
Behind the Adaptive and Resistance Mechanisms of Cancer Stem Cells to TRAIL

 , ,
, ,
Abstract
:1. Introduction
2. Recombinant TRAIL
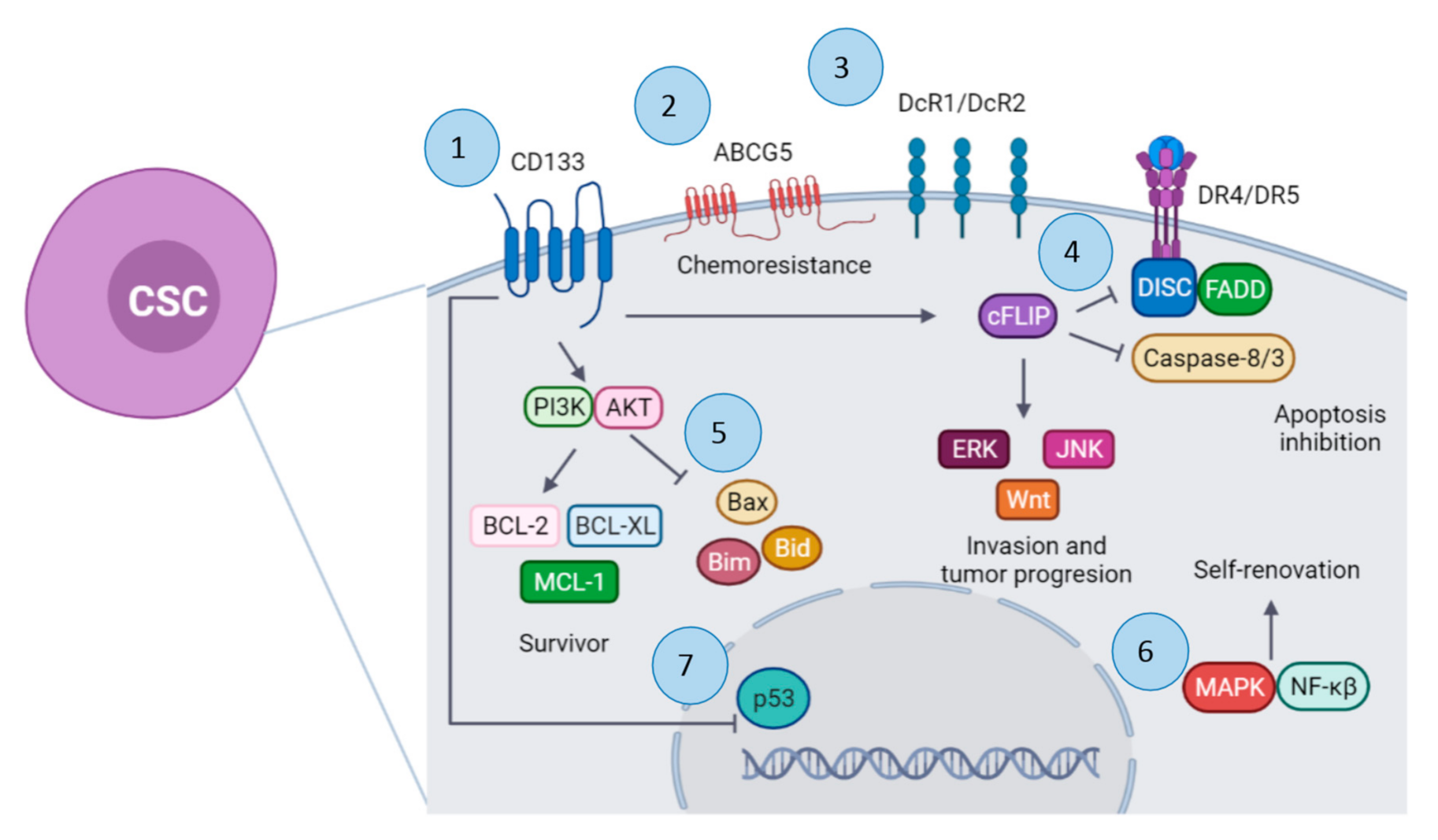
3. Cancer Stem Cells and TRAIL
4. TRAIL Resistance Mechanism
5. Microenvironment and TRAIL Activity
6. TRAIL Activity in Angiogenesis
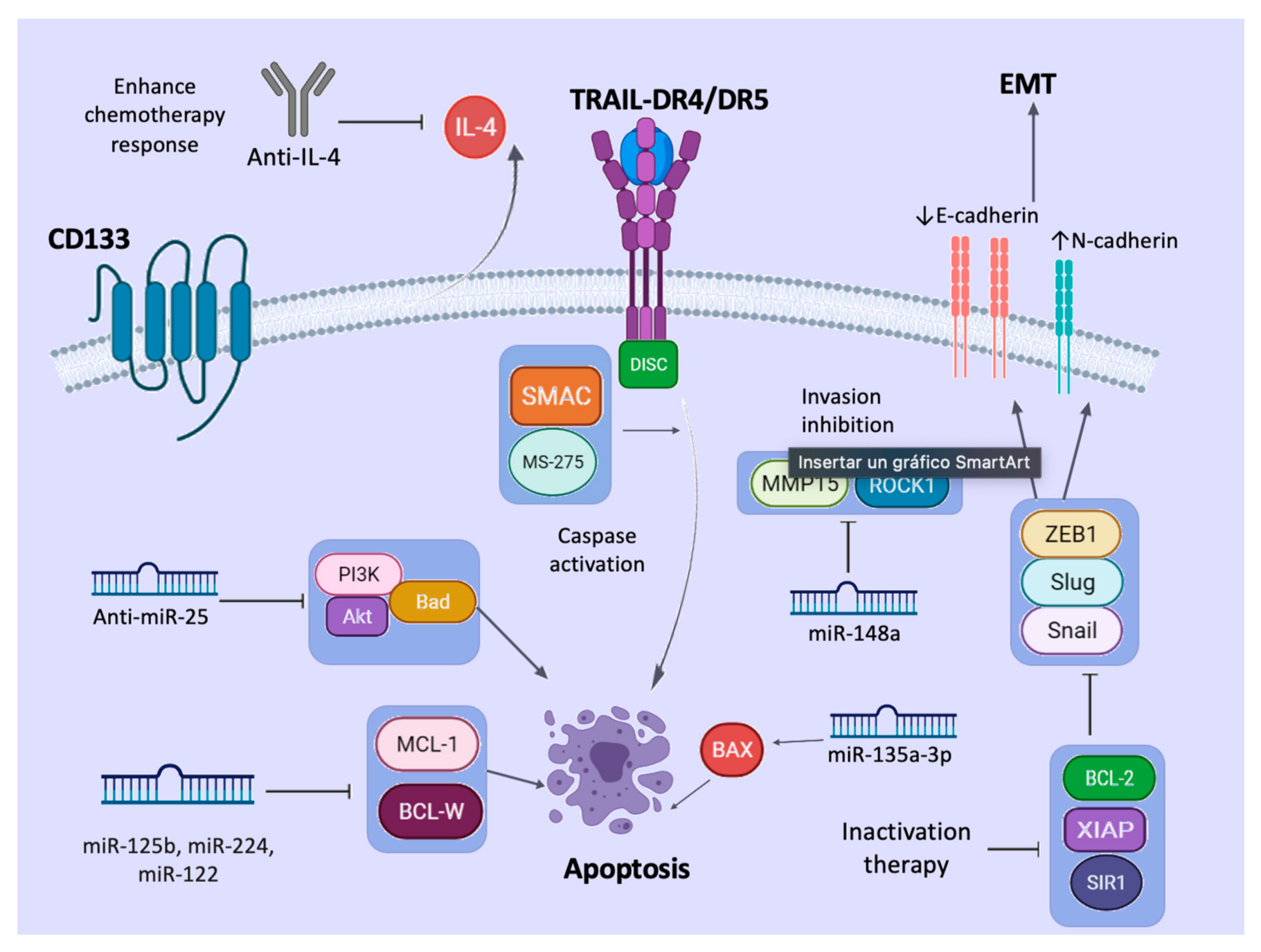
7. Regulation Mechanism
8. Mechanism against TRAIL Resistance
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCG5 | ATP-binding cassette transporter |
| APAF-1 | Apoptotic protease activating factor 1 |
| AP1 | Activator protein 1 |
| Apo2L | Apo-2 ligand |
| Bcl-2 | B-cell lymphoma 2 |
| BPM | Bone morphogenetic protein |
| CAD | Caspase-activated DNase |
| CAF | Cancer-associated fibroblasts |
| CRC | Colorectal cancer |
| CRD | Cysteine-rich domain |
| CSCs | Cancer stem cells |
| CTLs | Cytotoxic T cells |
| DAMPs | Damage-associated molecular patterns |
| DCs | Dendritic cells |
| DED | Death-inducing signaling complex |
| DCRs | Decoy receptors |
| DD | Death domain |
| DISC | Death-inducing signaling complex |
| DKK-1 | Dickkopf-1 |
| DR | Death receptor |
| EGF | Epidermal growth factor |
| ECM | Extracellular matrix |
| EMT | Epithelial–mesenchymal transition |
| EMILIN2 | Elastin microfibril interface-located protein 2 |
| FADD | Fas-associated death domain |
| FLIP | FLICE-like inhibitory protein |
| FOX | Forkhead Box |
| HGF | Hepatocyte growth factor |
| HIF-1α | Hypoxia-inducible 1alpha |
| ICAD | Inhibitor of caspase-activated DNase |
| IFN-β | Interferon-β |
| IL8 | Interleukin 8 |
| MAPK | mitogen-activated protein kinase |
| MDP | membrane-proximal domain |
| MDSC | myeloid-derived suppressor cells |
| MET | mesenchymal–epithelial transition |
| miRNAs or miRs | microRNAs |
| MMP12 | matrix metallopeptidase 12 |
| MSCs | Mesenchymal stem cells |
| NFAT | Nuclear factor of activated T cells |
| NK | Natural killer |
| NO | Nitric oxide |
| NSCLC | Non-small-cell lung cancer |
| OPG | Osteoprotegerin |
| PDGF | Platelet-derived growth factor |
| PI3K | Phosphatidylinositol-3-kinase |
| PTEN | Phosphatase and tensin homolog |
| rhTRAIL | Recombinant human TRAIL |
| sTRAIL | Soluble TRAIL |
| TAMs | Tumor-associated macrophages |
| TGF-β | Transforming growth factor-beta |
| Tregs | Regulatory T cells |
| TILs | Tumor-infiltrating lymphocytes |
| VEGF | Vascular endothelial growth factor |
| TNF | Tumor necrosis factor |
| TRAF2 | Tumor receptor-associated factor 2 |
| TRAIL | Tumor necrosis factor (TNF)-related apoptosis-inducing ligand |
| TNF-α | Tumor necrosis factor-alpha |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| VEGF | Vascular endothelial growth factor |
References
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.M.; Kong, W.Y.; Fang, C.-M.; Loh, H.-S.; Chuah, L.-H.; Abdullah, S.; Ngai, S.C. The TRAIL to cancer therapy: Hindrances and potential solutions. Crit. Rev. Oncol. 2019, 143, 81–94. [Google Scholar] [CrossRef]
- Teringova, E.; Tousek, P. Apoptosis in ischemic heart disease. J. Transl. Med. 2017, 15, 87. [Google Scholar] [CrossRef] [Green Version]
- Holoch, P.A.; Griffith, T.S. TNF-related apoptosis-inducing ligand (TRAIL): A new path to anti-cancer therapies. Eur. J. Pharmacol. 2009, 625, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labsch, S.; Liu, L.; Bauer, N.; Zhang, Y.; Aleksandrowicz, E.; Gladkich, J.; Schönsiegel, F.; Herr, I. Sulforaphane and TRAIL induce a synergistic elimination of advanced prostate cancer stem-like cells. Int. J. Oncol. 2014, 44, 1470–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naval, J.; de Miguel, D.; Gallego-Lleyda, A.; Anel, A.; Martinez-Lostao, L. Importance of TRAIL molecular anatomy in receptor oligomerization and signaling. Implications for cancer therapy. Cancers 2019, 11, 444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino, D.; Lalaoui, N.; Morizot, A.; Solary, E.; Micheau, O. TRAIL in cancer therapy: Present and future challenges. Expert Opin. Ther. Targets 2007, 11, 1299–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Gao, Q.; Xie, T.; Liu, Y.; Luo, L.; Xu, C.; Shen, L.; Wan, F.; Lei, T.; Ye, F. Synergistic effect of TRAIL and irradiation in elimination of glioblastoma stem-like cells. Clin. Exp. Med. 2018, 18, 399–411. [Google Scholar] [CrossRef]
