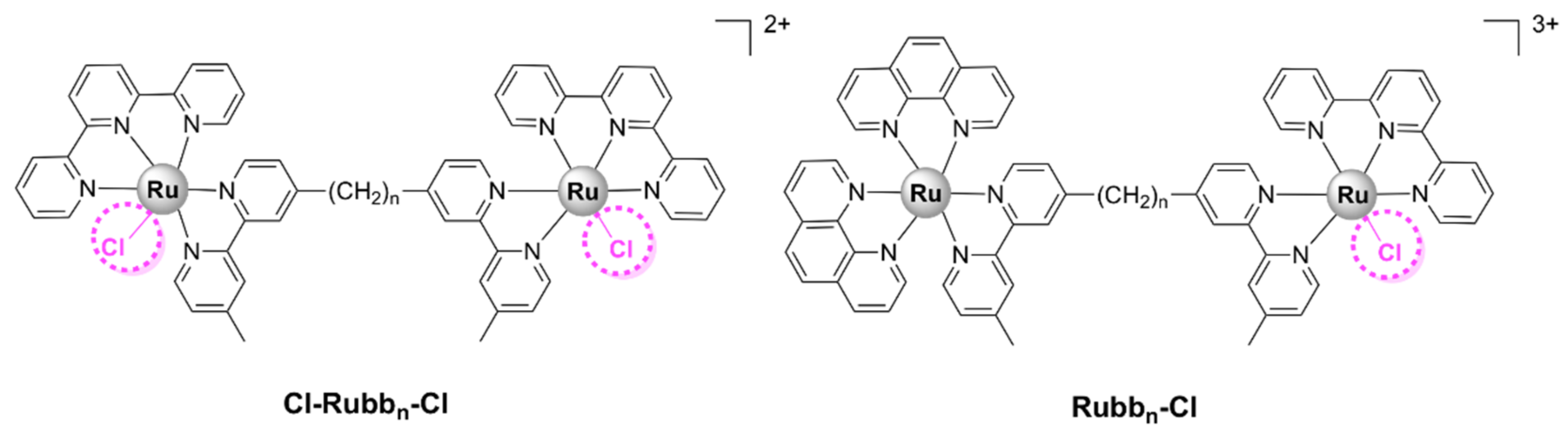
5.2.2. Chlorido Dinuclear Polypyridylruthenium (II) Complexes
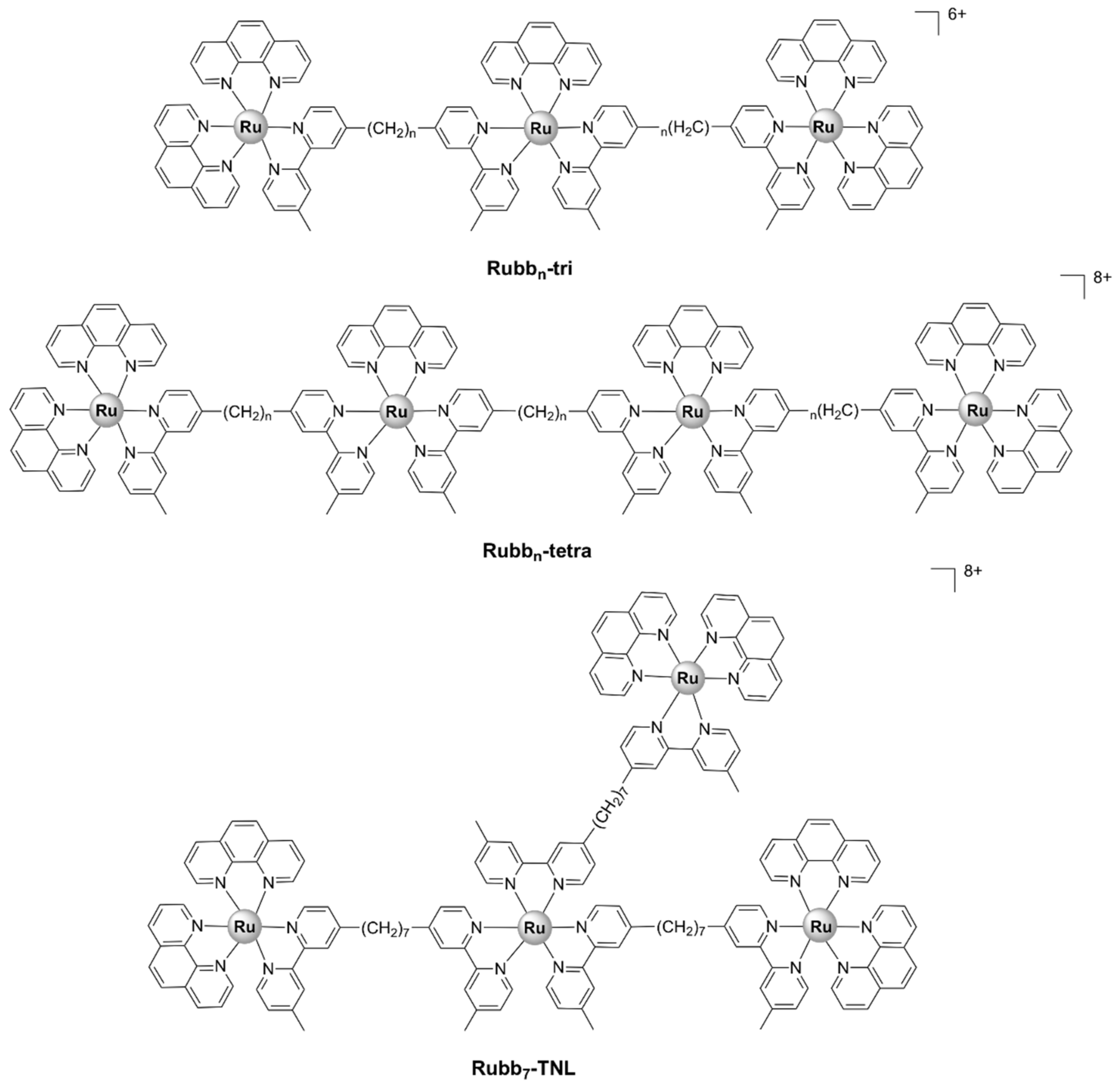
5.2.3. Tri-/Tetra-Nuclear Polypyridylruthenium(II) Complexes
5.2.4. Other Polynuclear Complexes
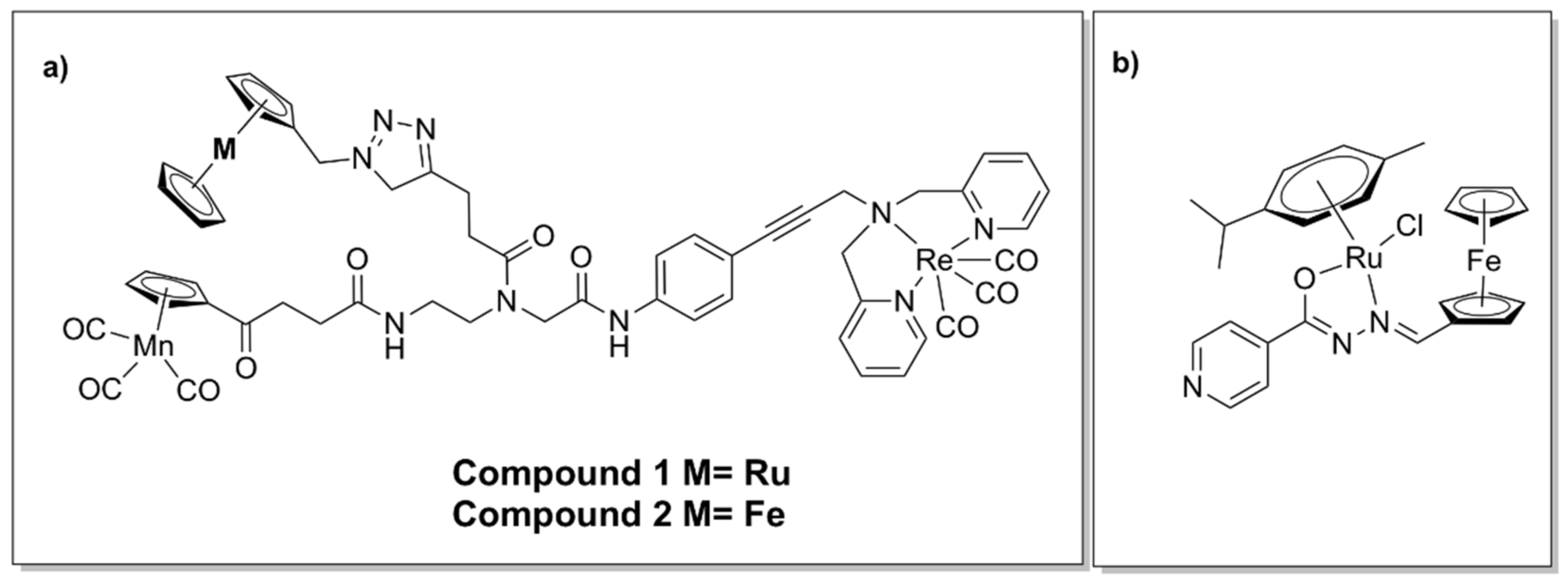
5.3. Hetero-bi/tri-Metallic Complexes
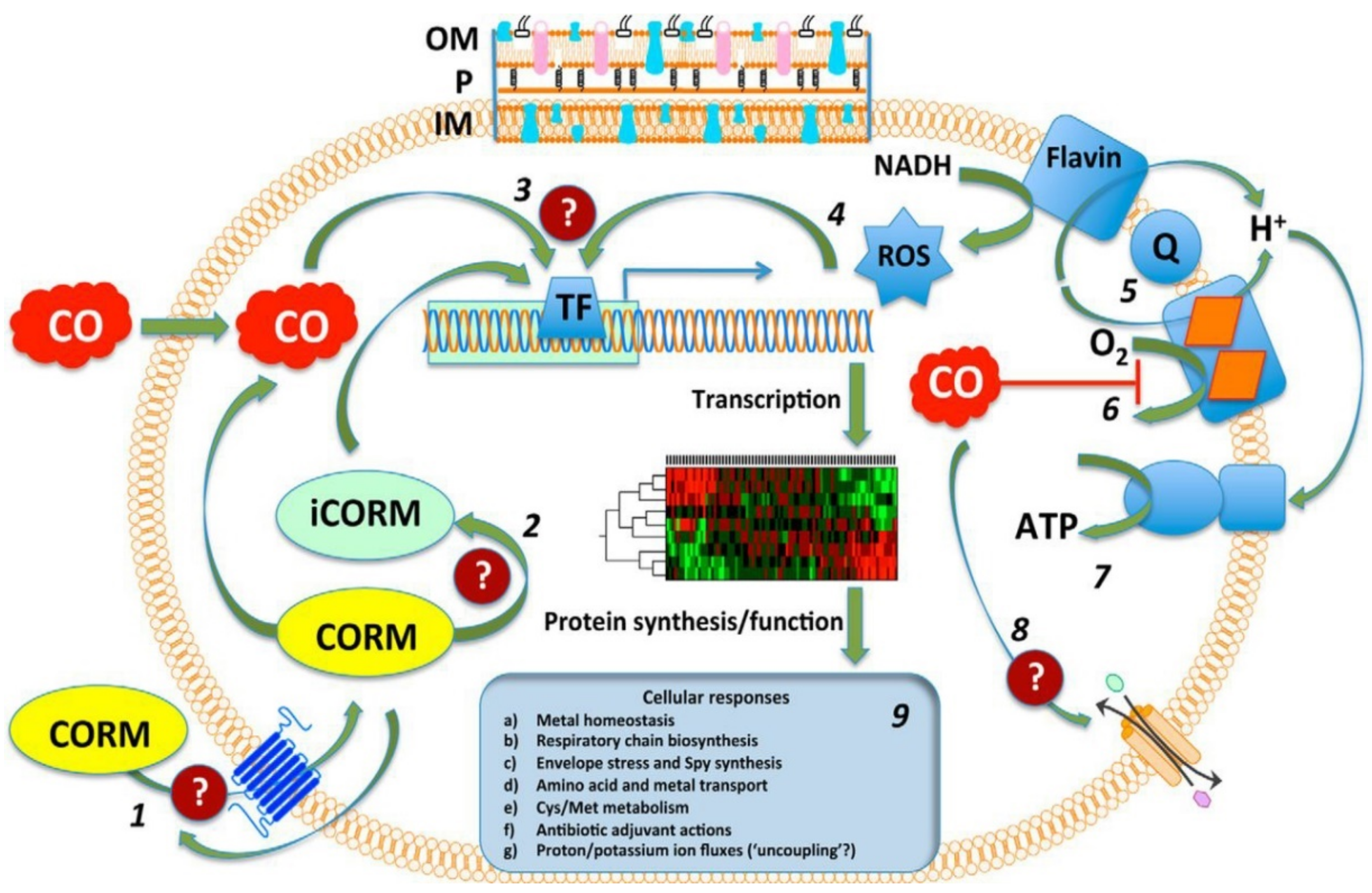
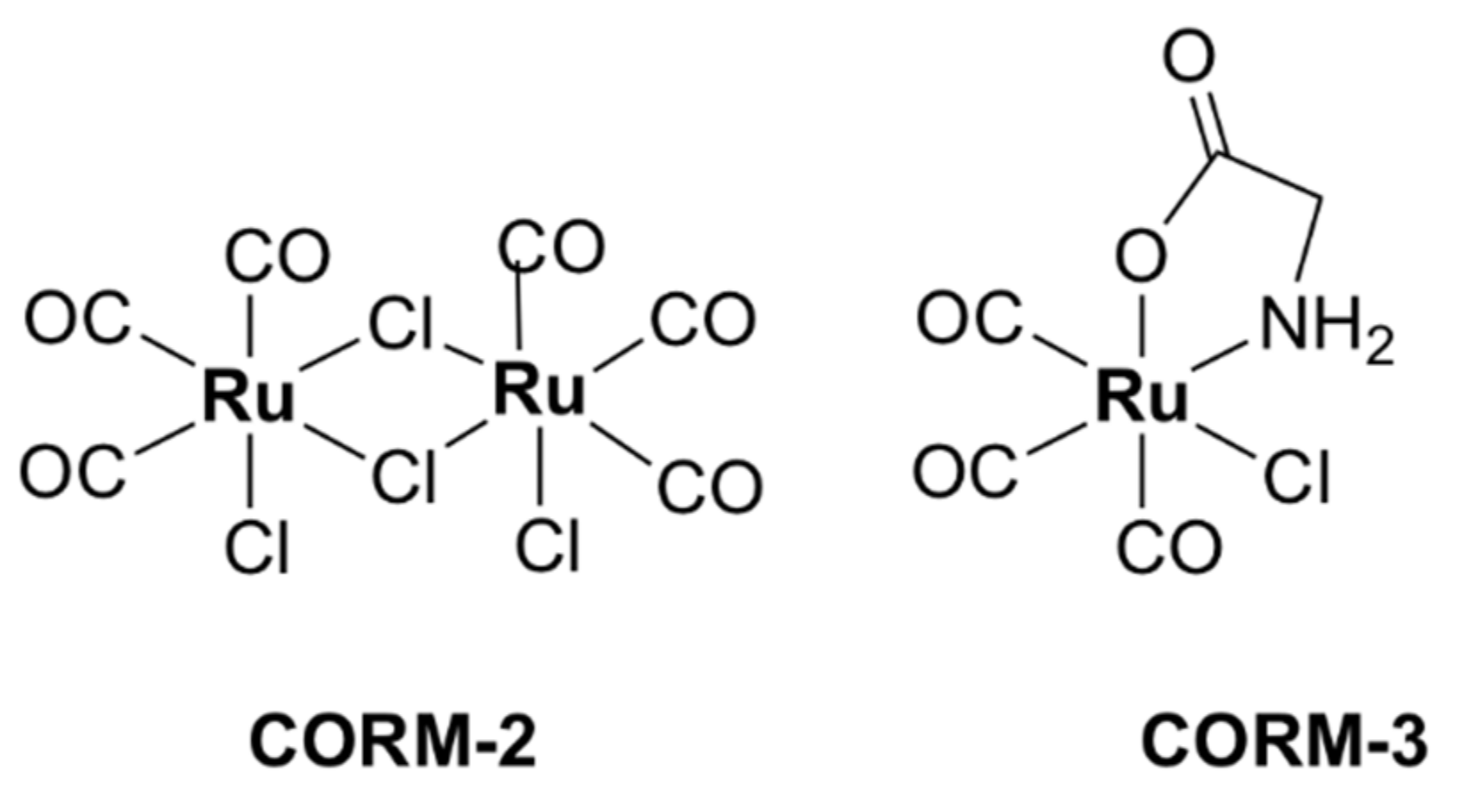
5.4. Ruthenium-Based Carbon-Monoxide-Releasing Molecules (CORMs)
5.4.1. Mechanisms of Action
The Role of CO
ROS Generation
Membrane Damage
The Role of the Ru(II) ion Interactions with Proteins and DNA
Effects on Gene Expression
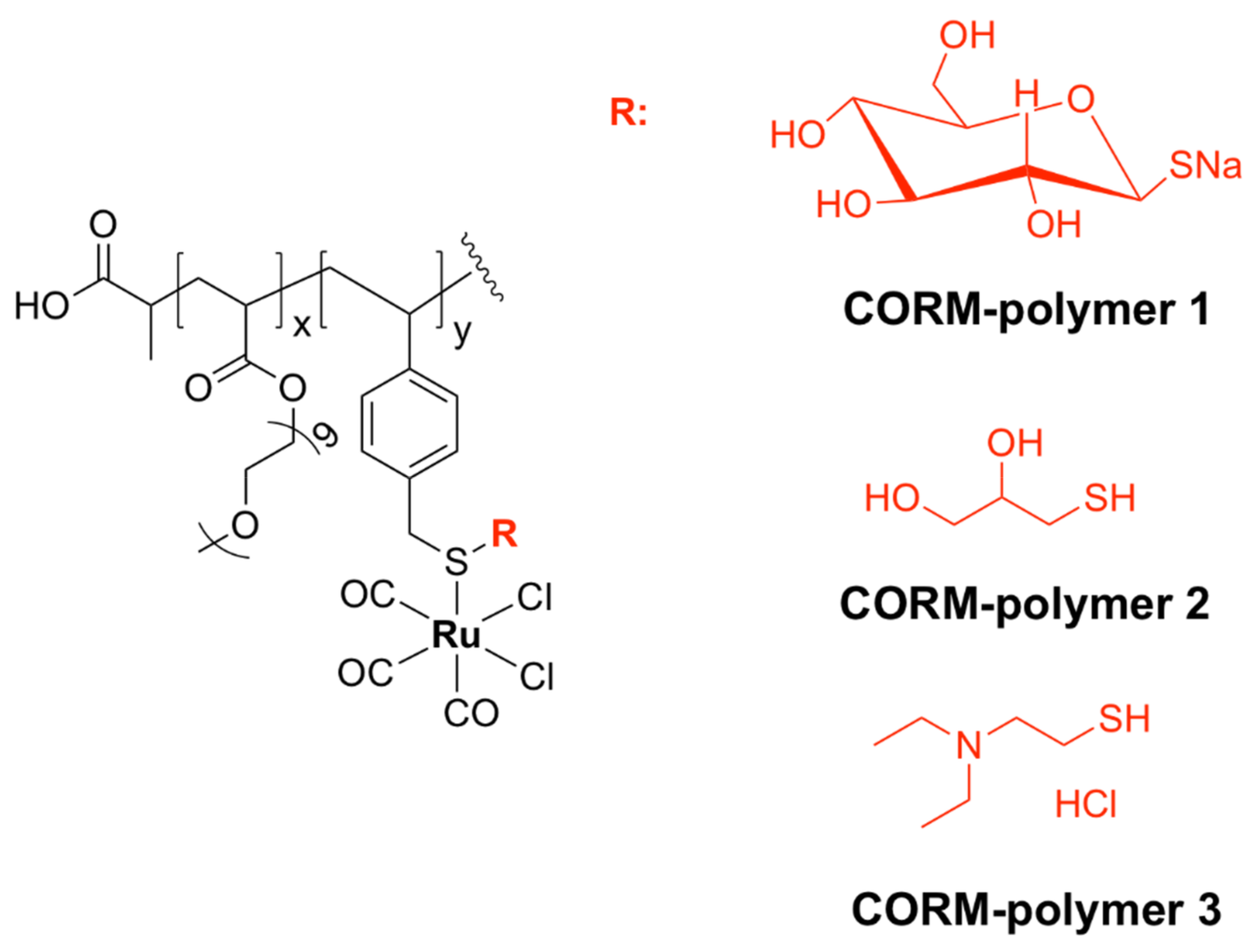
5.4.2. Ruthenium-Based CORM Polymers
5.4.3. Cellular Uptake
5.4.4. Toxicity and Pharmacokinetics
5.4.5. In Vivo Studies Regarding the Antibacterial Activity of CORMs
5.5. Ruthenium Complexes in Antimicrobial Photodynamic Therapy
6. Antiparasitic Activity of Ruthenium Complexes
6.1. Antiplasmodial Activity
6.2. Antitrypanosomal Activity
6.3. Antileishmaniasis Activity
7. Antiviral Activity of Ruthenium Complexes
7.1. Anti-HIV Activity
7.2. Anti-SARS-Cov-2 Activity
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, F.; Collins, J.G.; Keene, F.R. Ruthenium complexes as antimicrobial agents. Chem. Soc. Rev. 2015, 44, 2529–2542. [Google Scholar] [CrossRef] [Green Version]
- Frei, A.; Zuegg, J.; Elliott, A.G.; Baker, M.; Braese, S.; Brown, C.; Chen, F.G.; Dowson, C.; Dujardin, G.; Jung, N.; et al. Metal complexes as a promising source for new antibiotics. Chem. Sci. 2020, 11, 2627–2639. [Google Scholar] [CrossRef] [Green Version]
- Keogan, D.M.; Griffith, D.M. Current and potential applications of bismuth-based drugs. Molecules 2014, 19, 15258–15297. [Google Scholar] [CrossRef] [Green Version]
- Silver, S. Bacterial silver resistance: Molecular biology and uses and misuses of silver compounds. FEMS Microbiol. Rev. 2003, 27, 341–353. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-López, E.; Gomes, D.; Esteruelas, G.; Bonilla, L.; Lopez-Machado, A.L.; Galindo, R.; Cano, A.; Espina, M.; Ettcheto, M.; Camins, A.; et al. Metal-based nanoparticles as antimicrobial agents: An overview. Nanomaterials 2020, 10, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, C.N.; Prosser, K.E.; Stokes, R.W.; Cordes, A.; Metzler-Nolte, N.; Cohen, S.M. Expanding medicinal chemistry into 3D space: Metallofragments as 3D scaffolds for fragment-based drug discovery. Chem. Sci. 2020, 11, 1216–1225. [Google Scholar] [CrossRef] [Green Version]
- Hung, A.W.; Ramek, A.; Wang, Y.; Kaya, T.; Wilson, J.A.; Clemons, P.A.; Young, D.W. Route to three-dimensional fragments using diversity-oriented synthesis. Proc. Natl. Acad. Sci. USA 2011, 108, 6799–6804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galloway, W.R.J.D.; Isidro-Llobet, A.; Spring, D.R. Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 2010, 1, 80. [Google Scholar] [CrossRef] [Green Version]
- Frei, A. Metal complexes, an untapped source of antibiotic potential? Antibiotics 2020, 9, 90. [Google Scholar] [CrossRef] [Green Version]
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.L.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs are unique: Opportunities and challenges of discovery and development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef]
- Claudel, M.; Schwarte, J.V.; Fromm, K.M. New Antimicrobial Strategies Based on Metal Complexes. Chemistry 2020, 2, 849–899. [Google Scholar]
- Munteanu, A.-C.; Notaro, A.; Jakubaszek, M.; Cowell, J.; Tharaud, M.; Goud, B.; Uivarosi, V.; Gasser, G. Synthesis, Characterization, Cytotoxic Activity, and Metabolic Studies of Ruthenium(II) Polypyridyl Complexes Containing Flavonoid Ligands. Inorg. Chem. 2020, 59, 4424–4434. [Google Scholar] [CrossRef]
- Rademaker-Lakhai, J.M.; van den Bongard, D.; Pluim, D.; Beijnen, J.H.; Schellens, J.H.M. A Phase I and Pharmacological Study with Imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a Novel Ruthenium Anticancer Agent. Clin. Cancer Res. 2004, 10, 3717–3727. [Google Scholar] [CrossRef] [Green Version]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; Van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J.H.M. Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Invest. New Drugs 2015, 33, 201–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, A New Redox-Active Anticancer Agent – Preclinical Development and Results of a Clinical Phase I Study in Tumor Patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef] [PubMed]
- Lentz, F.; Drescher, A.; Lindauer, A.; Henke, M.; Hilger, R.A.; Hartinger, C.G.; Scheulen, M.E.; Dittrich, C.; Keppler, B.K.; Jaehde, U. Pharmacokinetics of a novel anticancer ruthenium complex (KP1019, FFC14A) in a phase I dose-escalation study. Anticancer. Drugs 2009, 20, 97–103. [Google Scholar] [CrossRef]
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Berger, W.; Keppler, B.K. NKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci. 2014, 5, 2925–2932. [Google Scholar] [CrossRef] [Green Version]
- Monro, S.; Colón, K.L.; Yin, H.; Roque, J.; Konda, P.; Gujar, S.; Thummel, R.P.; Lilge, L.; Cameron, C.G.; McFarland, S.A. Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev. 2019, 119, 797–828. [Google Scholar] [CrossRef]
- Antibiotic Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance (accessed on 20 April 2021).
- Silhavy, T.J.; Kahne, D.; Walker, S. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef] [PubMed]
- Pizarro-Cerdá, J.; Cossart, P. Bacterial Adhesion and Entry into Host Cells. Cell 2006, 124, 715–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef] [PubMed]
- Uivarosi, V.; Munteanu, A.-C.; Nițulescu, G.M. Chapter 2—An Overview of Synthetic and Semisynthetic Flavonoid Derivatives and Analogues: Perspectives in Drug Discovery. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 60, pp. 29–84. ISBN 1572-5995. [Google Scholar]
- Provenzani, A.; Hospodar, A.R.; Meyer, A.L.; Leonardi Vinci, D.; Hwang, E.Y.; Butrus, C.M.; Polidori, P. Multidrug-resistant gram-negative organisms: A review of recently approved antibiotics and novel pipeline agents. Int. J. Clin. Pharm. 2020, 42, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Kim, E.S.; Yoo, Y.-J.; Bae, H.-W.; Chung, I.-Y.; Cho, Y.-H. Phage-Derived Antibacterials: Harnessing the Simplicity, Plasticity, and Diversity of Phages. Viruses 2019, 11, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfei, S.; Schito, A.M. Positively charged polymers as promising devices against multidrug resistant gram-negative bacteria: A Review. Polymers 2020, 12, 1195. [Google Scholar] [CrossRef]
- Guo, C.; Mandalapu, D.; Ji, X.; Gao, J.; Zhang, Q. Chemistry and Biology of Teixobactin. Chem. – A Eur. J. 2018, 24, 5406–5422. [Google Scholar] [CrossRef]
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef]
- Raafat, D.; Otto, M.; Reppschläger, K.; Iqbal, J.; Holtfreter, S. Fighting Staphylococcus aureus Biofilms with Monoclonal Antibodies. Trends Microbiol. 2019, 27, 303–322. [Google Scholar] [CrossRef]
- Stewart, P.S. Mechanisms of antibiotic resistance in bacterial biofilms. Int. J. Med. Microbiol. 2002, 292, 107–113. [Google Scholar] [CrossRef]
- Gholizadeh, P.; Köse, Ş.; Dao, S.; Ganbarov, K.; Tanomand, A.; Dal, T.; Aghazadeh, M.; Ghotaslou, R.; Ahangarzadeh Rezaee, M.; Yousefi, B.; et al. How CRISPR-Cas System Could Be Used to Combat Antimicrobial Resistance. Infect. Drug Resist. 2020, 13, 1111–1121. [Google Scholar] [CrossRef] [Green Version]
- Southam, H.M.; Butler, J.A.; Chapman, J.A.; Poole, R.K. The Microbiology of Ruthenium Complexes. In Advances in Microbial Physiology; Poole, R.K., Ed.; Elsevier: London, UK, 2017; Volume 71, pp. 1–96. ISBN 0065-2911. [Google Scholar]
- Dwyer, F.P.; Gyarfas, E.C.; Rogers, W.P.; Koch, J.H. Biological Activity of Complex Ions. Nature 1952, 170, 190–191. [Google Scholar] [CrossRef]
- Dwyer, F.P.; Mayhew, E.; Roe, E.M.F.; Shulman, A. Inhibition of Landschütz Ascites Tumour Growth by Metal Chelates Derived from 3,4,7,8-Tetramethyl-1,10-phenanthroline. Br. J. Cancer 1965, 19, 195–199. [Google Scholar] [CrossRef]
- Abd-El-Aziz, A.S.; Agatemor, C.; Etkin, N. Antimicrobial resistance challenged with metal-based antimicrobial macromolecules. Biomaterials 2017, 118, 27–50. [Google Scholar] [CrossRef]
- Shulman, A.; Dwyer, F.P. Metal Chelates in Biological Systems. In Chelating Agents and Metal Chelates; Dwyer, F.P., Mellor, D.P., Eds.; Academic Press: Cambridge, MA, USA, 1964; ISBN 978-0-12-395499-2. [Google Scholar]
- Li, F.; Mulyana, Y.; Feterl, M.; Warner, J.M.; Collins, J.G.; Keene, F.R. The antimicrobial activity of inert oligonuclear polypyridylruthenium(II) complexes against pathogenic bacteria, including MRSA. Dalt. Trans. 2011, 40, 5032–5038. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Feterl, M.; Mulyana, Y.; Warner, J.M.; Collins, J.G.; Keene, F.R. In vitro susceptibility and cellular uptake for a new class of antimicrobial agents: Dinuclear ruthenium(II) complexes. J. Antimicrob. Chemother. 2012, 67, 2686–2695. [Google Scholar] [CrossRef] [Green Version]
- Dwyer, F.P.; Gyarfas, E.C.; Wright, R.D.; Shulman, A. Effect of Inorganic Complex Ions on Transmission at a Neuromuscular Junction. Nature 1957, 179, 425–426. [Google Scholar] [CrossRef]
- Bolhuis, A.; Hand, L.; Marshall, J.E.; Richards, A.D.; Rodger, A.; Aldrich-Wright, J. Antimicrobial activity of ruthenium-based intercalators. Eur. J. Pharm. Sci. 2011, 42, 313–317. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.-Y.; Sun, B.; Zhang, L.; Li, N.; Han, J.; Zhang, J.; Sun, X.; He, Q.-Y. Chemical Interference with Iron Transport Systems to Suppress Bacterial Growth of Streptococcus pneumoniae. PLoS One 2014, 9, e105953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.-Y.; Zhang, L.; Liu, J.; Li, N.; Yu, G.; Cao, K.; Han, J.; Zeng, G.; Pan, Y.; Sun, X.; et al. Proteomic analysis on the antibacterial activity of a Ru(II) complex against Streptococcus pneumoniae. J. Proteomics 2015, 115, 107–116. [Google Scholar] [CrossRef]
- de Sousa, A.P.; Ellena, J.; Gondim, A.C.S.; Lopes, L.G.F.; Sousa, E.H.S.; de Vasconcelos, M.A.; Teixeira, E.H.; Ford, P.C.; Holanda, A.K.M. Antimicrobial activity of cis-[Ru(bpy)2(L)(L′)]n+ complexes, where L = 4-(4-chlorobenzoyl)pyridine or 4-(benzoyl)pyridine and L′ = Cl− or CO. Polyhedron 2018, 144, 88–94. [Google Scholar] [CrossRef]
