
Mimicking Pathogens to Augment the Potency of Liposomal Cancer Vaccines

 and
and
Abstract
:1. Introduction
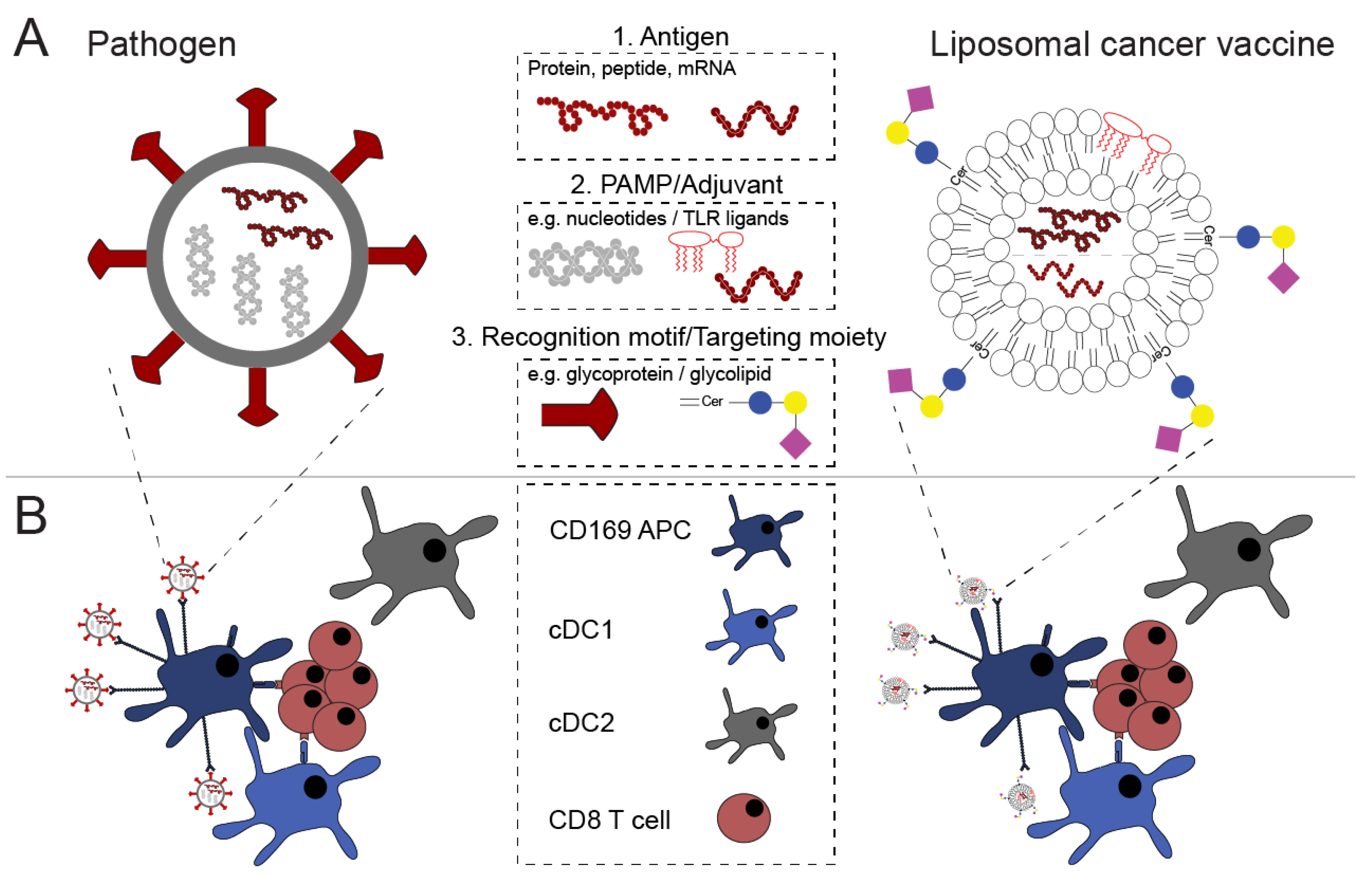
2. Antigen and Adjuvant Are the Basic Ingredients of Vaccines
2.1. Classification of Tumor Antigens
2.2. Vaccine Antigen Can Be Incorporated in Various Forms
2.3. Cancer Vaccination Aims to Induce CD8+ T Cell Responses via Cross-Presenting DCs
2.4. Vaccine Adjuvants Are Essential to Induce an Immune Response
3. Liposomes as Delivery System
3.1. Liposomal Antigen Accumulation in Lymph Nodes

{kind=link}
| Size | Charge | Administration Route | Effect | References |
|---|---|---|---|---|
| ~100 nm | Cationic | i.m. | Depot formation | [46] |
| ~100 nm | Neutral and anionic | i.m. | Draining via the regional lymphatics to the LN | [47] |
| ‘Large’ 500–900 nm | Cationic | s.c. | Retention at the site of injection | [47,58] |
| ‘Small’ 100–140 nm | Cationic | s.c. | Enhanced draining to the LN as compared to larger counterparts | [47,58] |
| ~250 nm | Cationic | s.c. | Liposomal surface decoration with PEG enhanced draining to the LN | [52] |
| ~300 nm | Neutral or anionic | i.v. | Antigen uptake in the spleen | [20] |
| ~160 nm | Cationic | i.v. | Antigen sequestration in the lung | [60] |
3.2. Liposomal Antigen Uptake in the Spleen
3.3. The Route of Vaccine Administration Dictates Subsequent Responses
3.4. Antigen Encapsulation Augments Antigen Immunogenicity
| Administration Route | Antigen | Adjuvant | Targeting (Moiety) | Targeted or Activated Cell | Effect | References |
|---|---|---|---|---|---|---|
| i.p. | OVA protein | CpG | Passive | N/A | Enhanced responses compared to soluble vaccine components | [87] |
| i.p. | OVA protein | CpG or Poly IC | Passive | DC and macrophage | Enhanced responses upon combination of TLR ligands | [88] |
| i.d. | OVA protein | CpG | Passive | N/A | Enhanced responses compared to soluble vaccine components | [89] |
| Intratumoral | N/A | aCD40 and CpG | Antibody mediated | Intratumoral macrophage and DC | Enhanced responses compared to soluble vaccine components | [90] |
| Inhalation | N/A | Cyclic dinucleotides | PS | DC and macrophage | Synergized anti-tumor responses when combined with irradiation | [91] |
| Hypodermic (not specified) | Melanoma peptides | CpG | Mannose | DC | Enhanced responses compared to soluble vaccine components | [92] |
| s.c. | OVA peptide | Poly IC and Pam3CSK4 | Passive | N/A | Enhanced responses compared to soluble peptide | [78] |