- De Looff, M.; de Jong, S.; Kruyt, F.A.E. Multiple interactions between cancer cells and the tumor microenvironment modulate trail signaling: Implications for TRAIL receptor targeted therapy. Front. Immunol. 2019, 10, 1530. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Gajan, A.; Chu, Q.; Xiong, H.; Wu, K.; Wu, G.S. Developing TRAIL/TRAIL death receptor-based cancer therapies. Cancer Metastasis Rev. 2018, 37, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Behrooz, A.B.; Syahir, A.; Ahmad, S. CD133: Beyond a cancer stem cell biomarker. J. Drug Target. 2019, 27, 257–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Xu, J.; Zhao, J.; Bai, J. Knockdown of miR-27a sensitizes colorectal cancer stem cells to TRAIL by promoting the formation of Apaf-1-caspase-9 complex. Oncotarget 2017, 8, 45213–45223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Miguel, D.; Lemke, J.; Anel, A.; Walczak, H.; Martinez-Lostao, L. Onto better TRAILs for cancer treatment. Cell Death Differ. 2016, 23, 733–747. [Google Scholar] [CrossRef] [Green Version]
- Sahlberg, S.H.; Spiegelberg, D.; Glimelius, B.; Stenerlöw, B.; Nestor, M. Evaluation of cancer stem cell markers CD133, CD44, CD24: Association with AKT isoforms and radiation resistance in colon cancer cells. PLoS ONE 2014, 9, e94621. [Google Scholar] [CrossRef]
- Kim, W.-T.; Ryu, A.C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Yelle, N.; Venugopal, C.; Singh, S.K. EMT: Mechanisms and therapeutic implications. Pharmacol. Ther. 2018, 182, 80–94. [Google Scholar] [CrossRef]
- Kim, T.; Veronese, A.; Pichiorri, F.; Lee, T.J.; Jeon, Y.-J.; Volinia, S.; Pineau, P.; Marchio, A.; Palatini, J.; Suh, S.-S.; et al. p53 regulates epithelial–mesenchymal transition through microRNAs targeting ZEB1 and ZEB. J. Exp. Med. 2011, 208, 875–883. [Google Scholar] [CrossRef]
- Babaei, G.; Aziz, S.G.-G.; Jaghi, N.Z.Z. EMT, cancer stem cells and autophagy; The three main axes of metastasis. Biomed. Pharmacother. 2021, 133, 110909. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, B.; Sun, D.; Liu, T.; Che, N.; Gu, Q.; Dong, X.; Li, R.; Liu, Y.; Li, J. Slug promotes hepatocellular cancer cell progression by increasing sox2 and nanog expression. Oncol. Rep. 2014, 33, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Javaeed, A.; Ghauri, S.K. Metastatic potential and prognostic significance of SOX2: A meta-analysis. World J. Clin. Oncol. 2019, 10, 234–246. [Google Scholar] [CrossRef]
- Zhou, P.; Li, B.; Liu, F.; Zhang, M.; Wang, Q.; Liu, Y.; Yao, Y.; Li, D. The epithelial to mesenchymal transition (EMT) and cancer stem cells: Implication for treatment resistance in pancreatic cancer. Mol. Cancer 2017, 16, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Stantic, M.; Zobalova, R.; Prokopova, K.; Neuzil, J.; Dong, L.-F. Cancer cells with high expression of CD133 exert FLIP upregulation and resistance to TRAIL-induced apoptosis. BioFactors 2008, 34, 231–235. [Google Scholar] [CrossRef]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—a clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Shanehbandi, D.; Asadi, M.; Shomali, N.; Faraji, A.; Khaze, V.; Pakdel, A.; Mokhtarzadeh, A.; Ebrahimi, A.A.; Shabani, A.; et al. Effects of CD133 silencing on survival and migration of HT-29 colorectal cancer cells. Iran. J. Immunol. 2019, 16, 246–257. [Google Scholar]