- De Sousa, A.P.; Gondim, A.C.S.; Sousa, E.H.S.; de Vasconcelos, M.A.; Teixeira, E.H.; Bezerra, B.P.; Ayala, A.P.; Martins, P.H.R.; Lopes, L.G.d.F.; Holanda, A.K.M. An unusual bidentate methionine ruthenium(II) complex: Photo-uncaging and antimicrobial activity. J. Biol. Inorg. Chem. 2020, 25, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Jiang, G.; Wang, J.; Duan, X.; Liao, Z.; Lin, X.; Shen, J.; Xiong, Y.; Jiang, G. Two ruthenium polypyridyl complexes functionalized with thiophen: Synthesis and antibacterial activity against Staphylococcus aureus. New J. Chem. 2020, 44, 17215–17221. [Google Scholar] [CrossRef]
- Bu, S.; Jiang, G.; Jiang, G.; Liu, J.; Lin, X.; Shen, J.; Xiong, Y.; Duan, X.; Wang, J.; Liao, X. Antibacterial activity of ruthenium polypyridyl complexes against Staphylococcus aureus and biofilms. J. Biol. Inorg. Chem. 2020, 25, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.; Shukla, M.; Kaul, G.; Chopra, S.; Patra, A.K. Rationally designed curcumin based ruthenium(ii) antimicrobials effective against drug-resistant Staphylococcus aureus. Dalt. Trans. 2019, 48, 11822–11828. [Google Scholar] [CrossRef] [PubMed]
- Gorle, A.K.; Feterl, M.; Warner, J.M.; Primrose, S.; Constantinoiu, C.C.; Keene, F.R.; Collins, J.G. Mononuclear Polypyridylruthenium(II) Complexes with High Membrane Permeability in Gram-Negative Bacteria—in particular Pseudomonas aeruginosa. Chem. – A Eur. J. 2015, 21, 10472–10481. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, B.; Kell, R.E.M.; Southam, H.M.; Butler, J.A.; Li, X.; Poole, R.K.; Keene, F.R.; Collins, J.G. The Antimicrobial Activity of Mononuclear Ruthenium(II) Complexes Containing the dppz Ligand. Chempluschem 2018, 83, 643–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smitten, K.L.; Thick, E.J.; Southam, H.M.; Bernardino de la Serna, J.; Foster, S.J.; Thomas, J.A. Mononuclear ruthenium(ii) theranostic complexes that function as broad-spectrum antimicrobials in therapeutically resistant pathogens through interaction with DNA. Chem. Sci. 2020, 11, 8828–8838. [Google Scholar] [CrossRef]
- De Grandis, R.A.; Resende, F.A.; da Silva, M.M.; Pavan, F.R.; Batista, A.A.; Varanda, E.A. In vitro evaluation of the cyto-genotoxic potential of Ruthenium(II) SCAR complexes: a promising class of antituberculosis agents. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2016, 798–799, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Andrade, A.L.; de Vasconcelos, M.A.; Arruda, F.V.d.F.; do Nascimento Neto, L.G.; Carvalho, J.M.d.S.; Gondim, A.C.S.; Lopes, L.G.d.F.; Sousa, E.H.S.; Teixeira, E.H. Antimicrobial activity and antibiotic synergy of a biphosphinic ruthenium complex against clinically relevant bacteria. Biofouling 2020, 36, 442–454. [Google Scholar] [CrossRef]
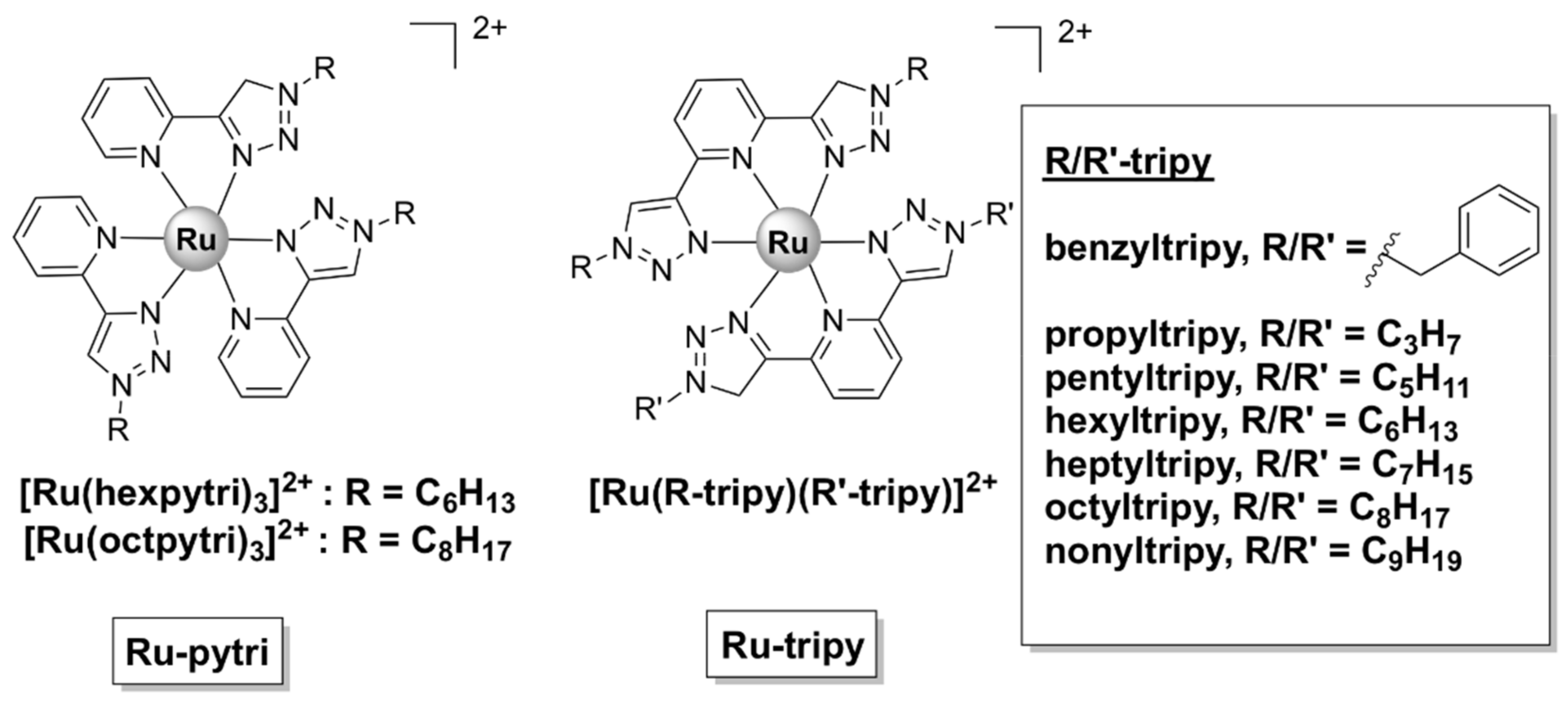
- Kumar, S.V.; Scottwell, S.O.; Waugh, E.; McAdam, C.J.; Hanton, L.R.; Brooks, H.J.L.; Crowley, J.D. Antimicrobial Properties of Tris(homoleptic) Ruthenium(II) 2-Pyridyl-1,2,3-triazole “click” Complexes against Pathogenic Bacteria, Including Methicillin-Resistant Staphylococcus aureus (MRSA). Inorg. Chem. 2016, 55, 9767–9777. [Google Scholar] [CrossRef]
- van Hilst, Q.V.C.; Vasdev, R.A.S.; Preston, D.; Findlay, J.A.; Scottwell, S.; Giles, G.I.; Brooks, H.J.L.; Crowley, J.D. Synthesis, Characterisation and Antimicrobial Studies of some 2,6-bis(1,2,3-Triazol-4-yl)Pyridine Ruthenium(II) “Click” Complexes. Asian J. Org. Chem. 2019, 8, 496–505. [Google Scholar] [CrossRef]
- Gorle, A.K.; Li, X.; Primrose, S.; Li, F.; Feterl, M.; Kinobe, R.T.; Heimann, K.; Warner, J.M.; Keene, F.R.; Collins, J.G. Oligonuclear polypyridylruthenium(II) complexes: Selectivity between bacteria and eukaryotic cells. J. Antimicrob. Chemother. 2016, 71, 1547–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Heimann, K.; Li, F.; Warner, J.M.; Richard Keene, F.; Grant Collins, J. Dinuclear ruthenium(II) complexes containing one inert metal centre and one coordinatively-labile metal centre: Syntheses and biological activities. Dalt. Trans. 2016, 45, 4017–4029. [Google Scholar] [CrossRef] [Green Version]
- Smitten, K.L.; Southam, H.M.; de la Serna, J.B.; Gill, M.R.; Jarman, P.J.; Smythe, C.G.W.; Poole, R.K.; Thomas, J.A. Using Nanoscopy To Probe the Biological Activity of Antimicrobial Leads That Display Potent Activity against Pathogenic, Multidrug Resistant, Gram-Negative Bacteria. ACS Nano 2019, 13, 5133–5146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varney, A.M.; Smitten, K.L.; Thomas, J.A.; McLean, S. Transcriptomic Analysis of the Activity and Mechanism of Action of a Ruthenium(II)-Based Antimicrobial That Induces Minimal Evolution of Pathogen Resistance. ACS Pharmacol. Transl. Sci. 2021, 4, 168–178. [Google Scholar] [CrossRef]
- Smitten, K.L.; Fairbanks, S.D.; Robertson, C.C.; Bernardino de la Serna, J.; Foster, S.J.; Thomas, J.A. Ruthenium based antimicrobial theranostics – using nanoscopy to identify therapeutic targets and resistance mechanisms in Staphylococcus aureus. Chem. Sci. 2020, 11, 70–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandrala, M.; Li, F.; Feterl, M.; Mulyana, Y.; Warner, J.M.; Wallace, L.; Keene, F.R.; Collins, J.G. Chlorido-containing ruthenium(II) and iridium(III) complexes as antimicrobial agents. Dalt. Trans. 2013, 42, 4686–4694. [Google Scholar] [CrossRef]
- Gorle, A.K.; Feterl, M.; Warner, J.M.; Wallace, L.; Keene, F.R.; Collins, J.G. Tri- and tetra-nuclear polypyridyl ruthenium(II) complexes as antimicrobial agents. Dalt. Trans. 2014, 43, 16713–16725. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Sundaraneedi, M.K.; Southam, H.M.; Poole, R.K.; Musgrave, I.F.; Keene, F.R.; Collins, J.G. Synthesis and biological properties of tetranuclear ruthenium complexes containing the bis[4(4′-methyl-2,2′-bipyridyl)]-1,7-heptane ligand. Dalt. Trans. 2019, 48, 14505–14515. [Google Scholar] [CrossRef]
- Nobre, L.S.; Seixas, J.D.; Romão, C.C.; Saraiva, L.M. Antimicrobial Action of Carbon Monoxide-Releasing Compounds. Antimicrob. Agents Chemother. 2007, 51, 4303–4307. [Google Scholar] [CrossRef] [Green Version]
- Tavares, A.F.; Parente, M.R.; Justino, M.C.; Oleastro, M.; Nobre, L.S.; Saraiva, L.M. The bactericidal activity of carbon monoxide-releasing molecules against Helicobacter pylori. PLoS One 2013, 8, e83157. [Google Scholar] [CrossRef] [Green Version]
- Sahlberg Bang, C.; Demirel, I.; Kruse, R.; Persson, K. Global gene expression profiling and antibiotic susceptibility after repeated exposure to the carbon monoxide-releasing molecule-2 (CORM-2) in multidrug-resistant ESBL-producing uropathogenic Escherichia coli. PLoS One 2017, 12, e0178541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmard, M.; Davidge, K.S.; Bouvet, O.; Morin, D.; Roux, D.; Foresti, R.; Ricard, J.D.; Denamur, E.; Poole, R.K.; Montravers, P.; et al. A carbon monoxide-releasing molecule (CORM-3) exerts bactericidal activity against Pseudomonas aeruginosa and improves survival in an animal model of bacteraemia. FASEB J. 2009, 23, 1023–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, S.M.; Marques, J.; Romão, C.C.; Saraiva, L.M. Metabolomics of Escherichia coli Treated with the Antimicrobial Carbon Monoxide-Releasing Molecule CORM-3 Reveals Tricarboxylic Acid Cycle as Major Target. Antimicrob. Agents Chemother. 2019, 63, e00643–e00719. [Google Scholar] [CrossRef]
- Donnelly, R.F.; Fletcher, N.C.; McCague, P.J.; Donnelly, J.; McCarron, P.A.; Tunney, M.M. Design, Synthesis and Photodynamic Antimicrobial Activity of Ruthenium Trischelate Diimine Complexes. Lett. Drug Des. Discov. 2007, 4, 175–179. [Google Scholar] [CrossRef]
- Smith, N.A.; Zhang, P.; Greenough, S.E.; Horbury, M.D.; Clarkson, G.J.; McFeely, D.; Habtemariam, A.; Salassa, L.; Stavros, V.G.; Dowson, C.G.; et al. Combatting AMR: Photoactivatable ruthenium(II)-isoniazid complex exhibits rapid selective antimycobacterial activity. Chem. Sci. 2017, 8, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, S.L.; Stepanyan, L.; Vahidi, N.; Jain, A.; Winkel, B.S.J.; Brewer, K.J. Visible light induced antibacterial properties of a Ru(II)–Pt(II) bimetallic complex. Inorganica Chim. Acta 2017, 454, 229–233. [Google Scholar] [CrossRef]
- Ghosh, S.; Amariei, G.; Mosquera, M.E.G.; Rosal, R. Polymeric ruthenium precursor as a photoactivated antimicrobial agent. J. Hazard. Mater. 2021, 402, 123788. [Google Scholar] [CrossRef]
- Yang, X.-Y.; He, K.; Du, G.; Wu, X.; Yu, G.; Pan, Y.; Zhang, G.; Sun, X.; He, Q.-Y. Integrated Translatomics with Proteomics to Identify Novel Iron–Transporting Proteins in Streptococcus pneumoniae. Front. Microbiol. 2016, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Srishailam, A.; Gabra, N.M.; Kumar, Y.P.; Reddy, K.L.; Devi, C.S.; Anil Kumar, D.; Singh, S.S.; Satyanarayana, S. Synthesis, characterization; DNA binding and antitumor activity of ruthenium(II) polypyridyl complexes. J. Photochem. Photobiol. B Biol. 2014, 141, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.R.; Reddy, P.V.; Kumar, Y.P.; Srishailam, A.; Nambigari, N.; Satyanarayana, S. Synthesis, Characterization, DNA Binding, Light Switch “On and Off”, Docking Studies and Cytotoxicity, of Ruthenium(II) and Cobalt(III) Polypyridyl Complexes. J. Fluoresc. 2014, 24, 803–817. [Google Scholar] [CrossRef]
- Mallepally, R.R.; Putta, V.R.; Chintakuntla, N.; Vuradi, R.K.; Kotha, L.R.; Sirasani, S. DNA Binding Behavior, Sensor Studies, Antimicrobial, Photocleavage and In vitro Cytotoxicity of Synthesized Ru(II) Complexes with Assorted Intercalating Polypyridyl Ligands. J. Fluoresc. 2016, 26, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhang, W.; Lv, M.; Yang, E.; Zhao, Q.; Wang, W. Antibacterial activity of ruthenium(II) polypyridyl complex manipulated by membrane permeability and cell morphology. Bioorg. Med. Chem. Lett. 2015, 25, 2068–2073. [Google Scholar] [CrossRef] [PubMed]
- Putta, V.R.; Chintakuntla, N.; Mallepally, R.R.; Avudoddi, S.; K, N.; Nancherla, D.; V V N, Y.; R S, P.; Surya, S.S.; Sirasani, S. Synthesis and Evaluation of In Vitro DNA/Protein Binding Affinity, Antimicrobial, Antioxidant and Antitumor Activity of Mononuclear Ru(II) Mixed Polypyridyl Complexes. J. Fluoresc. 2016, 26, 225–240. [Google Scholar] [CrossRef]