| s.c. | OVA protein | Cyclic dinucleotides | Passive | DC and macrophage | Enhanced responses compared to soluble vaccine adjuvant | [93] |
| s.c. | Human papilloma virus peptide | CpG | Mannose | DC | Enhanced responses compared to soluble vaccine components | [82] |
| s.c. | Melanoma antigen mRNA | N/A | mannose-mimicking head group | DC | Protection from tumor challenge | [94] |
| s.c. | ErbB2 protein | Pam(3)CAG, Pam(2)CAG and Pam(2)CGD | Mannose | DC | Enhanced responses upon targeting | [95] |
| i.v., s.c., i.m. | OVA-encoding mRNA | N/A | Passive | DC and macrophage | Enhanced responses upon i.v. immunization | [70] |
| i.v., s.c. | OVA peptide | Soluble poly IC and aCD40 | GM3 | CD169-expressing macrophages | Enhanced immune responses upon i.v. immunization, targeting and encapsulation of antigen | [54] |
| i.v. | Human papilloma virus peptide | CpG | Passive | cDC and pDC | Enhanced responses compared to soluble vaccine components | [96] |
| i.v. | OVA protein | Soluble poly IC and aCD40 | GM3 | CD169-expressing macrophages | Enhanced immune responses upon targeting and encapsulation of antigen | [83] |
| i.v. | OVA protein | α-Galactosylceramide | GM3 | CD169-expressing macrophages | Enhanced CD8 T cell responses upon targeting | [97] |
3.5. Adjuvant Encapsulation Augments Its Potency
4. Enhanced Delivery of Antigen to APCs Is a Third Component to Improve Cancer Vaccines
4.1. Different APCs for T Cell Priming
4.2. Active Targeting Leads to Uptake by APCs of Choice
4.3. Liposomal Vaccine Targeting to CD169-Expressing Cells in Mice and Men
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Tran, S.; DeGiovanni, P.; Piel, B.; Rai, P. Cancer nanomedicine: A review of recent success in drug delivery. Clin. Transl. Med. 2017, 6, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, J.S.O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Melief, C.J.; van Hall, T.; Arens, R.; Ossendorp, F.; Van Der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef] [PubMed]
- van der Burg, S.H.; Arens, R.; Ossendorp, F.; van Hall, T.; Melief, C.J.M. Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16, 219–233. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nat. Cell Biol. 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultan, H.; Kumai, T.; Nagato, T.; Wu, J.; Salazar, A.M.; Celis, E. The route of administration dictates the immunogenicity of pep-tide-based cancer vaccines in mice. Cancer Immunol. Immunother. 2019, 68, 455–466. [Google Scholar] [CrossRef]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 1–10. [Google Scholar] [CrossRef]