- Garofalo, M.; di Leva, G.; Romano, G.; Nuovo, G.; Suh, S.-S.; Ngankeu, A.; Taccioli, C.; Pichiorri, F.; Alder, H.; Secchiero, P.; et al. miR-221&222 Regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell 2009, 16, 498–509. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, T.; Montinaro, A.; von Karstedt, S.; Sevko, A.; Surinova, S.; Chakravarthy, A.; Taraborrelli, L.; Draber, P.; Lafont, E.; Vargas, F.A.; et al. The TRAIL-induced cancer secretome promotes a tumor-supportive immune microenvironment via CCR. Mol. Cell 2017, 65, 730–742.e5. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, D.; Liang, S.; Wang, J.; Liu, C.; Nie, C.; Shan, Z.; Wang, L.; Fan, Q.; Wang, F. miR-106b promotes cell invasion and metastasis via PTEN mediated EMT in ESCC. Oncol. Lett. 2018, 15, 4619–4626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhu, V.V.; Allen, J.E.; Dicker, D.T.; El-Deiry, W.S. Small molecule ONC201/TIC10 targets chemotherapy-resistant colorectal cancer stem-like cells in an Akt/Foxo3a/TRAILdependent manner. Cancer Res. 2015, 75, 1423–1432. [Google Scholar] [CrossRef] [Green Version]
- French, R.; Hayward, O.; Jones, S.; Yang, W.; Clarkson, R. Cytoplasmic levels of cFLIP determine a broad susceptibility of breast cancer stem/progenitor-like cells to TRAIL. Mol. Cancer 2015, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jalving, M.; Heijink, D.M.; Koornstra, J.J.; Ek, W.B.-V.; Zwart, N.; Wesseling, J.; Sluiter, W.J.; de Vries, E.G.E.; Kleibeuker, J.H.; de Jong, S. Regulation of TRAIL receptor expression by -catenin in colorectal tumours. Carcinogenesis 2013, 35, 1092–1099. [Google Scholar] [CrossRef] [Green Version]
- Coelho, B.P.; Fernandes, C.F.D.L.; Boccacino, J.M.; Souza, M.C.D.S.; Melo-Escobar, M.I.; Alves, R.N.; Prado, M.B.; Iglesia, R.P.; Cangiano, G.; Mazzaro, G.L.R.; et al. Multifaceted WNT signaling at the crossroads between epithelial-mesenchymal transition and autophagy in glioblastoma. Front. Oncol. 2020, 10, 597743. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, J.-Y.; Wei, W.-Z.; Wu, G.S. Activation of the akt survival pathway contributes to TRAIL resistance in cancer cells. PLoS ONE 2010, 5, e10226. [Google Scholar] [CrossRef] [Green Version]
- Sophonnithiprasert, T.; Nilwarangkoon, S.; Nakamura, Y.; Watanapokasin, R. Goniothalamin enhances TRAIL-induced apoptosis in colorectal cancer cells through DR5 upregulation and cFLIP downregulation. Int. J. Oncol. 2015, 47, 2188–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pretzsch, E.; Bösch, F.; Neumann, J.; Ganschow, P.; Bazhin, A.; Guba, M.; Werner, J.; Angele, M. Mechanisms of metastasis in colorectal cancer and metastatic organotropism: Hematogenous versus peritoneal spread. J. Oncol. 2019, 2019, 7407190. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef]
- Zwirner, N.W.; Ziblat, A. Regulation of NK cell activation and effector functions by the IL-12 family of cytokines: The case of IL-27. Front. Immunol. 2017, 8, 25. [Google Scholar] [CrossRef] [Green Version]
- Griffith, B.T.S.; Wiley, S.R.; Kubin, M.Z.; Sedger, L.M.; Maliszewski, C.R.; Fanger, N.A. Monocyte-mediated tumoricidal activity via the tumor necrosis factor-related cytokine, TRAIL. Cell 1999, 189, 1343–1353. [Google Scholar] [CrossRef]
- Tecchio, C.; Huber, V.; Scapini, P.; Calzetti, F.; Margotto, D.; Todeschini, G.; Pilla, L.; Martinelli, G.; Pizzolo, G.; Rivoltini, L.; et al. IFNα-stimulated neutrophils and monocytes release a soluble form of TNF-related apoptosis-inducing ligand (TRAIL/Apo-2 ligand) displaying apoptotic activity on leukemic cells. Blood 2004, 103, 3837–3844. [Google Scholar] [CrossRef] [Green Version]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Secchiero, P.; Gonelli, A.; Mirandola, P.; Melloni, E.; Zamai, L.; Celeghini, C.; Milani, D.; Zauli, G. Tumor necrosis factor-related apoptosis-inducing ligand induces monocytic maturation of leukemic and normal myeloid precursors through a caspase-dependent pathway. Blood 2002, 100, 2421–2429. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.A.; Drake, V.; Huang, A.; Chiu, S.; Zheng, L. Reprogramming the tumor microenvironment: Tumor-induced immunosuppressive factors paralyze T cells. OncoImmunology 2015, 4, e1016700. [Google Scholar] [CrossRef]