- Ravi Kumar, V.; Nagababu, P.; Srinivas, G.; Rajender Reddy, M.; Vinoda Rani, M.; Ravi, M.; Satyanarayana, S. Investigation of DNA/BSA binding of three Ru(II) complexes by various spectroscopic methods, molecular docking and their antimicrobial activity. J. Coord. Chem. 2017, 70, 3790–3809. [Google Scholar] [CrossRef]
- Vadivel, T.; Dhamodaran, M. Synthesis, characterization and antibacterial studies of ruthenium(III) complexes derived from chitosan schiff base. Int. J. Biol. Macromol. 2016, 90, 44–52. [Google Scholar] [CrossRef]
- Ashwini Kumar, K.; Laxma Reddy, K.; Satyanarayana, S. Study of the interaction between ruthenium(II) complexes and CT-DNA: synthesis, characterisation, photocleavage and antimicrobial activity studies. Supramol. Chem. 2010, 22, 629–643. [Google Scholar] [CrossRef]
- Ashwini Kumar, K.; Reddy, K.L.; Vidhisha, S.; Satyanarayana, S. Synthesis, characterization and DNA binding and photocleavage studies of [Ru(bpy)2BDPPZ]2+, [Ru(dmb)2BDPPZ]2+ and [Ru(phen)2BDPPZ]2+ complexes and their antimicrobial activity. Appl. Organomet. Chem. 2009, 23, 409–420. [Google Scholar] [CrossRef]
- Garner, R.N.; Gallucci, J.C.; Dunbar, K.R.; Turro, C. [Ru(bpy)2(5-cyanouracil)2]2+ as a Potential Light-Activated Dual-Action Therapeutic Agent. Inorg. Chem. 2011, 50, 9213–9215. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.-B.; Zhang, W.-Y.; He, M.; Gu, Y.-Y.; Bai, L.; Wang, Y.-J.; Yi, Q.-Y.; Du, F. New ruthenium polypyridyl complexes functionalized with fluorine atom or furan: Synthesis, DNA-binding, cytotoxicity and antitumor mechanism studies. Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 2020, 227, 117534. [Google Scholar] [CrossRef]
- Jiang, G.-B.; Zhang, W.-Y.; He, M.; Gu, Y.-Y.; Bai, L.; Wang, Y.-J.; Yi, Q.-Y.; Du, F. Design and synthesis of new ruthenium polypyridyl complexes with potent antitumor activity in vitro. Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 2019, 220, 117132. [Google Scholar] [CrossRef]
- Jiang, G.-B.; Zhang, W.-Y.; He, M.; Gu, Y.-Y.; Bai, L.; Wang, Y.-J.; Yi, Q.-Y.; Du, F. Development of four ruthenium polypyridyl complexes as antitumor agents: Design, biological evaluation and mechanism investigation. J. Inorg. Biochem. 2020, 208, 111104. [Google Scholar] [CrossRef]
- Jiang, G.-B.; Zhang, W.-Y.; He, M.; Gu, Y.-Y.; Bai, L.; Wang, Y.-J.; Yi, Q.-Y.; Du, F. Anticancer activity of two ruthenium(II) polypyridyl complexes toward Hepatocellular carcinoma HepG-2 cells. Polyhedron 2019, 169, 209–218. [Google Scholar] [CrossRef]
- Mulyana, Y.; Weber, D.K.; Buck, D.P.; Motti, C.A.; Collins, J.G.; Keene, F.R. Oligonuclear polypyridylruthenium(II) complexes incorporating flexible polar and non-polar bridges: synthesis, DNA-binding and cytotoxicity. Dalt. Trans. 2011, 40, 1510–1523. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Feterl, M.; Warner, J.M.; Keene, F.R.; Collins, J.G. Dinuclear polypyridylruthenium(II) complexes: Flow cytometry studies of their accumulation in bacteria and the effect on the bacterial membrane. J. Antimicrob. Chemother. 2013, 68, 2825–2833. [Google Scholar] [CrossRef] [Green Version]
- Pisani, M.J.; Fromm, P.D.; Mulyana, Y.; Clarke, R.J.; Körner, H.; Heimann, K.; Collins, J.G.; Keene, F.R. Mechanism of cytotoxicity and cellular uptake of lipophilic inert dinuclear polypyridylruthenium(II) complexes. ChemMedChem 2011, 6, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Harry, E.J.; Bottomley, A.L.; Edstein, M.D.; Birrell, G.W.; Woodward, C.E.; Keene, F.R.; Collins, J.G. Dinuclear ruthenium(II) antimicrobial agents that selectively target polysomes in vivo. Chem. Sci. 2014, 5, 685–693. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Gorle, A.K.; Ainsworth, T.D.; Heimann, K.; Woodward, C.E.; Collins, J.G.; Keene, F.R. RNA and DNA binding of inert oligonuclear ruthenium(II) complexes in live eukaryotic cells. Dalt. Trans. 2015, 44, 3594–3603. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Southam, H.M.; Butler, J.A.; Poole, R.K.; Burgun, A.; Tarzia, A.; Keene, F.R.; Collins, J.G. Synthesis, isomerisation and biological properties of mononuclear ruthenium complexes containing the bis[4(4′-methyl-2,2′-bipyridyl)]-1,7-heptane ligand. Dalt. Trans. 2018, 47, 2422–2434. [Google Scholar] [CrossRef] [Green Version]
- Liao, G.; Ye, Z.; Liu, Y.; Fu, B.; Fu, C. Octahedral ruthenium (II) polypyridyl complexes as antimicrobial agents against mycobacterium. PeerJ 2017, 5, e3252. [Google Scholar] [CrossRef] [Green Version]
- Allardyce, C.S.; Dyson, P.J.; Ellis, D.J.; Salter, P.A.; Scopelliti, R. Synthesis and characterisation of some water soluble ruthenium(II)–arene complexes and an investigation of their antibiotic and antiviral properties. J. Organomet. Chem. 2003, 668, 35–42. [Google Scholar] [CrossRef]
- Beckford, F.A.; Stott, A.; Gonzalez-Sarrías, A.; Seeram, N.P. Novel microwave synthesis of half-sandwich [(η6-C6H6)Ru] complexes and an evaluation of the biological activity and biochemical reactivity. Appl. Organomet. Chem. 2013, 27, 425–434. [Google Scholar] [CrossRef]
- Gichumbi, J.M.; Friedrich, H.B.; Omondi, B.; Singh, M.; Naicker, K.; Chenia, H.Y. Synthesis, characterization, and cytotoxic and antimicrobial activities of ruthenium(II) arene complexes with N,N-bidentate ligands. J. Coord. Chem. 2016, 69, 3531–3544. [Google Scholar] [CrossRef]
- Singh, K.S.; Devi, P.; Sawant, S.G.; Kaminsky, W. Arene ruthenium(II) complexes with 2-acetamidothiazole derived ligands: Synthesis, structural studies, antifouling and antibacterial properties. Polyhedron 2015, 100, 321–325. [Google Scholar] [CrossRef]
- Obradović, D.; Nikolić, S.; Milenković, I.; Milenković, M.; Jovanović, P.; Savić, V.; Roller, A.; Đorđić Crnogorac, M.; Stanojković, T.; Grgurić-Šipka, S. Synthesis, characterization, antimicrobial and cytotoxic activity of novel half-sandwich Ru(II) arene complexes with benzoylthiourea derivatives. J. Inorg. Biochem. 2020, 210, 111164. [Google Scholar] [CrossRef] [PubMed]
- Turan, N.; Buldurun, K.; Alan, Y.; Savci, A.; Çolak, N.; Mantarcı, A. Synthesis, characterization, antioxidant, antimicrobial and DNA binding properties of ruthenium(II), cobalt(II) and nickel(II) complexes of Schiff base containing o-vanillin. Res. Chem. Intermed. 2019, 45, 3525–3540. [Google Scholar] [CrossRef]
- Shadap, L.; Banothu, V.; Adepally, U.; Adhikari, S.; Kollipara, M.R. Variable structural bonding modes and antibacterial studies of thiosemicarbazone ligands of ruthenium, rhodium, and iridium metal complexes. J. Coord. Chem. 2020, 73, 175–187. [Google Scholar] [CrossRef]
- Lapasam, A.; Banothu, V.; Addepally, U.; Kollipara, M.R. Synthesis, structural and antimicrobial studies of half-sandwich ruthenium, rhodium and iridium complexes containing nitrogen donor Schiff-base ligands. J. Mol. Struct. 2019, 1191, 314–322. [Google Scholar] [CrossRef]
- Nobre, L.S.; Jeremias, H.; Romão, C.C.; Saraiva, L.M. Examining the antimicrobial activity and toxicity to animal cells of different types of CO-releasing molecules. Dalt. Trans. 2016, 45, 1455–1466. [Google Scholar] [CrossRef]
- Dkhar, L.; Banothu, V.; Kaminsky, W.; Kollipara, M.R. Synthesis of half sandwich platinum group metal complexes containing pyridyl benzothiazole hydrazones: Study of bonding modes and antimicrobial activity. J. Organomet. Chem. 2020, 914, 121225. [Google Scholar] [CrossRef]
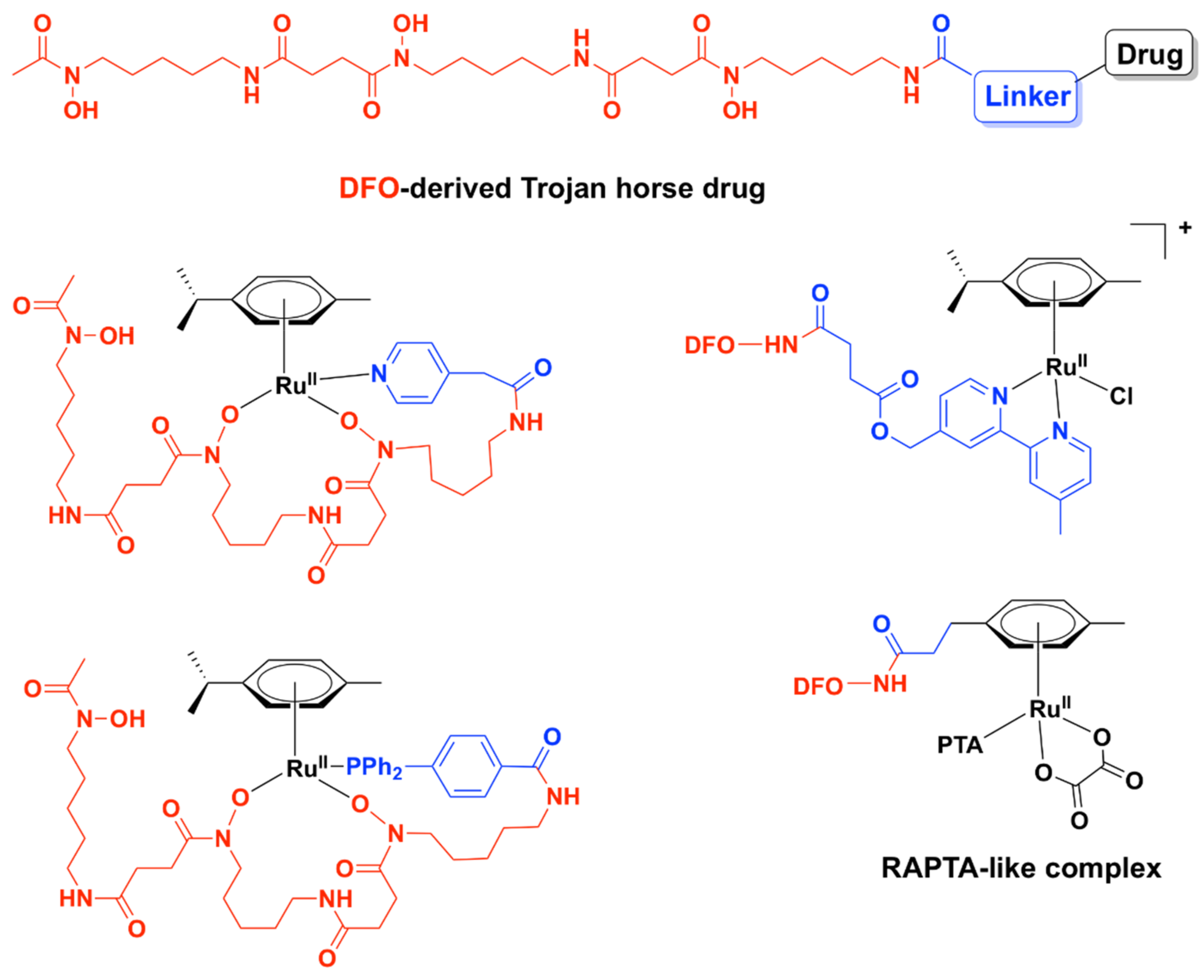
- Laurent, Q.; Batchelor, L.K.; Dyson, P.J. Applying a Trojan Horse Strategy to Ruthenium Complexes in the Pursuit of Novel Antibacterial Agents. Organometallics 2018, 37, 915–923. [Google Scholar] [CrossRef]
- Chhabra, R.; Saha, A.; Chamani, A.; Schneider, N.; Shah, R.; Nanjundan, M. Iron Pathways and Iron Chelation Approaches in Viral, Microbial, and Fungal Infections. Pharmaceuticals 2020, 13, 275. [Google Scholar] [CrossRef] [PubMed]
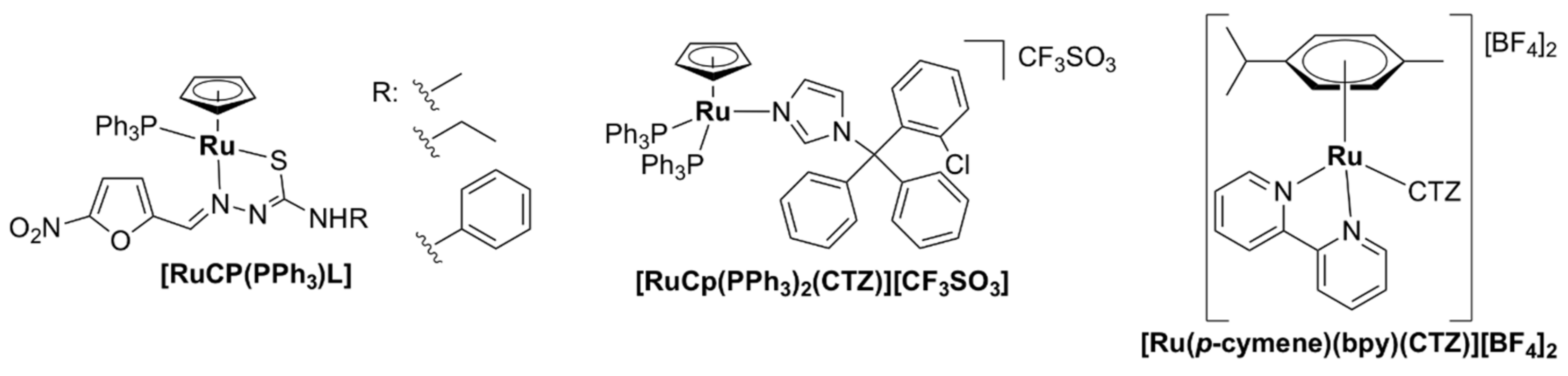
- Yildirim, H.; Guler, E.; Yavuz, M.; Ozturk, N.; Kose Yaman, P.; Subasi, E.; Sahin, E.; Timur, S. Ruthenium (II) complexes of thiosemicarbazone: Synthesis, biosensor applications and evaluation as antimicrobial agents. Mater. Sci. Eng. C 2014, 44, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Namiecińska, E.; Sadowska, B.; Więckowska-Szakiel, M.; Dołęga, A.; Pasternak, B.; Grazul, M.; Budzisz, E. Anticancer and antimicrobial properties of novel η6-p-cymene ruthenium(ii) complexes containing a N,S-type ligand, their structural and theoretical characterization. RSC Adv. 2019, 9, 38629–38645. [Google Scholar] [CrossRef] [Green Version]
- Felício, M.R.; Silva, O.N.; Gonçalves, S.; Santos, N.C.; Franco, O.L. Peptides with Dual Antimicrobial and Anticancer Activities. Front. Chem. 2017, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appelt, P.; Fagundes, F.D.; Facchin, G.; Gabriela Kramer, M.; Back, D.F.; Cunha, M.A.A.; Sandrino, B.; Wohnrath, K.; de Araujo, M.P. Ruthenium (II) complexes containing 2-mercaptothiazolinates as ligands and evaluation of their antimicrobial activity. Inorganica Chim. Acta 2015, 436, 152–158. [Google Scholar] [CrossRef]
- Beloglazkina, E.K.; Manzheliy, E.A.; Moiseeva, A.A.; Maloshitskaya, O.A.; Zyk, N.V.; Skvortsov, D.A.; Osterman, I.A.; Sergiev, P.V.; Dontsova, O.A.; Ivanenkov, Y.A.; et al. Synthesis, characterisation, cytotoxicity and antibacterial activity of ruthenium(II) and rhodium(III) complexes with sulfur-containing terpyridines. Polyhedron 2016, 107, 27–37. [Google Scholar] [CrossRef]
- Lapasam, A.; Dkhar, L.; Joshi, N.; Poluri, K.M.; Kollipara, M.R. Antimicrobial selectivity of ruthenium, rhodium, and iridium half sandwich complexes containing phenyl hydrazone Schiff base ligands towards B. thuringiensis and P. aeruginosa bacteria. Inorganica Chim. Acta 2019, 484, 255–263. [Google Scholar] [CrossRef]
- Elnagar, M.M.; Samir, S.; Shaker, Y.M.; Abdel-Shafi, A.A.; Sharmoukh, W.; Abdel-Aziz, M.S.; Abou-El-Sherbini, K.S. Synthesis, characterization, and evaluation of biological activities of new 4′-substituted ruthenium (II) terpyridine complexes: Prospective anti-inflammatory properties. Appl. Organomet. Chem. 2021, 35, 1–16. [Google Scholar] [CrossRef]