- de Winde, C.M.; Munday, C.; Acton, S.E. Molecular mechanisms of dendritic cell migration in immunity and cancer. Med. Microbiol. Immunol. 2020, 209, 515–529. [Google Scholar] [CrossRef]
- Fanger, N.A.; Maliszewski, C.R.; Schooley, K.; Griffith, T.S. Human dendritic cells mediate cellular apoptosis via tumor necrosis factor–related apoptosis-inducing ligand (trail). J. Exp. Med. 1999, 190, 1155–1164. [Google Scholar] [CrossRef]
- Rossin, A.; Miloro, G.; Hueber, A.-O. TRAIL and FasL functions in cancer and autoimmune diseases: Towards an increasing complexity. Cancers 2019, 11, 639. [Google Scholar] [CrossRef] [Green Version]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Facciabene, A.; Motz, G.T.; Coukos, G. T-Regulatory Cells: Key players in tumor immune escape and angiogenesis: Figure. Cancer Res. 2012, 72, 2162–2171. [Google Scholar] [CrossRef] [Green Version]
- Hallett, M.A.; Venmar, K.T.; Fingleton, B. Cytokine stimulation of epithelial cancer cells: The similar and divergent functions of IL-4 and IL-13. Cancer Res. 2012, 72, 6338–6343. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.-H.; Ojha, U.; Lee, Y.M. Pathological angiogenesis and inflammation in tissues. Arch. Pharmacal. Res. 2021, 44, 1–15. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2019, 77, 1745–1770. [Google Scholar] [CrossRef] [Green Version]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Annan, D.A.-M.; Kikuchi, H.; Maishi, N.; Hida, Y.; Hida, K. Tumor endothelial cell—A biological tool for translational cancer research. Int. J. Mol. Sci. 2020, 21, 3238. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bao, Q.; Renner, A.; Camaj, P.; Eichhorn, M.; Ischenko, I.; Angele, M.; Kleespies, A.; Jauch, K.-W.; Bruns, C. Cancer stem cells and angiogenesis. Int. J. Dev. Biol. 2011, 55, 477–482. [Google Scholar] [CrossRef] [Green Version]
- Ping, Y.-F.; Zhang, X.; Bian, X.-W. Cancer stem cells and their vascular niche: Do they benefit from each other? Cancer Lett. 2016, 380, 561–567. [Google Scholar] [CrossRef]
- Jun, J.C.; Rathore, A.; Younas, H.; Gilkes, D.; Polotsky, V.Y. Hypoxia-inducible factors and cancer. Curr. Sleep Med. Rep. 2017, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Vallée, A.; Guillevin, R.; Vallée, J.-N. Vasculogenesis and angiogenesis initiation under normoxic conditions through Wnt/β-catenin pathway in gliomas. Rev. Neurosci. 2017, 29, 71–91. [Google Scholar] [CrossRef]
- Rivera, L.B.; Bergers, G. Myeloid cell-driven angiogenesis and immune regulation in tumors. Trends Immunol. 2015, 36, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-L.; Easton, A.S. Evidence that tumor necrosis factor-related apoptosis inducing ligand (TRAIL) inhibits angiogenesis by inducing vascular endothelial cell apoptosis. Biochem. Biophys. Res. Commun. 2010, 391, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Cartland, S.P.; Genner, S.W.; Zahoor, A.; Kavurma, M.M. Comparative Evaluation of TRAIL, FGF-2 and VEGF-A-induced angiogenesis in vitro and in vivo. Int. J. Mol. Sci. 2016, 17, 2025. [Google Scholar] [CrossRef] [Green Version]
- Patil, M.S.; Cartland, S.P.; Kavurma, M.M. TRAIL signals, extracellular matrix and vessel remodelling. Vasc. Biol. 2020, 2, R73–R84. [Google Scholar] [CrossRef]