- Nyawade, E.A.; Friedrich, H.B.; Omondi, B.; Chenia, H.Y.; Singh, M.; Gorle, S. Synthesis and characterization of new α,α′-diaminoalkane-bridged dicarbonyl(η5-cyclopentadienyl)ruthenium(II) complex salts: Antibacterial activity tests of η5-cyclopentadienyl dicarbonyl ruthenium(II) amine complexes. J. Organomet. Chem. 2015, 799–800, 138–146. [Google Scholar] [CrossRef]
- Rani, S.; Kumar, S.; Chandra, S. Synthesis, structural, spectral, thermal and antimicrobial studies of palladium(II), platinum(II), ruthenium(III) and iridium(III) complexes derived from N,N,N,N-tetradentate macrocyclic ligand. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 78, 1507–1514. [Google Scholar] [CrossRef]
- Balasubramian, K.P.; Raju, V.V.; Chinnusamy, V. Synthesis, characteristic, catalytic and antimicrobial activities of imidazolo substituted benzylidene imines with ruthenium(II) complexes. J. Indian Chem. Soc. 2009, 86, 570–576. [Google Scholar]
- Thilagavathi, N.; Manimaran, A.; Priya, N.P.; Sathya, N.; Jayabalakrishnan, C. Synthesis, characterization, electrochemical, catalytic and antimicrobial activity studies of hydrazone Schiff base ruthenium(II) complexes. Appl. Organomet. Chem. 2010, 24, 301–307. [Google Scholar] [CrossRef]
- Thilagavathi, N.; Manimaran, A.; Padma Priya, N.; Sathya, N.; Jayabalakrishnan, C. Synthesis, spectroscopic, redox, catalytic and antimicrobial properties of some ruthenium(II) Schiff base complexes. Transit. Met. Chem. 2009, 34, 725–732. [Google Scholar] [CrossRef]
- Thilagavathi, N.; Jayabalakrishnan, C. Synthesis, characterization, catalytic and antimicrobial studies of ruthenium(III) complexes. Cent. Eur. J. Chem. 2010, 8, 842–851. [Google Scholar] [CrossRef]
- Thangadurai, T.D.; Ihm, S.-K. Ruthenium(II) Complexes Derived from Substituted Cyclobutane and Substituted Thiazole Schiff Base Ligands: Synthetic, Spectral, Catalytic and Antimicrobial Studies. Synth. React. Inorganic, Met. Nano-Metal Chem. 2005, 35, 499–507. [Google Scholar] [CrossRef]
- Matshwele, J.T.P.; Nareetsile, F.; Mapolelo, D.; Matshameko, P.; Leteane, M.; Nkwe, D.O.; Odisitse, S. Synthesis of Mixed Ligand Ruthenium (II/III) Complexes and Their Antibacterial Evaluation on Drug-Resistant Bacterial Organisms. J. Chem. 2020, 2020, 2150419. [Google Scholar] [CrossRef]
- Pavan, F.R.; Poelhsitz, G.V.; Barbosa, M.I.F.; Leite, S.R.A.; Batista, A.A.; Ellena, J.; Sato, L.S.; Franzblau, S.G.; Moreno, V.; Gambino, D.; et al. Ruthenium(II) phosphine/diimine/picolinate complexes: Inorganic compounds as agents against tuberculosis. Eur. J. Med. Chem. 2011, 46, 5099–5107. [Google Scholar] [CrossRef] [PubMed]
- Pavan, F.R.; Poelhsitz, G.V.; da Cunha, L.V.P.; Barbosa, M.I.F.; Leite, S.R.A.; Batista, A.A.; Cho, S.H.; Franzblau, S.G.; de Camargo, M.S.; Resende, F.A.; et al. In Vitro and In Vivo Activities of Ruthenium(II) Phosphine/Diimine/Picolinate Complexes (SCAR) against Mycobacterium tuberculosis. PLoS One 2013, 8, e64242. [Google Scholar]
- Rolston, K.V.; Wang, W.; Nesher, L.; Smith, J.R.; Rybak, M.J.; Prince, R.A. Time-kill determination of the bactericidal activity of telavancin and vancomycin against clinical methicillin-resistant Staphylococcus aureus isolates from cancer patients. Diagn. Microbiol. Infect. Dis. 2017, 87, 338–342. [Google Scholar] [CrossRef]
- Demirezen, N.; Tarınç, D.; Polat, D.; Çeşme, M.; Gölcü, A.; Tümer, M. Synthesis of trimethoprim metal complexes: Spectral, electrochemical, thermal, DNA-binding and surface morphology studies. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 94, 243–255. [Google Scholar] [CrossRef]
- Ude, Z.; Romero-Canelón, I.; Twamley, B.; Fitzgerald Hughes, D.; Sadler, P.J.; Marmion, C.J. A novel dual-functioning ruthenium(II)–arene complex of an anti-microbial ciprofloxacin derivative — Anti-proliferative and anti-microbial activity. J. Inorg. Biochem. 2016, 160, 210–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colina-Vegas, L.; Dutra, J.L.; Villarreal, W.; Neto, J.H.d.A.; Cominetti, M.R.; Pavan, F.; Navarro, M.; Batista, A.A. Ru(II)/clotrimazole/diphenylphosphine/bipyridine complexes: Interaction with DNA, BSA and biological potential against tumor cell lines and Mycobacterium tuberculosis. J. Inorg. Biochem. 2016, 162, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Naglah, A.M.; Al-Omar, M.A.; Almehizia, A.A.; Obaidullah, A.J.; Bhat, M.A.; Al-Shakliah, N.S.; Belgacem, K.; Majrashi, B.M.; Refat, M.S.; Adam, A.M.A. Synthesis, Spectroscopic, and Antimicrobial Study of Binary and Ternary Ruthenium(III) Complexes of Ofloxacin Drug and Amino Acids as Secondary Ligands. Crystals 2020, 10, 225. [Google Scholar] [CrossRef] [Green Version]
- Măciucă, A.-M.; Munteanu, A.-C.; Uivarosi, V. Quinolone Complexes with Lanthanide Ions: An Insight into their Analytical Applications and Biological Activity. Molecules 2020, 25, 1347. [Google Scholar] [CrossRef] [Green Version]
- Pisani, M.J.; Weber, D.K.; Heimann, K.; Collins, J.G.; Keene, F.R. Selective mitochondrial accumulation of cytotoxic dinuclear polypyridyl ruthenium(II) complexes. Metallomics 2010, 2, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.K.; Sani, M.-A.; Downton, M.T.; Separovic, F.; Keene, F.R.; Collins, J.G. Membrane Insertion of a Dinuclear Polypyridylruthenium(II) Complex Revealed by Solid-State NMR and Molecular Dynamics Simulation: Implications for Selective Antibacterial Activity. J. Am. Chem. Soc. 2016, 138, 15267–15277. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.L.; Spillane, C.B.; Smith, J.A.; Buck, D.P.; Collins, J.G.; Keene, F.R. Dinuclear ruthenium(II) complexes with flexible bridges as non-duplex DNA binding agents. Dalt. Trans. 2007, 4333–4342. [Google Scholar] [CrossRef]
- Li, F.; Weber, D.K.; Morgan, J.L.; Collins, J.G.; Keene, F.R. An approach to therapeutic agents through selective targeting of destabilised nucleic acid duplex sequences. Dalt. Trans. 2012, 41, 6528–6535. [Google Scholar] [CrossRef]
- Gill, M.R.; Garcia-Lara, J.; Foster, S.J.; Smythe, C.; Battaglia, G.; Thomas, J.A. A ruthenium(II) polypyridyl complex for direct imaging of DNA structure in living cells. Nat. Chem. 2009, 1, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Millimouno, F.M.; Dong, J.; Yang, L.; Li, J.; Li, X. Targeting Apoptosis Pathways in Cancer and Perspectives with Natural Compounds from Mother Nature. Cancer Prev. Res. 2014, 7, 1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, D.P.; Paul, J.A.; Pisani, M.J.; Collins, J.G.; Keene, F.R. Binding of a Flexibly-linked Dinuclear Ruthenium(II) Complex to Adenine-bulged DNA Duplexes. Aust. J. Chem. 2010, 63, 1365–1375. [Google Scholar] [CrossRef]
- Recht, M.I.; Douthwaite, S.; Puglisi, J.D. Basis for prokaryotic specificity of action of aminoglycoside antibiotics. EMBO J. 1999, 18, 3133–3138. [Google Scholar] [CrossRef] [PubMed]
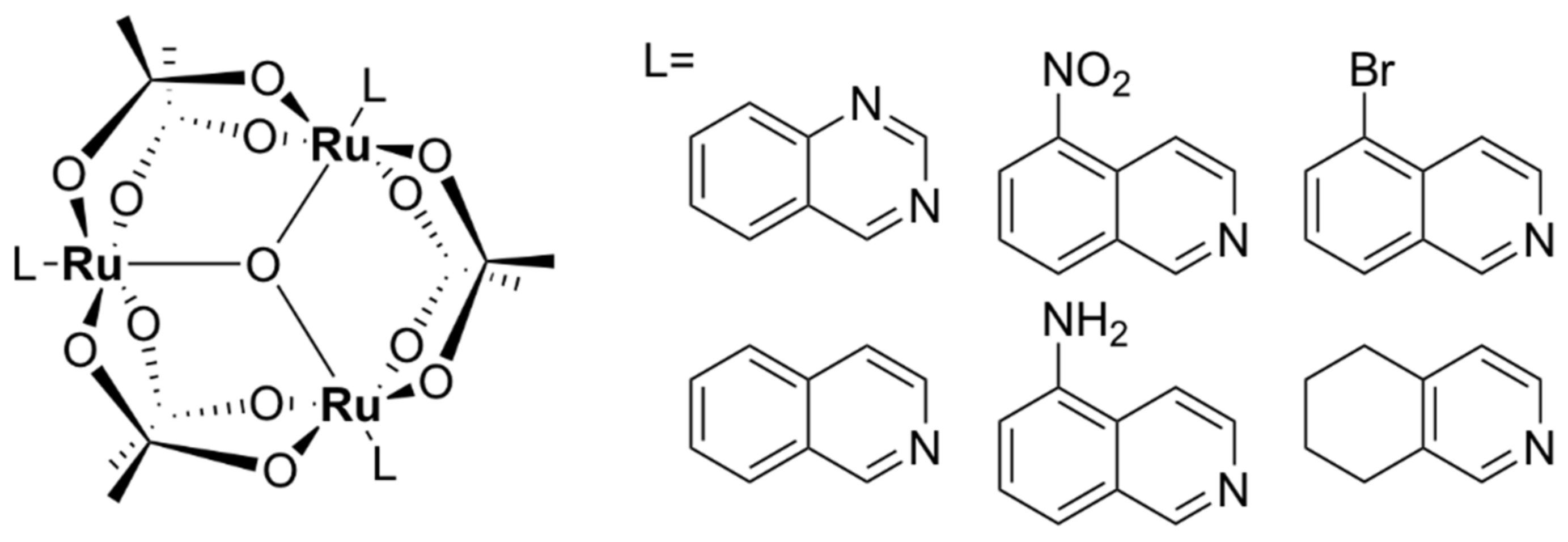
- Li, F.; Gorle, A.K.; Ranson, M.; Vine, K.L.; Kinobe, R.; Feterl, M.; Warner, J.M.; Keene, F.R.; Collins, J.G.; Day, A.I. Probing the pharmacokinetics of cucurbit[7, 8 and 10]uril: And a dinuclear ruthenium antimicrobial complex encapsulated in cucurbit[10]uril. Org. Biomol. Chem. 2017, 15, 4172–4179. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.G.; May-Dracka, T.L.; Gagnon, M.M.; Tommasi, R. Trends and exceptions of physical properties on antibacterial activity for Gram-positive and Gram-negative pathogens. J. Med. Chem. 2014, 57, 10144–10161. [Google Scholar] [CrossRef]
- Kitamura, Y.; Ihara, T.; Tsujimura, Y.; Osawa, Y.; Sasahara, D.; Yamamoto, M.; Okada, K.; Tazaki, M.; Jyo, A. Template-directed formation of luminescent lanthanide complexes: Versatile tools for colorimetric identification of single nucleotide polymorphism. J. Inorg. Biochem. 2008, 102, 1921–1931. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.V.; Lo, W.K.C.; Brooks, H.J.L.; Crowley, J.D. Synthesis, structure, stability and antimicrobial activity of a ruthenium(II) helicate derived from a bis-bidentate “click” pyridyl-1,2,3-triazole ligand. Inorganica Chim. Acta 2015, 425, 1–6. [Google Scholar] [CrossRef]
- Roopashree, B.; Gayathri, V.; Gopi, A.; Devaraju, K.S. Syntheses, characterizations, and antimicrobial activities of binuclear ruthenium(III) complexes containing 2-substituted benzimidazole derivatives. J. Coord. Chem. 2012, 65, 4023–4040. [Google Scholar] [CrossRef]
- Wenzel, M.; Patra, M.; Senges, C.H.R.; Ott, I.; Stepanek, J.J.; Pinto, A.; Prochnow, P.; Vuong, C.; Langklotz, S.; Metzler-Nolte, N.; et al. Analysis of the mechanism of action of potent antibacterial hetero-tri-organometallic compounds: a structurally new class of antibiotics. ACS Chem. Biol. 2013, 8, 1442–1450. [Google Scholar] [CrossRef]
- Stringer, T.; Seldon, R.; Liu, N.; Warner, D.F.; Tam, C.; Cheng, L.W.; Land, K.M.; Smith, P.J.; Chibale, K.; Smith, G.S. Antimicrobial activity of organometallic isonicotinyl and pyrazinyl ferrocenyl-derived complexes. Dalt. Trans. 2017, 46, 9875–9885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Winkel, B.S.J.; Brewer, K.J. In vivo inhibition of E. coli growth by a Ru(II)/Pt(II) supramolecule [(tpy)RuCl(dpp)PtCl2](PF6). J. Inorg. Biochem. 2007, 101, 1525–1528. [Google Scholar] [CrossRef]
- Jain, A.; Wang, J.; Mashack, E.R.; Winkel, B.S.J.; Brewer, K.J. Multifunctional DNA Interactions of Ru−Pt Mixed Metal Supramolecular Complexes with Substituted Terpyridine Ligands. Inorg. Chem. 2009, 48, 9077–9084. [Google Scholar] [CrossRef]
- Wareham, L.K.; Poole, R.K.; Tinajero-Trejo, M. CO-releasing Metal Carbonyl Compounds as Antimicrobial Agents in the Post-antibiotic Era. J. Biol. Chem. 2015, 290, 18999–19007. [Google Scholar] [CrossRef] [Green Version]
- Ling, K.; Men, F.; Wang, W.-C.; Zhou, Y.-Q.; Zhang, H.-W.; Ye, D.-W. Carbon Monoxide and Its Controlled Release: Therapeutic Application, Detection, and Development of Carbon Monoxide Releasing Molecules (CORMs). J. Med. Chem. 2018, 61, 2611–2635. [Google Scholar] [CrossRef]
- Adach, W.; Błaszczyk, M.; Olas, B. Carbon monoxide and its donors - Chemical and biological properties. Chem. Biol. Interact. 2020, 318, 108973. [Google Scholar] [CrossRef]
- Yan, H.; Du, J.; Zhu, S.; Nie, G.; Zhang, H.; Gu, Z.; Zhao, Y. Emerging Delivery Strategies of Carbon Monoxide for Therapeutic Applications: From CO Gas to CO Releasing Nanomaterials. Small 2019, 15, 1904382. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.; Mann, B.E.; Motterlini, R.; Poole, R.K. The carbon monoxide-releasing molecule, CORM-3 (Ru(CO)3Cl(glycinate)), targets respiration and oxidases in Campylobacter jejuni, generating hydrogen peroxide. IUBMB Life 2011, 63, 363–371. [Google Scholar] [CrossRef] [PubMed]
- McLean, S.; Begg, R.; Jesse, H.E.; Mann, B.E.; Sanguinetti, G.; Poole, R.K. Analysis of the bacterial response to Ru(CO)3Cl(Glycinate) (CORM-3) and the inactivated compound identifies the role played by the ruthenium compound and reveals sulfur-containing species as a major target of CORM-3 action. Antioxid. Redox Signal. 2013, 19, 1999–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.L.; Jesse, H.E.; Hughes, B.; Lund, V.; Naylor, K.; Davidge, K.S.; Cook, G.M.; Mann, B.E.; Poole, R.K. Ru(CO)3Cl(Glycinate) (CORM-3): A carbon monoxide-releasing molecule with broad-spectrum antimicrobial and photosensitive activities against respiration and cation transport in Escherichia coli. Antioxidants Redox Signal. 2013, 19, 497–509. [Google Scholar] [CrossRef] [Green Version]
- Rana, N.; McLean, S.; Mann, B.E.; Poole, R.K. Interaction of the carbon monoxide-releasing molecule Ru(CO)3Cl(glycinate) (CORM-3) with Salmonella enterica serovar Typhimurium: In situ measurements of carbon monoxide binding by integrating cavity dual-beam spectrophotometry. Microbiology 2014, 160, 2771–2779. [Google Scholar] [CrossRef]
- Johnson, T.R.; Mann, B.E.; Teasdale, I.P.; Adams, H.; Foresti, R.; Green, C.J.; Motterlini, R. Metal carbonyls as pharmaceuticals? [Ru(CO)3Cl(glycinate)], a CO-releasing molecule with an extensive aqueous solution chemistry. Dalt. Trans. 2007, 1500–1508. [Google Scholar] [CrossRef]
- Jesse, H.E.; Nye, T.L.; McLean, S.; Green, J.; Mann, B.E.; Poole, R.K. Cytochrome bd-I in Escherichia coli is less sensitive than cytochromes bd-II or bo′ to inhibition by the carbon monoxide-releasing molecule, CORM-3: N-acetylcysteine reduces CO-RM uptake and inhibition of respiration. Biochim. Biophys. Acta - Proteins Proteomics 2013, 1834, 1693–1703. [Google Scholar] [CrossRef] [Green Version]
- Davidge, K.S.; Sanguinetti, G.; Yee, C.H.; Cox, A.G.; McLeod, C.W.; Monk, C.E.; Mann, B.E.; Motterlini, R.; Poole, R.K. Carbon monoxide-releasing antibacterial molecules target respiration and global transcriptional regulators. J. Biol. Chem. 2009, 284, 4516–4524. [Google Scholar] [CrossRef] [Green Version]
- Desmard, M.; Foresti, R.; Morin, D.; Dagouassat, M.; Berdeaux, A.; Denamur, E.; Crook, S.H.; Mann, B.E.; Scapens, D.; Montravers, P.; et al. Differential antibacterial activity against Pseudomonas aeruginosa by carbon monoxide-releasing molecules. Antioxid. Redox Signal. 2012, 16, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaves-Ferreira, M.; Albuquerque, I.S.; Matak-Vinkovic, D.; Coelho, A.C.; Carvalho, S.M.; Saraiva, L.M.; Romão, C.C.; Bernardes, G.J.L. Spontaneous CO Release from RuII(CO)2–Protein Complexes in Aqueous Solution, Cells, and Mice. Angew. Chemie Int. Ed. 2015, 54, 1172–1175. [Google Scholar] [CrossRef] [Green Version]
- Southam, H.M.; Smith, T.W.; Lyon, R.L.; Liao, C.; Trevitt, C.R.; Middlemiss, L.A.; Cox, F.L.; Chapman, J.A.; El-Khamisy, S.F.; Hippler, M.; et al. A thiol-reactive Ru(II) ion, not CO release, underlies the potent antimicrobial and cytotoxic properties of CO-releasing molecule-3. Redox Biol. 2018, 18, 114–123. [Google Scholar] [CrossRef]
- Tavares, A.F.N.; Teixeira, M.; Romão, C.C.; Seixas, J.D.; Nobre, L.S.; Saraiva, L.M. Reactive oxygen species mediate bactericidal killing elicited by carbon monoxide-releasing molecules. J. Biol. Chem. 2011, 286, 26708–26717. [Google Scholar] [CrossRef] [Green Version]
- Marazioti, A.; Bucci, M.; Coletta, C.; Vellecco, V.; Baskaran, P.; Szabó, C.; Cirino, G.; Marques, A.R.; Guerreiro, B.; Gonçalves, A.M.L.; et al. Inhibition of nitric oxide-stimulated vasorelaxation by carbon monoxide-releasing molecules. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2570–2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taillé, C.; El-Benna, J.; Lanone, S.; Boczkowski, J.; Motterlini, R. Mitochondrial respiratory chain and NAD(P)H oxidase are targets for the antiproliferative effect of carbon monoxide in human airway smooth muscle. J. Biol. Chem. 2005, 280, 25350–25360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuckerbraun, B.S.; Chin, B.Y.; Bilban, M.; D’Avila, J.d.C.; Rao, J.; Billiar, T.R.; Otterbein, L.E. Carbon monoxide signals via inhibition of cytochrome c oxidase and generation of mitochondrial reactive oxygen species. FASEB J. 2007, 21, 1099–1106. [Google Scholar] [CrossRef] [Green Version]
- Murray, T.S.; Okegbe, C.; Gao, Y.; Kazmierczak, B.I.; Motterlini, R.; Dietrich, L.E.P.; Bruscia, E.M. The Carbon Monoxide Releasing Molecule CORM-2 Attenuates Pseudomonas aeruginosa Biofilm Formation. PLoS One 2012, 7, e35499. [Google Scholar] [CrossRef]
- Seixas, J.D.; Chaves-Ferreira, M.; Montes-Grajales, D.; Gonçalves, A.M.; Marques, A.R.; Saraiva, L.M.; Olivero-Verbel, J.; Romão, C.C.; Bernardes, G.J.L. An N-Acetyl Cysteine Ruthenium Tricarbonyl Conjugate Enables Simultaneous Release of CO and Ablation of Reactive Oxygen Species. Chem. Eur. J. 2015, 21, 14708–14712. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.L.; Wareham, L.K.; McLean, S.; Begg, R.; Greaves, S.; Mann, B.E.; Sanguinetti, G.; Poole, R.K. CO-Releasing Molecules Have Nonheme Targets in Bacteria: Transcriptomic, Mathematical Modeling and Biochemical Analyses of CORM-3 [Ru(CO)3Cl(glycinate)] Actions on a Heme-Deficient Mutant of Escherichia coli. Antioxid. Redox Signal. 2015, 23, 148–162. [Google Scholar]
- Santos-Silva, T.; Mukhopadhyay, A.; Seixas, J.D.; Bernardes, G.J.L.; Romão, C.C.; Romão, M.J. CORM-3 Reactivity toward Proteins: The Crystal Structure of a Ru(II) Dicarbonyl−Lysozyme Complex. J. Am. Chem. Soc. 2011, 133, 1192–1195. [Google Scholar] [CrossRef] [PubMed]
- Santos-Silva, T.; Mukhopadhyay, A.; Seixas, J.D.; Bernardes, G.J.L.; Romão, C.C.; Romão, M.J. Towards improved therapeutic CORMs: Understanding the reactivity of CORM-3 with proteins. Curr. Med. Chem. 2011, 18, 3361–3366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seixas, J.D.; Santos, M.F.A.; Mukhopadhyay, A.; Coelho, A.C.; Reis, P.M.; Veiros, L.F.; Marques, A.R.; Penacho, N.; Gonçalves, A.M.L.; Romão, M.J.; et al. A contribution to the rational design of Ru(CO)3Cl2L complexes for in vivo delivery of CO. Dalt. Trans. 2015, 44, 5058–5075. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, H.; Zhao, Q.; Chen, Y.; Liu, B.; Zhang, B.; Zheng, Q. Syntheses and evaluation of drug-like properties of CO-releasing molecules containing ruthenium and group 6 metal. Eur. J. Med. Chem. 2014, 74, 199–215. [Google Scholar] [CrossRef]
- Winburn, I.C.; Gunatunga, K.; McKernan, R.D.; Walker, R.J.; Sammut, I.A.; Harrison, J.C. Cell damage following carbon monoxide releasing molecule exposure: Implications for therapeutic applications. Basic Clin. Pharmacol. Toxicol. 2012, 111, 31–41. [Google Scholar] [CrossRef]
- Stingl, K.; De Reuse, H. Staying alive overdosed: How does Helicobacter pylori control urease activity? Int. J. Med. Microbiol. 2005, 295, 307–315. [Google Scholar] [CrossRef]
- Lemire, J.A.; Harrison, J.J.; Turner, R.J. Antimicrobial activity of metals: Mechanisms, molecular targets and applications. Nat. Rev. Microbiol. 2013, 11, 371–384. [Google Scholar] [CrossRef]
- Arnett, C.H.; Chalkley, M.J.; Agapie, T. A Thermodynamic Model for Redox-Dependent Binding of Carbon Monoxide at Site-Differentiated, High Spin Iron Clusters. J. Am. Chem. Soc. 2018, 140, 5569–5578. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.L.; McLean, S.; Begg, R.; Sanguinetti, G.; Poole, R.K. Analysis of transcript changes in a heme-deficient mutant of Escherichia coli in response to CORM-3 [Ru(CO)3Cl(glycinate)]. Genomics Data 2015, 5, 231–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobre, L.S.; Al-Shahrour, F.; Dopazo, J.; Saraiva, L.M. Exploring the antimicrobial action of a carbon monoxide-releasing compound through whole-genome transcription profiling of Escherichia coli. Microbiology 2009, 155, 813–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wareham, L.K.; Begg, R.; Jesse, H.E.; Van Beilen, J.W.A.; Ali, S.; Svistunenko, D.; McLean, S.; Hellingwerf, K.J.; Sanguinetti, G.; Poole, R.K. Carbon Monoxide Gas Is Not Inert, but Global, in Its Consequences for Bacterial Gene Expression, Iron Acquisition, and Antibiotic Resistance. Antioxid. Redox Signal. 2016, 24, 1013–1028. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-Z.; Nikaido, H. Efflux-mediated drug resistance in bacteria: An update. Drugs 2009, 69, 1555–1623. [Google Scholar] [CrossRef]
- Villemin, E.; Ong, Y.C.; Thomas, C.M.; Gasser, G. Polymer encapsulation of ruthenium complexes for biological and medicinal applications. Nat. Rev. Chem. 2019, 3, 261–282. [Google Scholar] [CrossRef]
- Nguyen, D.; Nguyen, T.-K.; Rice, S.A.; Boyer, C. CO-Releasing Polymers Exert Antimicrobial Activity. Biomacromolecules 2015, 16, 2776–2786. [Google Scholar] [CrossRef]
- Bang, C.S.; Kruse, R.; Demirel, I.; Önnberg, A.; Söderquist, B.; Persson, K. Multiresistant uropathogenic extended-spectrum β-lactamase (ESBL)-producing Escherichia coli are susceptible to the carbon monoxide releasing molecule-2 (CORM-2). Microb. Pathog. 2014, 66, 29–35. [Google Scholar] [CrossRef]
- Chung, S.W.; Liu, X.; Macias, A.A.; Baron, R.M.; Perrella, M.A. Heme oxygenase-1-derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J. Clin. Invest. 2008, 118, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josefsen, L.B.; Boyle, R.W. Photodynamic Therapy and the Development of Metal-Based Photosensitisers. Met. Based. Drugs 2008, 2008, 276109. [Google Scholar] [CrossRef] [Green Version]
- Le Gall, T.; Lemercier, G.; Chevreux, S.; Tücking, K.-S.; Ravel, J.; Thétiot, F.; Jonas, U.; Schönherr, H.; Montier, T. Ruthenium(II) Polypyridyl Complexes as Photosensitizers for Antibacterial Photodynamic Therapy: A Structure-Activity Study on Clinical Bacterial Strains. ChemMedChem 2018, 13, 2229–2239. [Google Scholar] [CrossRef]
- Soliman, N.; Sol, V.; Ouk, T.-S.; Thomas, C.M.; Gasser, G. Encapsulation of a Ru(II) Polypyridyl Complex into Polylactide Nanoparticles for Antimicrobial Photodynamic Therapy. Pharmaceutics 2020, 12, 961. [Google Scholar] [CrossRef]