- di Bartolo, B.; Cartland, S.; Prado-Lourenco, L.; Griffith, T.S.; Gentile, C.; Ravindran, J.; Azahri, N.S.M.; Thai, T.; Yeung, A.W.S.; Thomas, S.R.; et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) promotes angiogenesis and ischemia-induced neovascularization via NADPH oxidase 4 (NOX4) and nitric oxide-dependent mechanisms. J. Am. Hear. Assoc. 2015, 4, e002527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncalves, I.; Singh, P.; Tengryd, C.; Cavalera, M.; Mattisson, I.Y.; Nitulescu, M.; Persson, A.F.; Volkov, P.; Engström, G.; Orho-Melander, M.; et al. sTRAIL-R2 (soluble TNF [tumor necrosis factor]-related apoptosis-inducing ligand receptor 2) a marker of plaque cell apoptosis and cardiovascular events. Stroke 2019, 50, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, L.; Zhou, H.J.; Min, W. The role of NOX4 and TRX2 in angiogenesis and their potential cross-talk. Antioxidants 2017, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Islas, J.F.; Moreno-Cuevas, J.E. A microRNA perspective on cardiovascular development and diseases: An update. Int. J. Mol. Sci. 2018, 19, 2075. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Leng, H.; Shi, X.; Ji, J.; Fu, J. MiR-155 promotes cell proliferation and inhibits apoptosis by PTEN signaling pathway in the psoriasis. Biomed. Pharmacother. 2017, 90, 524–530. [Google Scholar] [CrossRef]
- Hamada, S.; Masamune, A.; Miura, S.; Satoh, K.; Shimosegawa, T. MiR-365 induces gemcitabine resistance in pancreatic cancer cells by targeting the adaptor protein SHC1 and pro-apoptotic regulator BAX. Cell. Signal. 2014, 26, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Lowe, S.W. The microcosmos of cancer. Nat. Cell Biol. 2012, 482, 347–355. [Google Scholar] [CrossRef]
- Lu, T.; Shao, N.; Ji, C. Targeting microRNAs to modulate TRAIL-induced apoptosis of cancer cells. Cancer Gene Ther. 2012, 20, 33–37. [Google Scholar] [CrossRef]
- Xiao, F.; Chen, J.; Lian, C.; Han, P.; Zhang, C. Tumor necrosis factor-related apoptosis-inducing ligand induces cytotoxicity specific to osteosarcoma by microRNA response elements. Mol. Med. Rep. 2014, 11, 739–745. [Google Scholar] [CrossRef]
- Zhou, R.; Yuan, P.; Wang, Y.; Hunsberger, J.G.; Elkahloun, A.; Wei, Y.; Damschroder-Williams, P.; Du, J.; Chen, G.; Manji, H.K. Evidence for selective microRNAs and their effectors as common long-term targets for the actions of mood stabilizers. Neuropsychopharmacology 2008, 34, 1395–1405. [Google Scholar] [CrossRef]
- Li, D.; Ji, L.; Liu, L.; Liu, Y.; Hou, H.; Yu, K.; Sun, Q.; Zhao, Z. Characterization of circulating microRNA expression in patients with a ventricular septal defect. PLoS ONE 2014, 9, e106318. [Google Scholar] [CrossRef]
- Villanova, L.; Careccia, S.; De Maria, R.; Fiori, M.E. Micro-economics of apoptosis in cancer: ncRNAs modulation of BCL-2 family members. Int. J. Mol. Sci. 2018, 19, 958. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Huang, Y.; Chen, L.; Wang, J. miR-221 regulates proliferation and apoptosis of ovarian cancer cells by targeting BMF. Oncol. Lett. 2018, 16, 6697–6704. [Google Scholar] [CrossRef] [Green Version]
- Voigt, S.; Philipp, S.; Davarnia, P.; Winoto-Morbach, S.; Röder, C.; Arenz, C.; Trauzold, A.; Kabelitz, D.; Schütze, S.; Kalthoff, H.; et al. TRAIL-induced programmed necrosis as a novel approach to eliminate tumor cells. BMC Cancer 2014, 14, 74. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Lu, H.; Wang, X.; Jin, H. MicroRNAs in hepatocellular carcinoma: Regulation, function, and clinical implications. Sci. World J. 2013, 2013, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Razumilava, N.; Bronk, S.F.; Smoot, R.L.; Fingas, C.D.; Werneburg, N.W.; Roberts, L.; Mott, J.L. miR-25 targets TNF-related apoptosis inducing ligand (TRAIL) death receptor-4 and promotes apoptosis resistance in cholangiocarcinoma. Hepatology 2012, 55, 465–475. [Google Scholar] [CrossRef]