- Rensmo, H.; Lunell, S.; Siegbahn, H. Absorption and electrochemical properties of ruthenium(II) dyes, studied by semiempirical quantum chemical calculations. J. Photochem. Photobiol. A Chem. 1998, 114, 117–124. [Google Scholar] [CrossRef]
- Lei, W.; Zhou, Q.; Jiang, G.; Zhang, B.; Wang, X. Photodynamic inactivation of Escherichia coli by Ru(II) complexes. Photochem. Photobiol. Sci. 2011, 10, 887–890. [Google Scholar] [CrossRef] [PubMed]
- Allardyce, C.; Dyson, P. Ruthenium in Medicine: Current Clinical Uses and Future Prospects. Platin. Met. Rev. 2001, 45, 62–69. [Google Scholar]
- Wassmer, S.C.; Grau, G.E.R. Severe malaria: What’s new on the pathogenesis front? Int. J. Parasitol. 2017, 47, 145–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baartzes, N.; Stringer, T.; Smith, G.S. Targeting Sensitive-Strain and Resistant-Strain Malaria Parasites Through a Metal-Based Approach. In Advances in Bioorganometallic Chemistry; Hirao, T., Moriuchi, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 193–213. ISBN 978-0-12-814197-7. [Google Scholar]
- Miotto, O.; Almagro-Garcia, J.; Manske, M.; Macinnis, B.; Campino, S.; Rockett, K.A.; Amaratunga, C.; Lim, P.; Suon, S.; Sreng, S.; et al. Multiple populations of artemisinin-resistant Plasmodium falciparum in Cambodia. Nat. Genet. 2013, 45, 648–655. [Google Scholar] [CrossRef]
- Martínez, A.; Rajapakse, C.S.K.; Naoulou, B.; Kopkalli, Y.; Davenport, L.; Sánchez-Delgado, R.A. The mechanism of antimalarial action of the ruthenium(II)-chloroquine complex [RuCl(2)(CQ)] (2). J. Biol. Inorg. Chem. 2008, 13, 703–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Delgado, R.A.; Navarro, M.; Pérez, H.; Urbina, J.A. Toward a Novel Metal-Based Chemotherapy against Tropical Diseases. 2. Synthesis and Antimalarial Activity in Vitro and in Vivo of New Ruthenium− and Rhodium−Chloroquine Complexes. J. Med. Chem. 1996, 39, 1095–1099. [Google Scholar] [CrossRef]
- Macedo, T.S.; Colina-Vegas, L.; DA Paixão, M.; Navarro, M.; Barreto, B.C.; Oliveira, P.C.M.; Macambira, S.G.; Machado, M.; Prudêncio, M.; D’Alessandro, S.; et al. Chloroquine-containing organoruthenium complexes are fast-acting multistage antimalarial agents. Parasitology 2016, 143, 1543–1556. [Google Scholar] [CrossRef] [Green Version]
- Adams, M.; de Kock, C.; Smith, P.J.; Land, K.M.; Liu, N.; Hopper, M.; Hsiao, A.; Burgoyne, A.R.; Stringer, T.; Meyer, M.; et al. Improved antiparasitic activity by incorporation of organosilane entities into half-sandwich ruthenium(II) and rhodium(III) thiosemicarbazone complexes. Dalt. Trans. 2015, 44, 2456–2468. [Google Scholar] [CrossRef] [Green Version]
- Rylands, L.-I.; Welsh, A.; Maepa, K.; Stringer, T.; Taylor, D.; Chibale, K.; Smith, G.S. Structure-activity relationship studies of antiplasmodial cyclometallated ruthenium(II), rhodium(III) and iridium(III) complexes of 2-phenylbenzimidazoles. Eur. J. Med. Chem. 2019, 161, 11–21. [Google Scholar] [CrossRef]
- Chellan, P.; Land, K.M.; Shokar, A.; Au, A.; An, S.H.; Taylor, D.; Smith, P.J.; Chibale, K.; Smith, G.S. Di- and Trinuclear Ruthenium-, Rhodium-, and Iridium-Functionalized Pyridyl Aromatic Ethers: A New Class of Antiparasitic Agents. Organometallics 2013, 32, 4793–4804. [Google Scholar] [CrossRef]
- Stringer, T.; Quintero, M.A.S.; Wiesner, L.; Smith, G.S.; Nordlander, E. Evaluation of PTA-derived ruthenium(II) and iridium(III) quinoline complexes against chloroquine-sensitive and resistant strains of the Plasmodium falciparum malaria parasite. J. Inorg. Biochem. 2019, 191, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Chellan, P.; Land, K.M.; Shokar, A.; Au, A.; An, S.H.; Taylor, D.; Smith, P.J.; Riedel, T.; Dyson, P.J.; Chibale, K.; et al. Synthesis and evaluation of new polynuclear organometallic Ru(ii), Rh(iii) and Ir(iii) pyridyl ester complexes as in vitro antiparasitic and antitumor agents. Dalt. Trans. 2014, 43, 513–526. [Google Scholar] [CrossRef] [Green Version]
- De Souza, N.B.; Aguiar, A.C.C.; de Oliveira, A.C.; Top, S.; Pigeon, P.; Jaouen, G.; Goulart, M.O.F.; Krettli, A.U. Antiplasmodial activity of iron(II) and ruthenium(II) organometallic complexes against Plasmodium falciparum blood parasites. Mem. Inst. Oswaldo Cruz 2015, 110, 981–988. [Google Scholar] [CrossRef] [Green Version]
- Lidani, K.C.F.; Andrade, F.A.; Bavia, L.; Damasceno, F.S.; Beltrame, M.H.; Messias-Reason, I.J.; Sandri, T.L. Chagas Disease: From Discovery to a Worldwide Health Problem. Front. public Heal. 2019, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.G.E. Update on human African trypanosomiasis (sleeping sickness). J. Neurol. 2019, 266, 2334–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
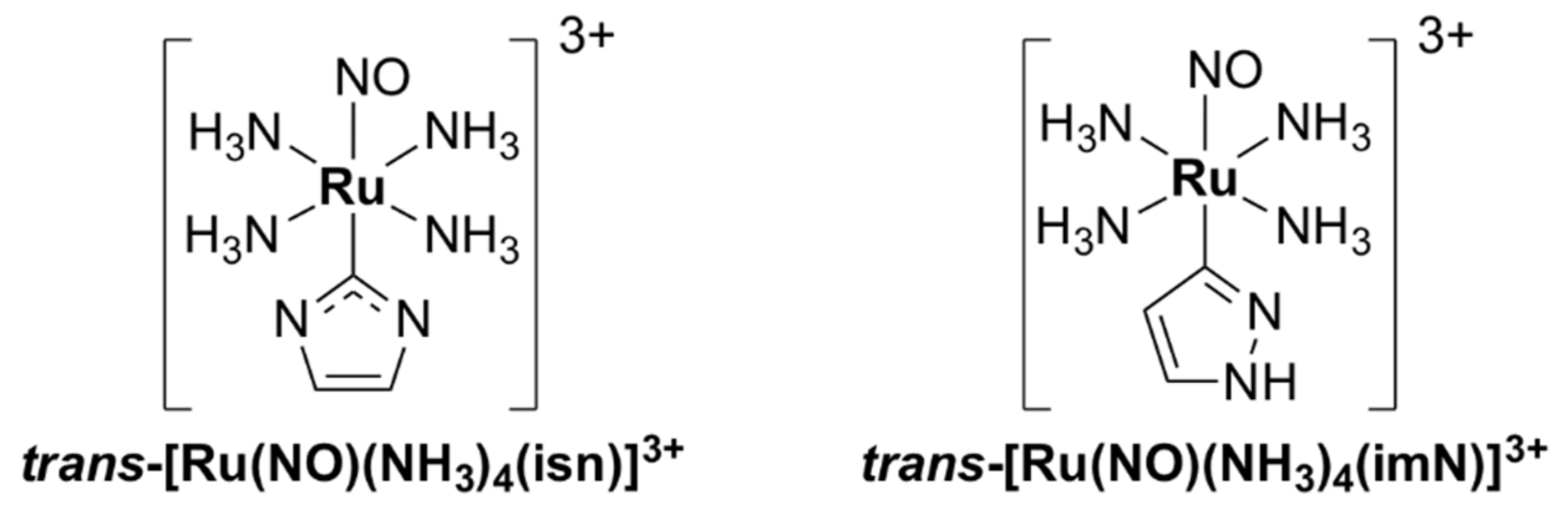
- Silva, J.J.N.; Osakabe, A.L.; Pavanelli, W.R.; Silva, J.S.; Franco, D.W. In vitro and in vivo antiproliferative and trypanocidal activities of ruthenium NO donors. Br. J. Pharmacol. 2007, 152, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.J.N.; Pavanelli, W.R.; Pereira, J.C.M.; Silva, J.S.; Franco, D.W. Experimental chemotherapy against Trypanosoma cruzi infection using ruthenium nitric oxide donors. Antimicrob. Agents Chemother. 2009, 53, 4414–4421. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.J.N.; Guedes, P.M.M.; Zottis, A.; Balliano, T.L.; Nascimento Silva, F.O.; França Lopes, L.G.; Ellena, J.; Oliva, G.; Andricopulo, A.D.; Franco, D.W.; et al. Novel ruthenium complexes as potential drugs for Chagas’s disease: Enzyme inhibition and in vitro/in vivo trypanocidal activity. Br. J. Pharmacol. 2010, 160, 260–269. [Google Scholar] [CrossRef] [Green Version]
- Bastos, T.M.; Barbosa, M.I.F.; da Silva, M.M.; Júnior, J.W.d.c.; Meira, C.S.; Guimaraes, E.T.; Ellena, J.; Moreira, D.R.M.; Batista, A.A.; Soares, M.B.P. Nitro/nitrosyl-ruthenium complexes are potent and selective anti-Trypanosoma cruzi agents causing autophagy and necrotic parasite death. Antimicrob. Agents Chemother. 2014, 58, 6044–6055. [Google Scholar] [CrossRef] [Green Version]
- Possato, B.; Carneiro, Z.A.; de Albuquerque, S.; Nikolaou, S. New uses for old complexes: The very first report on the trypanocidal activity of symmetric trinuclear ruthenium complexes. J. Inorg. Biochem. 2017, 176, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Arce, E.R.; Sarniguet, C.; Morais, T.S.; Tomaz, A.I.; Azar, C.O.; Figueroa, R.; Diego Maya, J.; Medeiros, A.; Comini, M.; et al. Novel ruthenium(II) cyclopentadienyl thiosemicarbazone compounds with antiproliferative activity on pathogenic trypanosomatid parasites. J. Inorg. Biochem. 2015, 153, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez Arce, E.; Sarniguet, C.; Moraes, T.S.; Vieites, M.; Tomaz, A.I.; Medeiros, A.; Comini, M.A.; Varela, J.; Cerecetto, H.; González, M.; et al. A new ruthenium cyclopentadienyl azole compound with activity on tumor cell lines and trypanosomatid parasites. J. Coord. Chem. 2015, 68, 2923–2937. [Google Scholar] [CrossRef]
- Martínez, A.; Carreon, T.; Iniguez, E.; Anzellotti, A.; Sánchez, A.; Tyan, M.; Sattler, A.; Herrera, L.; Maldonado, R.A.; Sánchez-Delgado, R.A. Searching for new chemotherapies for tropical diseases: Ruthenium-clotrimazole complexes display high in vitro activity against Leishmania major and Trypanosoma cruzi and low toxicity toward normal mammalian cells. J. Med. Chem. 2012, 55, 3867–3877. [Google Scholar] [CrossRef] [Green Version]
- Sundar, S.; Chakravarty, J. Leishmaniasis: an update of current pharmacotherapy. Expert Opin. Pharmacother. 2013, 14, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.I.F.; Corrêa, R.S.; de Oliveira, K.M.; Rodrigues, C.; Ellena, J.; Nascimento, O.R.; Rocha, V.P.C.; Nonato, F.R.; Macedo, T.S.; Barbosa-Filho, J.M.; et al. Antiparasitic activities of novel ruthenium/lapachol complexes. J. Inorg. Biochem. 2014, 136, 33–39. [Google Scholar] [CrossRef]
- Wong, E.L.-M.; Sun, R.W.-Y.; Chung, N.P.-Y.; Lin, C.-L.S.; Zhu, N.; Che, C.-M. A Mixed-Valent Ruthenium−Oxo Oxalato Cluster Na7[Ru4(μ3-O)4(C2O4)6] with Potent Anti-HIV Activities. J. Am. Chem. Soc. 2006, 128, 4938–4939. [Google Scholar] [CrossRef]
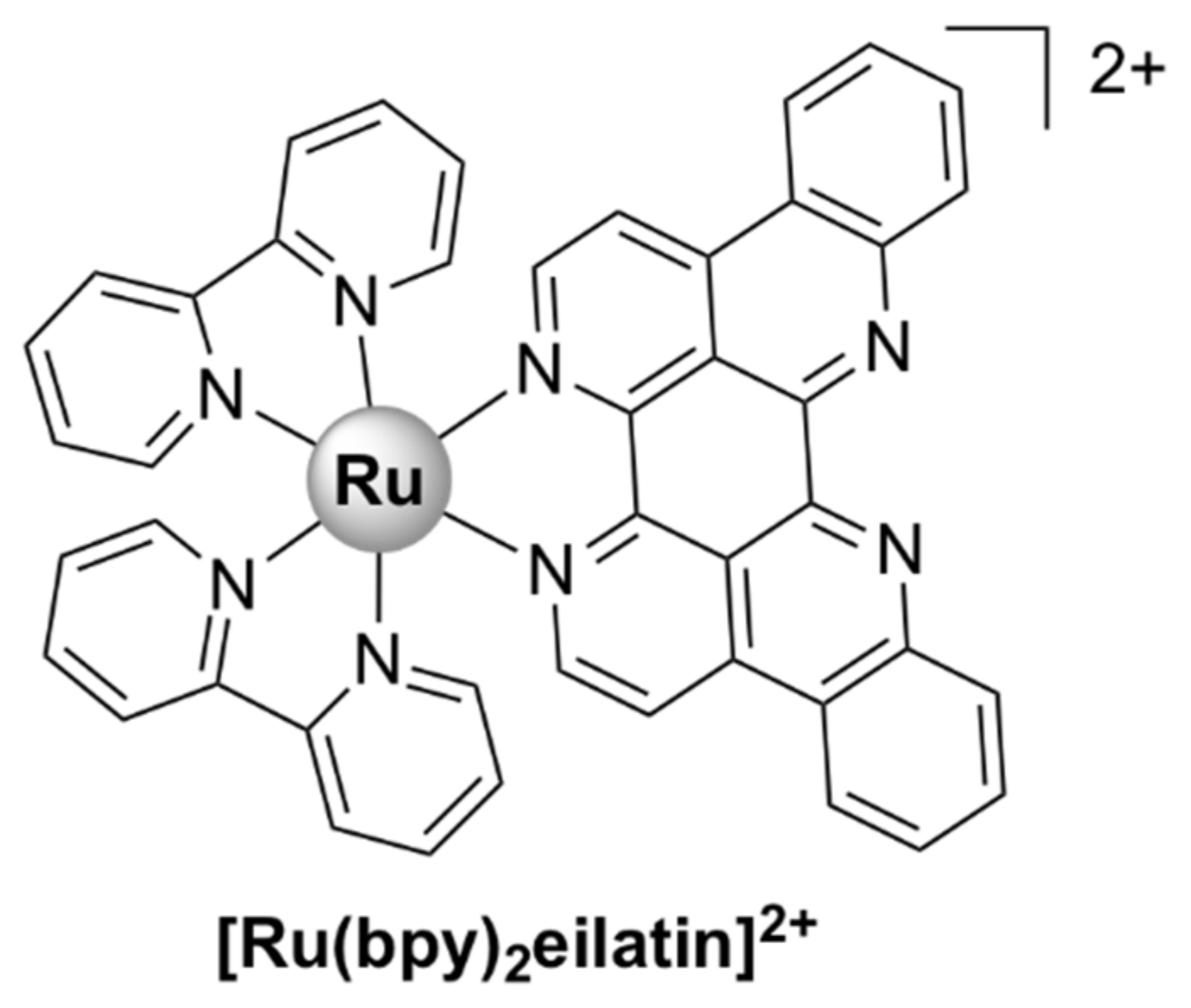
- Luedtke, N.W.; Hwang, J.S.; Glazer, E.C.; Gut, D.; Kol, M.; Tor, Y. Eilatin Ru(II) complexes display anti-HIV activity and enantiomeric diversity in the binding of RNA. Chembiochem 2002, 3, 766–771. [Google Scholar] [CrossRef]
- van Oosterhout, C.; Hall, N.; Ly, H.; Tyler, K.M. COVID-19 evolution during the pandemic – Implications of new SARS-CoV-2 variants on disease control and public health policies. Virulence 2021, 12, 507–508. [Google Scholar] [CrossRef]
- Mahase, E. Covid-19: What new variants are emerging and how are they being investigated? BMJ 2021, 372, n158. [Google Scholar] [CrossRef] [PubMed]
- Neuditschko, B.; Legin, A.A.; Baier, D.; Schintlmeister, A.; Reipert, S.; Wagner, M.; Keppler, B.K.; Berger, W.; Meier-Menches, S.M.; Gerner, C. Interaction with Ribosomal Proteins Accompanies Stress Induction of the Anticancer Metallodrug BOLD-100/KP1339 in the Endoplasmic Reticulum. Angew. Chem. Int. Ed. Engl. 2021, 60, 5063–5068. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Xiao, Y.; Li, D.; Jiang, Q.; Zhu, L.; Lin, D.; Jiang, H.; Chen, W.; Wang, L.; Liu, C.; et al. PGK1 and GRP78 overexpression correlates with clinical significance and poor prognosis in Chinese endometrial cancer patients. Oncotarget 2017, 9, 680–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakewell, S.J.; Rangel, D.F.; Ha, D.P.; Sethuraman, J.; Crouse, R.; Hadley, E.; Costich, T.L.; Zhou, X.; Nichols, P.; Lee, A.S. Suppression of stress induction of the 78-kilodalton glucose regulated protein (GRP78) in cancer by IT-139, an anti-tumor ruthenium small molecule inhibitor. Oncotarget 2018, 9, 29698–29714. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef]
- Ha, D.P.; Van Krieken, R.; Carlos, A.J.; Lee, A.S. The stress-inducible molecular chaperone GRP78 as potential therapeutic target for coronavirus infection. J. Infect. 2020, 81, 452–482. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elshahat, M.E.; Elfiky, A.A. COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 2020, 80, 554–562. [Google Scholar] [CrossRef]
- Bold Therapeutics Potential to Fight COVID-19. Available online: https://www.bold-therapeutics.com/COVID-19 (accessed on 18 April 2021).
- O’Kane, G.M.; Spratlin, J.L.; Kavan, P.; Goodwin, R.A.; McWhirter, E.; Thompson, D.; Jones, M.; McAllister, E.R.; Machado, A.; Lemmerick, Y.; et al. BOLD-100-001 (TRIO039): A phase Ib dose-escalation study of BOLD-100 in combination with FOLFOX chemotherapy in patients with advanced gastrointestinal solid tumors. J. Clin. Oncol. 2021, 39, TPS145. [Google Scholar] [CrossRef]
- Lippi, G.; Salvagno, G.L.; Pegoraro, M.; Militello, V.; Caloi, C.; Peretti, A.; De Nitto, S.; Bovo, C.; Lo Cascio, G. Preliminary evaluation of Roche Cobas Elecsys Anti-SARS-CoV-2 chemiluminescence immunoassay. Clin. Chem. Lab. Med. 2020, 58, e251–e253. [Google Scholar] [CrossRef]
- Riester, E.; Findeisen, P.; Hegel, J.K.; Kabesch, M.; Ambrosch, A.; Rank, C.M.; Langen, F.; Laengin, T.; Niederhauser, C. Performance evaluation of the Roche Elecsys Anti-SARS-CoV-2 S immunoassay. medRxiv 2021. [Google Scholar] [CrossRef]
- Rothan, H.A.; Stone, S.; Natekar, J.; Kumari, P.; Arora, K.; Kumar, M. The FDA-approved gold drug auranofin inhibits novel coronavirus (SARS-COV-2) replication and attenuates inflammation in human cells. Virology 2020, 547, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Marzo, T.; Messori, L. A Role for Metal-Based Drugs in Fighting COVID-19 Infection? The Case of Auranofin. ACS Med. Chem. Lett. 2020, 11, 1067–1068. [Google Scholar] [CrossRef] [PubMed]
- Karges, J.; Kalaj, M.; Gembicky, M.; Cohen, S.M. ReI Tricarbonyl Complexes as Coordinate Covalent Inhibitors for the SARS-CoV-2 Main Cysteine Protease. Angew. Chemie Int. Ed. 2021, 60, 10716. [Google Scholar] [CrossRef] [PubMed]






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex [Reference] | Activity Strain: MIC Values (µg/mL) | Toxicity to Healthy Mammalian Cells (IC50, µg/mL, 24 h, unless Stated Otherwise) | Modes of Action | |
|---|---|---|---|---|
| Gram-Positive Strains | Gram-Negative Strains | |||
| Polypyridylruthenium (II) complexes | ||||
| [Ru(2,9-Me2phen)2(dppz)]2+ [40] | S. aureus MRSA252: 2, MRSA41: 4, MSSA160: 8, B. subtilis 168: 4 | Not active on E. coli MC4100 | - | bactericidal; DNA intercalation |
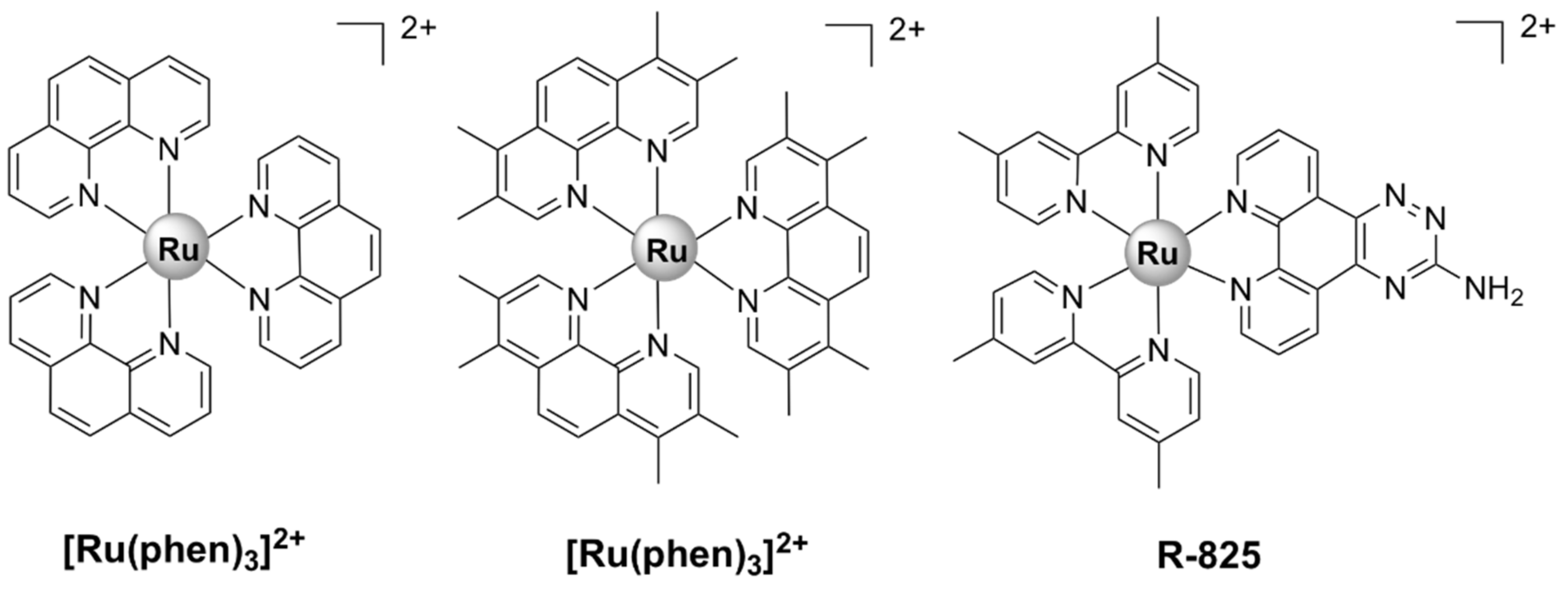
| R-825 [41] | S. pneumoniae D39 WT: 27.5 piuA mutant: 55 | - | Not toxic to human alveolar epithelial A549 cells up to 480 µM | interference with iron acquisition systems in S. pneumoniae cells |
| X-03 [42] | S. pneumoniae D39: 25, Streptococcus suis 05ZYH33: 100, S. pyogenes MGAS5005: 25, Listeria monocytogenes 19,117: 25, S. aureus 29,213: 50 | E. coli K12: > 200, Vibrio alginolyticus V12G01: > 200, Vibrio parahaemolyticus RIMD 2,210,633: > 200, A. baumanii 19,606: > 200 | Not toxic to human alveolar A549 and bronchial HBE epithelial cells up to 100 µg/mL | interference with iron acquisition systems in S. pneumoniae cells; oxidative stress, membrane damage |
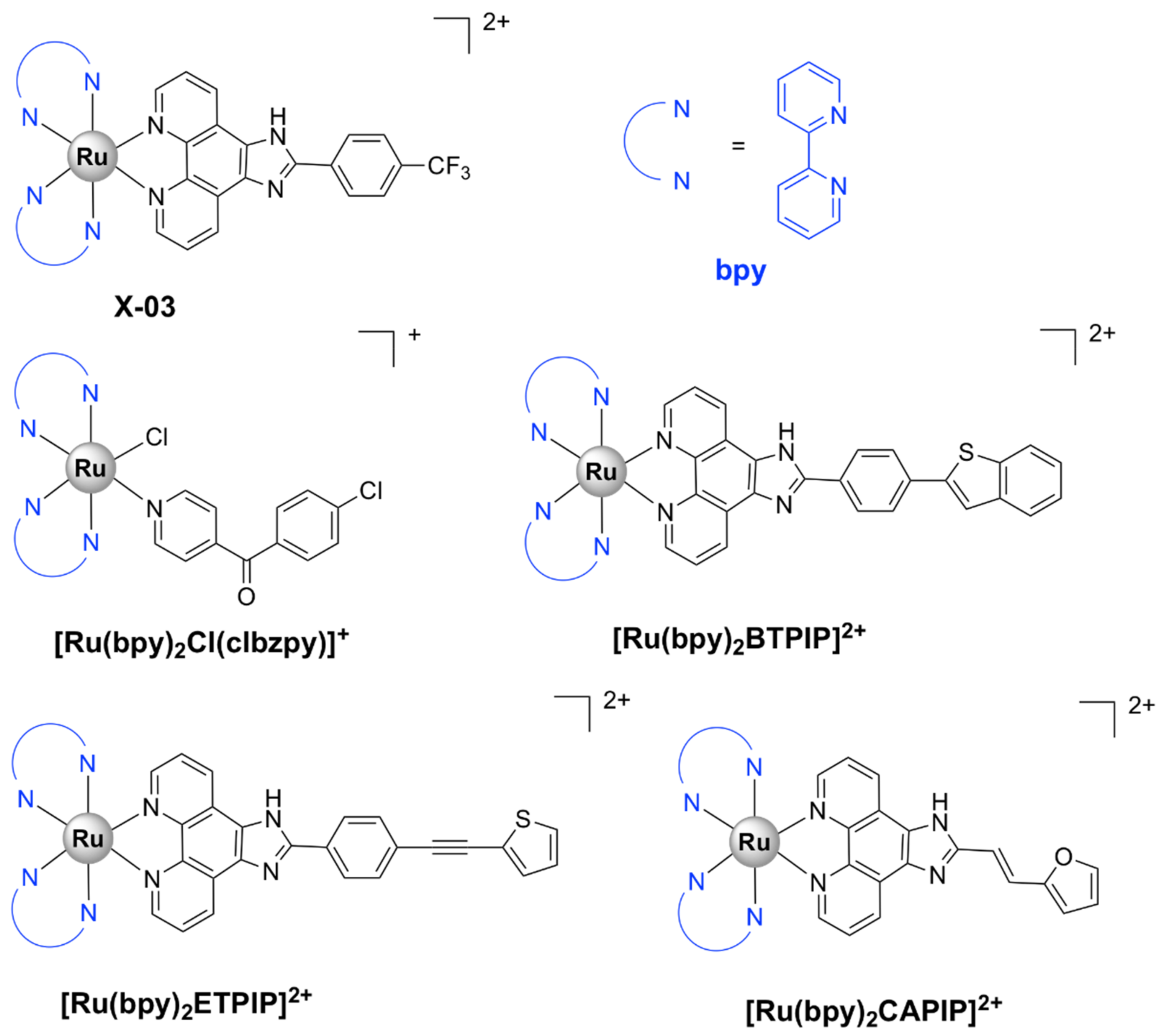
| [Ru(bpy)2Cl(clbzpy)]+ [43] | S. aureus ATCC 25,923: 500, S. epidermidis ATCC 12,228: 250 | P. aeruginosa ATCC 10,145: not active | - | membrane damage |
| [Ru(bpy)2(methionine)]2+ [44] | upon blue LED irradiation S. aureus ATCC 25,923: 62.5, S. epidermidis ATCC 12,228: 125 | P. aeruginosa ATCC 10,145: not active E. coli ATCC 11,303: 500 | - | DNA photodamage |
| [Ru(dmb)2(ETPIP)]2+ [45] | S. aureus Newman: 50 | - | - | - |
| [Ru(phen)2(ETPIP)]2+ [45] | S. aureus Newman: 25 | - | - | inhibits biofilm formation; interacts with intracellular thiols |
| [Ru(bpy)2(BTPIP)]2+ [46] | S. aureus Newman: 16 | - | - | inhibits biofilm formation |
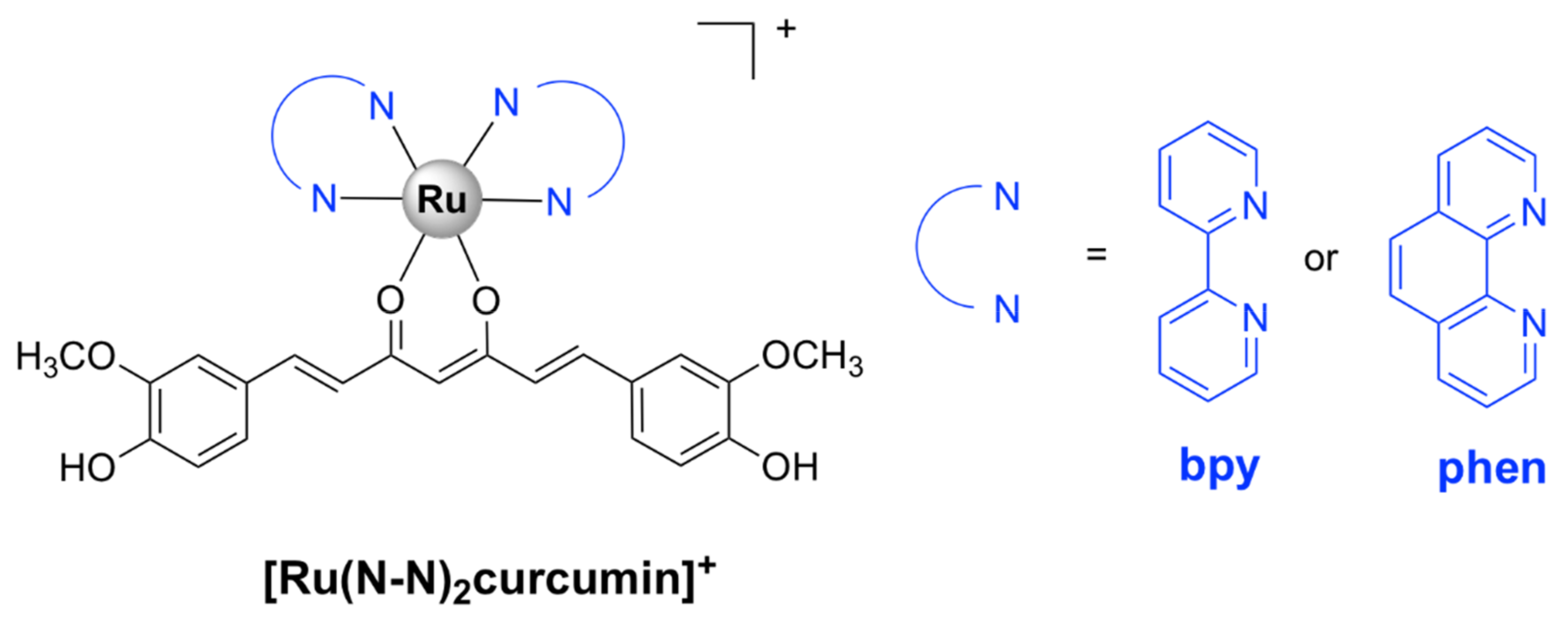
| [Ru(bpy)2curcumin]+ [47] | S. aureus ATCC 29,213: 1 | A. baumanii BAA-1605: > 64, E. coli ATCC 25,922: > 64, K. pneumoniae BAA-1705: > 64, P. aeruginosa ATCC 27,853: > 64 | Vero (African green monkey kidney epithelial) cells: > 80 | bactericidal; inhibits biofilm formation |
| [Ru(phen)2curcumin]+ [47] | S. aureus ATCC 29,213: 1 | A. baumanii BAA-1605: 8–16, E. coli ATCC 25,922: > 64, K. pneumoniae BAA-1705: > 64, P. aeruginosa ATCC 27,853: > 64 | Vero (African green monkey kidney epithelial) cells: > 80 | - |
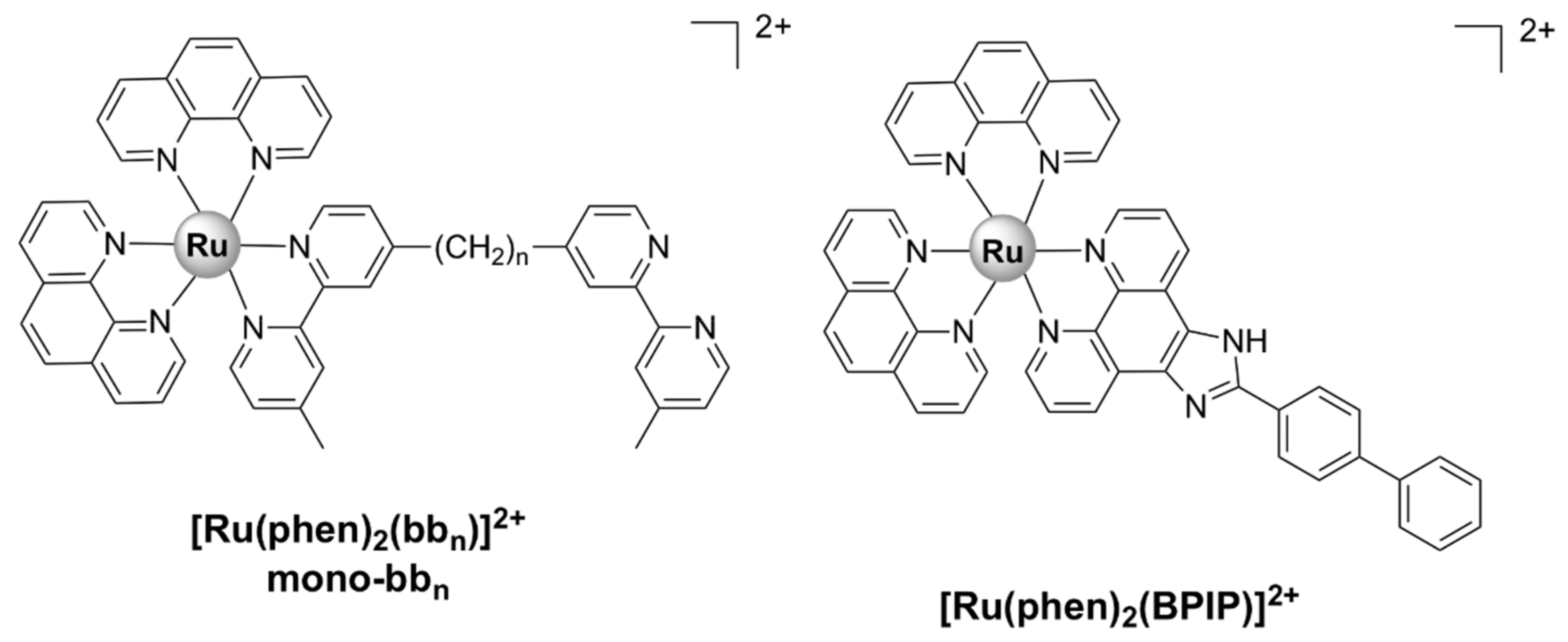
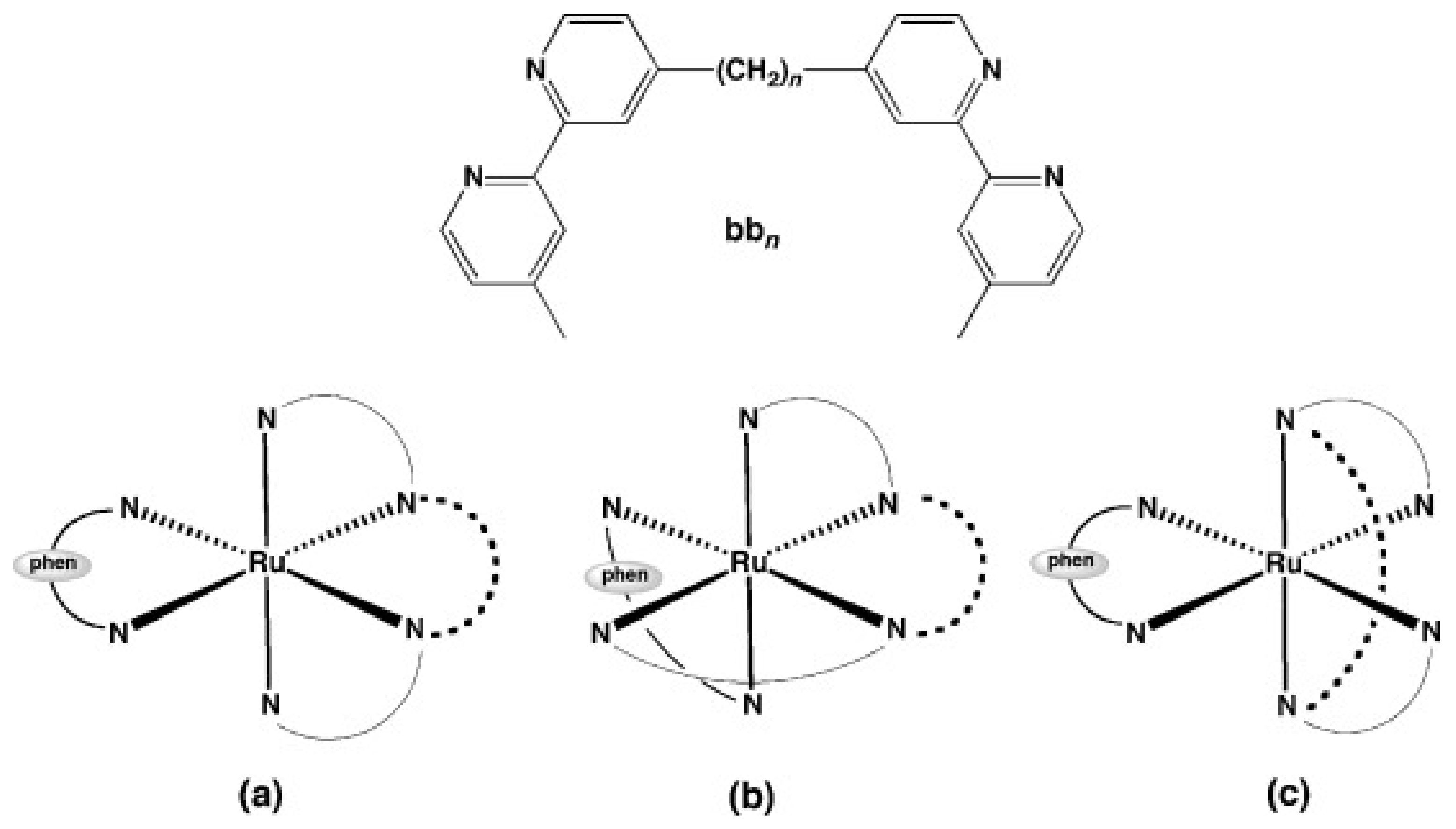
| Mono-bb7 [38] | S. aureus MSSA ATCC 25,923: 4 MRSA (JCU culture collection): 16 | E. coli ATCC 25,922: 16 P. aeruginosa ATCC 27,853: > 128 | - | bactericidal; membrane damage |
| Mono-bb10 [37,38] | S. aureus MSSA ATCC 25,923: 4 MRSA (JCU culture collection): 16 | E. coli ATCC 25,922: 16 P. aeruginosa ATCC 27,853: 32 | - | bactericidal |
| Mono-bb16 [37] | S. aureus MSSA ATCC 25,923: 16 MRSA (JCU culture collection): 16 | E. coli ATCC 25,922: 64 P. aeruginosa ATCC 27,853: 64 | - | - |
| cis-α-[Ru(phen)bb12]2+ [48] | S. aureus MSSA ATCC 25,923: 0.5 MRSA (JCU culture collection): 4 | E. coli ATCC 25,922: 8 P. aeruginosa ATCC 27,853: 8 | - | DNA binding |
| cis-β-[Ru(phen)(bb12)]2+ [48] | S. aureus MSSA ATCC 25,923: 0.5 MRSA (JCU culture collection): 4 | E. coli ATCC 25,922: 16 P. aeruginosa ATCC 27,853: 32 | - | DNA binding |
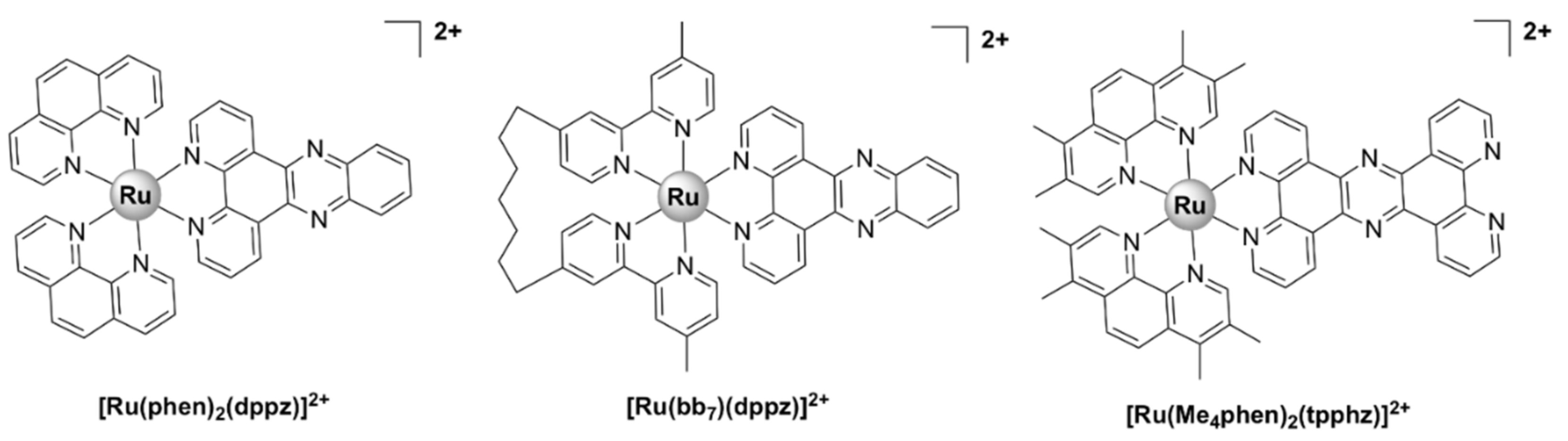
| [Ru(bb7)(dppz)]2+ [49] | S. aureus SH 1000: 2 MRSA USA 300 LAC JE2: 2 | E. coli avian pathogenic: 8 uropathogenic: 8 E. coli MG1655: 8 P. aeruginosa PAO1: 16 | human embryonic kidney HEK-293 cells: 27 (48 h), human fetal hepatocyte L02 cells: 64 (48 h) | bactericidal, DNA binding |
| [Ru(Me4phen)2(dppz)]2+ [50] | S. aureus SH1000: 9.7, E. faecalis V583: 38.8 | E. coli MG1655: 4.9, EC958: 4.9, P. aeruginosa PA2017: 9.7 A. baumannii AB184: 9.7 | - | bactericidal, chromosomal DNA binding |
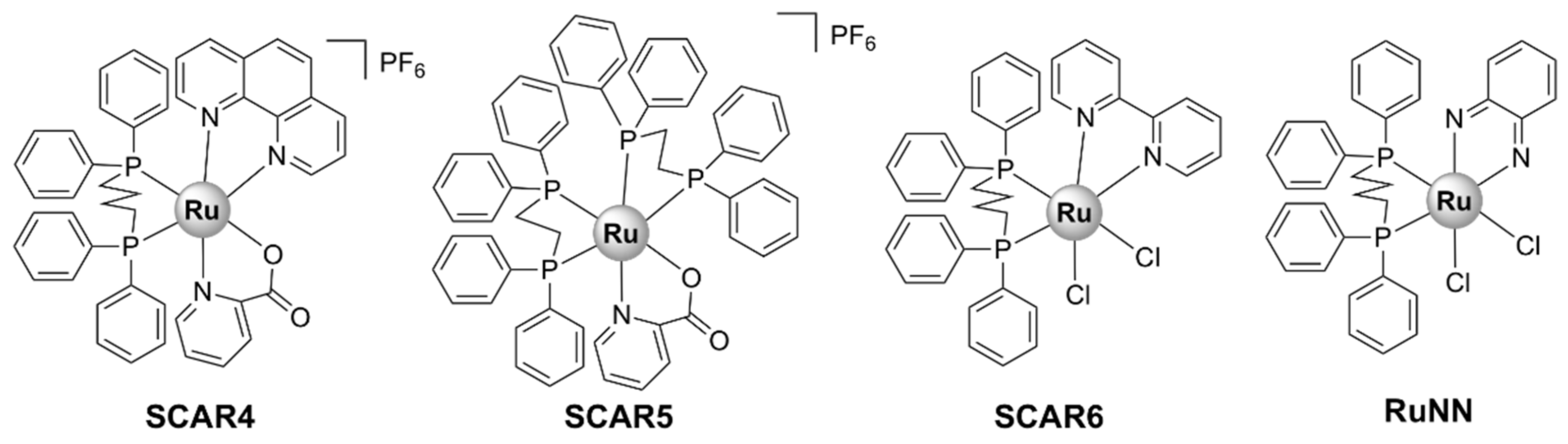
| SCAR4 [51] | M. tuberculosis H37Rv ATCC 27,294 (neither G+, nor G-): 0.63 | - | Mouse monocyte macrophage J774A.1 cell line: 19.5 | covalent binding to DNA |
| SCAR5 [51] | M. tuberculosis H37Rv ATCC 27,294 (neither G+, nor G-): 0.26 | - | J774A.1: 3.9 | covalent binding to DNA |
| SCAR6 [51] | M. tuberculosis H37Rv ATCC 27,294 (neither G+, nor G-): 3.90 | - | J774A.1: 78.2 | covalent binding to DNA |
| RuNN [52] | S. aureus ATCC 25,923: 15.6, S. aureus ATCC 700,698 (MRSA): 62.5 S. epidermidis ATCC 12,228: 31.2, S. epidermidis ATCC 358,983: 62.5 | - | no cytotoxic effect against human erythrocytes | bactericidal; inhibits biofilm formation |
| [Ru(hexpytri)3](PF6)2 [53] | S. aureus MSSA ATCC 25,923: 8, S. aureus MSSA NZRM 9653: 1, S. aureus MRSA MR 9519: 4, S. pyogenes: 4 | E. coli ATCC 25,922: non-active | Vero cells: IC50 > 128 (48h) | cell wall/cytoplasmic membrane damage |