- Zeng, Z.; Li, Y.; Pan, Y.; Lan, X.; Song, F.; Sun, J.; Zhou, K.; Liu, X.; Ren, X.; Wang, F.; et al. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurita, S.; Mott, J.L.; Almada, L.L.; Bronk, S.F.; Werneburg, N.W.; Sun, S.-Y.; Roberts, L.R.; Fernandez-Zapico, M.E.; Gores, G.J. GLI3-dependent repression of DR4 mediates hedgehog antagonism of TRAIL-induced apoptosis. Oncogene 2010, 29, 4848–4858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Jiang, J.; Shi, S.; Xie, H.; Zhou, L.; Zheng, S. Knockdown of miR-25 increases the sensitivity of liver cancer stem cells to TRAIL-induced apoptosis via PTEN/PI3K/Akt/Bad signaling pathway. Int. J. Oncol. 2016, 49, 2600–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panner, A.; Crane, C.A.; Weng, C.; Feletti, A.; Parsa, A.T.; Pieper, R.O. A novel PTEN-dependent link to ubiquitination controls FLIPS stability and TRAIL sensitivity in glioblastoma multiforme. Cancer Res. 2009, 69, 7911–7916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Sharkawi, F.Z.; Ewais, S.M.; Fahmy, R.H.; Rashed, L.A. PTEN and TRAIL genes loaded zein nanoparticles as potential therapy for hepatocellular carcinoma. J. Drug Target. 2017, 25, 513–522. [Google Scholar] [CrossRef]
- Wang, H.; Xu, C.; Kong, X.; Li, X.; Kong, X.; Wang, Y.; Ding, X.; Yang, Q. Trail resistance induces epithelial-mesenchymal transition and enhances invasiveness by suppressing PTEN via miR-221 in breast cancer. PLoS ONE 2014, 9, e99067. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Shan, Z.; Hong, J.; Yang, L. MicroRNA-92a promotes epithelial-mesenchymal transition through activation of PTEN/PI3K/AKT signaling pathway in non-small cell lung cancer metastasis. Int. J. Oncol. 2017, 51, 235–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, F.; Jiang, W.; Zhou, L.; Chen, Z. Circulating exosomal miR-17-5p and miR-92a-3p predict pathologic stage and grade of colorectal cancer. Transl. Oncol. 2018, 11, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, G.; Wang, H.; Liu, Z. MicroRNA-129-5p suppresses proliferation, migration and invasion of retinoblastoma cells through PI3K/AKT signaling pathway by targeting PAX6. Pathol. Res. Pr. 2019, 215, 152641. [Google Scholar] [CrossRef]
- Zi, Y.; Zhang, Y.; Wu, Y.; Zhang, L.; Yang, R.; Huang, Y. Downregulation of microRNA-25-3p inhibits the proliferation and promotes the apoptosis of multiple myeloma cells via targeting the PTEN/PI3K/AKT signaling pathway. Int. J. Mol. Med. 2020, 47, 1. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Jeon, Y.-J.; Laganà, A.; Middleton, J.; Secchiero, P.; Garofalo, M.; Croce, C.M. MicroRNA-148a reduces tumorigenesis and increases TRAIL-induced apoptosis in NSCLC. Proc. Natl. Acad. Sci. USA 2015, 112, 8650–8655. [Google Scholar] [CrossRef] [Green Version]
- Farooqi, A.A.; Gadaleta, C.D.; Ranieri, G.; Fayyaz, S.; Marech, I. New frontiers in promoting TRAIL-mediated cell death: Focus on natural sensitizers, miRNAs, and nanotechnological advancements. Cell Biophys. 2015, 74, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Chen, Q.; Zhong, Z.; Xu, C.; Wu, C.; Liu, B.; Chen, Y. Down-regulation of MiR-30c promotes the invasion of non-small cell lung cancer by targeting MTA1. Cell. Physiol. Biochem. 2013, 32, 476–485. [Google Scholar] [CrossRef]
- Xu, C.; Zeng, Q.; Xu, W.; Jiao, L.; Chen, Y.; Zhang, Z.; Wu, C.; Jin, T.; Pan, A.; Wei, R.; et al. miRNA-100 inhibits human bladder urothelial carcinogenesis by directly targeting mTOR. Mol. Cancer Ther. 2013, 12, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Shin, E.A.; Sohn, E.J.; Won, G.; Choi, J.-U.; Jeong, M.; Kim, B.; Kim, M.-J.; Kim, S.-H. Upregulation of microRNA135a-3p and death receptor 5 plays a critical role in Tanshinone I sensitized prostate cancer cells to TRAIL induced apoptosis. Oncotarget 2014, 5, 5624–5636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Schwabe, R.; Qian, T.; Lemasters., J.; Brenner, D. TRAIL-mediated apoptosis requires NF-kappaB inhibition and the mitochondrial permeability transition in human hepatoma cells. Hepatology 2002, 36, 1498–1508. [Google Scholar]
- Nguyen, P.N.N.; Huang, C.-J.; Sugii, S.; Cheong, S.K.; Choo, K.B. Selective activation of miRNAs of the primate-specific chromosome 19 miRNA cluster (C19MC) in cancer and stem cells and possible contribution to regulation of apoptosis. J. Biomed. Sci. 2017, 24, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ralff, M.D.; El-Deiry, W.S. TRAIL pathway targeting therapeutics. Expert Rev. Precis. Med. Drug Dev. 2018, 3, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Shi, S.; Gong, T.; Zhang, Z.; Sun, X. Cancer stem cells: Therapeutic implications and perspectives in cancer therapy. Acta Pharm. Sin. B 2013, 3, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Fakiruddin, K.S.; Ghazalli, N.; Lim, M.N.; Zakaria, Z.; Abdullah, S. Mesenchymal stem cell expressing TRAIL as targeted therapy against sensitised tumour. Int. J. Mol. Sci. 2018, 19, 2188. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Liu, B.; Chen, D.; Setroikromo, R.; Haisma, H.J.; Quax, W.J. Histone deacetylase inhibitors sensitize TRAIL-induced apoptosis in colon cancer cells. Cancers 2019, 11, 645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, R.K.; Kurzrock, R.; Shankar, S. MS-275 Sensitizes TRAIL-resistant breast cancer cells, inhibits angiogenesis and metastasis, and reverses epithelial-mesenchymal transition in vivo. Mol. Cancer Ther. 2010, 9, 3254–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Feng, X.; Han, H.; Guo, S.; Wang, G. Synergistic effects of combined treatment with histone deacetylase inhibitor suberoylanilide hydroxamic acid and TRAIL on human breast cancer cells. Sci. Rep. 2016, 6, 28004. [Google Scholar] [CrossRef] [Green Version]
- Stöhr, D.; Schmid, J.O.; Beigl, T.B.; Mack, A.; Maichl, D.S.; Cao, K.; Budai, B.; Fullstone, G.; Kontermann, R.E.; Mürdter, T.E.; et al. Stress-induced TRAILR2 expression overcomes TRAIL resistance in cancer cell spheroids. Cell Death Differ. 2020, 27, 3037–3052. [Google Scholar] [CrossRef]
- Vinogradov, S.; Wei, X. Cancer stem cells and drug resistance: The potential of nanomedicine. Nanomedicine 2012, 7, 597–615. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; YunXu, Z.; Hou, P.; Jiang, C.; Liu, L.; Tang, M.; Yang, X.; Zhang, Y.; Liu, Y. Icaritin sensitizes human glioblastoma cells to TRAIL-induced apoptosis. Cell Biophys. 2015, 72, 533–542. [Google Scholar] [CrossRef]
- Zhang, Z.; Patel, S.B.; King, M.R. Micelle-in-liposomes for sustained delivery of anticancer agents that promote potent TRAIL-induced cancer cell apoptosis. Molecules 2020, 26, 157. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.-G.; Kim, E.H.; Kim, J.Y.; Kim, S.U.; Kwon, T.K.; Yoon, A.-R.; Yun, C.-O.; Choi, K.S. Silibinin sensitizes human glioma cells to TRAIL-mediated apoptosis via DR5 up-regulation and down-regulation of c-FLIP and survivin. Cancer Res. 2007, 67, 8274–8284. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Liu, M.; Tang, W.; Li, Y.; Lian, J.; Lawrence, T.S.; Xu, L. A Smac-mimetic sensitizes prostate cancer cells to TRAIL-induced apoptosis via modulating both IAPs and NF-kappaB. BMC Cancer 2009, 9, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.W.; Kim, S.J.; Park, S.H.; Yang, H.G.; Kang, M.C.; Choi, Y.W.; Kim, S.M.; Jeun, S.-S.; Sung, Y.C. Complete regression of metastatic renal cell carcinoma by multiple injections of engineered mesenchymal stem cells expressing dodecameric TRAIL and HSV-TK. Clin. Cancer Res. 2013, 19, 415–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinnah, K.; Park, S.-Y. Duloxetine enhances TRAIL-mediated apoptosis via AMPK-mediated inhibition of autophagy flux in lung cancer cells. Anticancer Res. 2019, 39, 6621–6633. [Google Scholar] [CrossRef]
- Onoe-Takahashi, A.; Suzuki-Karasaki, M.; Suzuki-Karasaki, M.; Ochiai, T.; Suzuki-Karasaki, Y. Autophagy inhibitors regulate TRAIL sensitivity in human malignant cells by targeting the mitochondrial network and calcium dynamics. Int. J. Oncol. 2019, 54, 1734–1746. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Takimoto, R.; Dicker, D.; Chen, Y.; Gazitt, Y.; El-Deiry, W. Enhanced TRAIL sensitivity by p53 overexpression in human cancer but not normal cell lines. Int. J. Oncol. 2001, 18, 241–247. [Google Scholar] [CrossRef]