| [Ru(hexyltripy) (heptyltripy)]Cl2 [54] | S. aureus ATCC 25,923: 2 | E. coli ATCC 25,922: 8 | HDFa (skin cells): 16.4 | abnormal cellular division |
| ΔΔ-Rubb7 [37,38] | S. aureus MSSA ATCC 25,923: 16 MRSA (JCU culture collection): 16 | E. coli ATCC 25,922: 16 P. aeruginosa ATCC 27853: 128 | Red blood cells: > 1024 | bactericidal; membrane damage, interaction with ribosomal RNA |
| ΔΔ-Rubb12 [55,56] | S. aureus MSSA ATCC 25,923: 1 MRSA (JCU culture collection): 1 | E. coli ATCC 25,922: 2 P. aeruginosa ATCC 27,853: 16 | Baby hamster kidney (BHK): 113.9, HEK-293: 82.2 | bactericidal; membrane damage, interaction with ribosomal RNA |
| ΔΔ-Rubb16 [56] | S. aureus MSSA ATCC 25,923: 1 MRSA (JCU culture collection): 1 | E. coli ATCC 25,922: 4 P. aeruginosa ATCC 27,853: 8 | Red blood cells: 22, BHK: 49.8, HEK-293: 35.1 | bactericidal; membrane damage, interaction with ribosomal RNA |
| [Ru2(Me4phen)2(tpphz)]4+ [57,58,59] | S. aureus MSSA SH1000: 86, Enterococcus faecalis V583: 1 | E. coli WT G1655: 2.5, EC958 ST131 (multi-drug-resistant, clinical isolate): 3.5, P. aeruginosa (clinical isolate): 4, K. pneumoniae (clinical isolate): 3.5, A. baumannii (clinical isolate): 3.5 | HEK-293: 270 | membrane and DNA damage |
| Cl-Rubb7-Cl [55,60] | S. aureus MSSA ATCC 25,923: 8 MRSA (JCU culture collection): 8 | E. coli ATCC 25,922: 8 P. aeruginosa ATCC 27,853: 32 | - | bactericidal |
| Cl-Rubb12-Cl [55,60] | S. aureus MSSA ATCC 25,923: 1 MRSA (JCU culture collection): 1 | E. coli ATCC 25,922: 2 P. aeruginosa ATCC 27,853: 8 | - | bactericidal |
| Cl-Rubb16-Cl [55,60] | S. aureus MSSA ATCC 25,923: 8 MRSA (JCU culture collection): 8 | E. coli ATCC 25,922: 8 P. aeruginosa ATCC 27,853: > 128 | - | bactericidal |
| Rubb7-Cl [56] | S. aureus MSSA ATCC 25,923: 8 MRSA (JCU culture collection): 16 | E. coli ATCC 25,922: 1 P. aeruginosa ATCC 27,853: 16 | BHK: 337.5, HEK-293: 98 | interaction with chromosomal DNA and ribosomal RNA |
| Rubb12-Cl [56] | S. aureus MSSA ATCC 25,923: 1 MRSA (JCU culture collection): 1 | E. coli ATCC 25,922: 1 P. aeruginosa ATCC 27,853: 16 | BHK: 70.6, HEK-293: 87.3 | interaction with chromosomal DNA and ribosomal RNA |
| Rubb16-Cl [56] | S. aureus MSSA ATCC 25,923: 1 MRSA (JCU culture collection): 2 | E. coli ATCC 25,922: 4 P. aeruginosa ATCC 27,853: 64 | BHK: 34.9, HEK-293: 63.5 | interaction with chromosomal DNA and ribosomal RNA |
| Rubb7-tri [37,61] | S. aureus MSSA ATCC 25,923: 4 MRSA (JCU culture collection): 4 | E. coli ATCC 25,922: 16 P. aeruginosa ATCC 27,853: 2 | - | interaction with DNA |
| Rubb7-tetra (Rubb7-TL) [62] | S. aureus MSSA ATCC 25,923: 8 MRSA (JCU culture collection): 16 | E. coli avian pathogenic: 16 uropathogenic: 16 E. coli MG1655: 16 P. aeruginosa PAO1: 32 | BHK: 176 (24 h) BHK: 36.4 (72 h) | interaction with proteins |
| Rubb7-TNL [62] | S. aureus MSSA ATCC 25,923: 4 MRSA (JCU culture collection): 8 | E. coli avian pathogenic: 16 uropathogenic: 16 E. coli MG1655: 8 P. aeruginosa PAO1: 16 | BHK: 276 (24 h) BHK: 81.6 (72 h) | interaction with proteins |
| Rubb12-tri [37,55,61] | S. aureus: 1 MRSA (JCU culture collection): 1 | E. coli: 4 P. aeruginosa: 32 | BHK: 50.9 (72 h), HEK-293: 21.8 (72 h) | bactericidal, interaction with DNA |
| Rubb12-tetra [37,55,61] | S. aureus: 2 MRSA (JCU culture collection): 2 | E. coli: 2 P. aeruginosa: 16 | BHK: 43.7 (72 h), HEK-293: 21.3 (72 h) | bactericidal, interaction with DNA |
| Rubb16-tri [37,55,61] | S. aureus: 2 MRSA (JCU culture collection): 2 | E. coli: 8 P. aeruginosa: 32 | BHK: 25.1 (72 h), HEK-293: 20.2 (72 h) | bactericidal, interaction with DNA |
| Rubb16-tetra [37,55,61] | S. aureus: 2 MRSA (JCU culture collection): 2 | E. coli: 8 P. aeruginosa: 32 | BHK: 19.8 (72 h), HEK-293: 15.8 (72 h) | bactericidal, interaction with DNA |
| Ruthenium-based CORMs | ||||
| CORM-2 [63,64,65] | Growth inhibitory effects on S. aureus (MIC value not reported) | E. coli avian pathogenic: 250, uropathogenic: 250, E. coli MG1655: 250, P. aeruginosa PAO1: 3.8 H. pylori strains (including antibiotic resistant): 100–200 | Murine RAW264.7 monocyte macrophages: > 50 (DMEM culture medium) | Bactericidal, inhibition of aerobic respiration, inhibition of biofilm formation and disruption of mature biofilms, ROS generation, interaction with chromosomal DNA and intracellular proteins, interference with iron homeostasis |
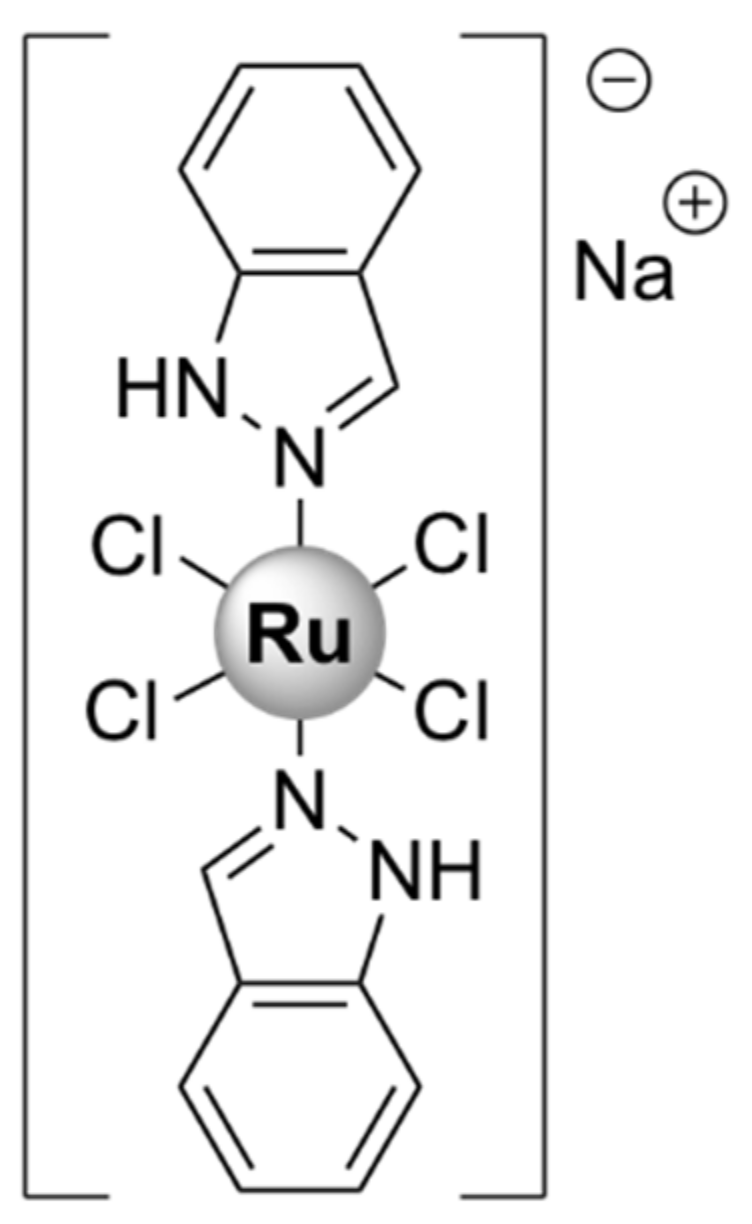
| CORM-3 [64,66,67] | Growth inhibitory effects on S. aureus, Lactobacillus lactis (MIC value not reported) | E. coli MG1655: 4 (minimal GDMM medium) and > 512 (in rich MH-II medium) H. pylori 26,695: 420 (antibiotic resistant strains) | L929 murine fibroblast cells: 63 (RPMI culture medium), RAW264.7: > 30 (DMEM culture medium) | |
| Ruthenium complexes in Antimicrobial Photodynamic Therapy | ||||
| [Ru(dmob)3]2+ [68] | S. aureus NCTC 10788: 12.5 | P. aeruginosa NCTC 8626: 50 | - | Light activation |
| cis-[Ru(bpy)2(INH)2]2+ [69] | Mycobacterium smegmatis: 4 | human lung fibroblast MRC-5 cell line: > 200 | 465 nm blue light activation | |
| [Ru(Ph2phen)2(dpp) PtCl2]2+ [70] | - | E. coli JM109: 8 | - | visible light activation, binding to chromosomal DNA |
| [Ru(CO)2Cl2]n [71] | S. aureus CETC 240, coincident with ATCC 6538 P: 0.033 | E. coli CET 516, coincident with ATCC 8739: 0.0066 | human dermal fibroblasts hDF: > 3.33 | 365 nm UV light activation, ROS generation, biofilm inhibition |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munteanu, A.-C.; Uivarosi, V. Ruthenium Complexes in the Fight against Pathogenic Microorganisms. An Extensive Review. Pharmaceutics 2021, 13, 874. https://doi.org/10.3390/pharmaceutics13060874
Munteanu A-C, Uivarosi V. Ruthenium Complexes in the Fight against Pathogenic Microorganisms. An Extensive Review. Pharmaceutics. 2021; 13(6):874. https://doi.org/10.3390/pharmaceutics13060874
Chicago/Turabian StyleMunteanu, Alexandra-Cristina, and Valentina Uivarosi. 2021. "Ruthenium Complexes in the Fight against Pathogenic Microorganisms. An Extensive Review" Pharmaceutics 13, no. 6: 874. https://doi.org/10.3390/pharmaceutics13060874
APA StyleMunteanu, A.-C., & Uivarosi, V. (2021). Ruthenium Complexes in the Fight against Pathogenic Microorganisms. An Extensive Review. Pharmaceutics, 13(6), 874. https://doi.org/10.3390/pharmaceutics13060874