- Hu, B.; Zhu, H.; Qiu, S.; Su, Y.; Ling, W.; Xiao, W.; Qi, Y. Enhanced TRAIL sensitivity by E1A expression in human cancer and normal cell lines: Inhibition by adenovirus E1B19K and E3 proteins. Biochem. Biophys. Res. Commun. 2004, 325, 1153–1162. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Liu, Y.; Huang, W.; Cheng, D. MiR-760 enhances TRAIL sensitivity in non-small cell lung cancer via targeting the protein FOXA1. Biomed. Pharmacother. 2018, 99, 523–529. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Treatment | Cancer | Effect | Reference |
|---|---|---|---|
| Goniothalamin plus TRAIL | Colorectal cancer | Enhance cytotoxicity and apoptosis | [35] |
| Icaritin plus TRAIL | Glioblastoma | Enhance apoptosis by c-FLIP downregulation and inhibition of NF-κB activity | [36] |
| Micelle-in-liposomes with piperlongumine plus TRAIL | Prostate cancer | Increase sensitization to TRAIL apoptosis in cancer cells | [37] |
| Silibinin plus TRAIL | Glioma | Enhance apoptosis by upregulation of DR5 and downregulation of cFLIP and survival | [38] |
| SH122 plus TRAIL | Prostate cancer | Enhanced TRAIL-induced apoptosis via D5R and the mitochondrial pathway | [39] |
| MSC/dTRAIL-TK gene therapy | Renal cell carcinoma | Enhance sensitization to TRAIL and increase apoptosis | [40] |
| Duloxetine plus TRAIL | Lung cancer | Enhance apoptosis of tumor cells through inhibition of autophagy | [41] |
| 3-Methyladenine and chloroquine plus TRAIL | Malignant melanoma and osteosarcoma | Enhance pro-apoptotic mitochondrial pathway of tumor cells through inhibition of autophagy | [42] |
| Adenovirus-p53 plus TRAIL | Ovarian and nasopharyngeal squamous cancer | Overexpression of DR5 receptor in cancer cells to increase apoptosis by TRAIL | [43] |
| Adenovirus E1A plus adenovirus-hTRAIL | Hepatic cancer | Enhance apoptosis by upregulation of TRAIL receptors | [44] |
| MiR-760 plus TRAIL | Non-small-cell lung cancer | Enhance apoptosis by targeting FOXA1 | [45] |
| Recombinant TRAIL | Disease | Phase | Clinical Trial |
|---|---|---|---|
| Recombinant human Apo-2 ligand for injection | Non-small-cell lung cancer (NSCLC) stage IV | 3 | NCT03083743 |
| Recombinant human TRAIL–trimer fusion protein (SCB-313) | Malignant pleural effusions | 1 | NCT038669697 |
| Peritoneal malignancies | 1 | NCT03443674 | |
| Peritoneal carcinomatosis | 1 | NCT04047771 | |
| rhApo2L/TRAIL (AMG 951) with chemotherapy bevacizumab | Non-small-cell lung cancer (NSCLC) | 2 | NCT00508625 |
| Dulanermin plus rituximab | Non-Hodgkin’s lymphoma | 1, 2 | NCT00400764 |
| Dulanermin plus Camptosar® /Erbitux® or FOLFIRI | Metastatic colorectal cancer | 1 | NCT00671372 |
| Dulanermin with FOLFOX and bevacizumab | Metastatic colorectal cancer | 1 | NCT00873756 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quiroz-Reyes, A.G.; Delgado-Gonzalez, P.; Islas, J.F.; Gallegos, J.L.D.; Martínez Garza, J.H.; Garza-Treviño, E.N. Behind the Adaptive and Resistance Mechanisms of Cancer Stem Cells to TRAIL. Pharmaceutics 2021, 13, 1062. https://doi.org/10.3390/pharmaceutics13071062
Quiroz-Reyes AG, Delgado-Gonzalez P, Islas JF, Gallegos JLD, Martínez Garza JH, Garza-Treviño EN. Behind the Adaptive and Resistance Mechanisms of Cancer Stem Cells to TRAIL. Pharmaceutics. 2021; 13(7):1062. https://doi.org/10.3390/pharmaceutics13071062
Chicago/Turabian StyleQuiroz-Reyes, Adriana G., Paulina Delgado-Gonzalez, Jose Francisco Islas, Juan Luis Delgado Gallegos, Javier Humberto Martínez Garza, and Elsa N. Garza-Treviño. 2021. "Behind the Adaptive and Resistance Mechanisms of Cancer Stem Cells to TRAIL" Pharmaceutics 13, no. 7: 1062. https://doi.org/10.3390/pharmaceutics13071062
APA StyleQuiroz-Reyes, A. G., Delgado-Gonzalez, P., Islas, J. F., Gallegos, J. L. D., Martínez Garza, J. H., & Garza-Treviño, E. N. (2021). Behind the Adaptive and Resistance Mechanisms of Cancer Stem Cells to TRAIL. Pharmaceutics, 13(7), 1062. https://doi.org/10.3390/pharmaceutics13071062