- Sahin, U.; Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Kreiter, S.; Castle, J.C.; Türeci, Ö.; Sahin, U. Targeting the tumor mutanome for personalized vaccination therapy. OncoImmunology 2012, 1, 768–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Hall, T.; van der Burg, S.H. Mechanisms of peptide vaccination in mouse models: Tolerance, immunity, and hyperreactivity. Adv. Immunol. 2012, 114, 51–76. [Google Scholar] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Grabbe, S.; Haas, H.; Diken, M.; Kranz, L.M.; Langguth, P.; Sahin, U. Translating nanoparticulate-personalized cancer vaccines into clinical applications: Case study with RNA-lipoplexes for the treatment of melanoma. Nanomedicine 2016, 11, 2723–2734. [Google Scholar] [CrossRef] [PubMed]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antivi-ral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Salomon, N.; Vascotto, F.; Selmi, A.; Vormehr, M.; Quinkhardt, J.; Bukur, T.; Schrörs, B.; Löewer, M.; Diken, M.; Türeci, Ö.; et al. A liposomal RNA vaccine inducing neoanti-gen-specific CD4(+) T cells augments the antitumor activity of local radiotherapy in mice. Oncoimmunology 2020, 9, 1771925. [Google Scholar] [CrossRef] [PubMed]
- Selmi, A.; Vascotto, F.; Kautz-Neu, K.; Türeci, Ö.; Sahin, U.; Von Stebut, E.; Diken, M.; Kreiter, S. Uptake of synthetic naked RNA by skin-resident dendritic cells via macropinocytosis allows antigen expression and induction of T-cell responses in mice. Cancer Immunol. Immunother. 2016, 65, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Selmi, A.; Diken, M.; Koslowski, M.; Britten, C.M.; Huber, C.; Türeci, Ö.; Sahin, U. Intranodal Vaccination with Naked Antigen-Encoding RNA Elicits Potent Prophylactic and Therapeutic Antitumoral Immunity. Cancer Res. 2010, 70, 9031–9040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böttcher, J.P.; e Sousa, C.R. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joffre, O.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Dudziak, D.; Kamphorst, A.O.; Heidkamp, G.F.; Buchholz, V.R.; Trumpfheller, C.; Yamazaki, S.; Cheong, C.; Liu, K.; Lee, H.-W.; Park, C.G.; et al. Differential Antigen Processing by Dendritic Cell Subsets in Vivo. Science 2007, 315, 107–111. [Google Scholar] [CrossRef]
- Grotzke, J.E.; Sengupta, D.; Lu, Q.; Cresswell, P. The ongoing saga of the mechanism(s) of MHC class I-restricted cross-presentation. Curr. Opin. Immunol. 2017, 46, 89–96. [Google Scholar] [CrossRef]
- Colbert, J.D.; Cruz, F.M.; Rock, K.L. Cross-presentation of exogenous antigens on MHC I molecules. Curr. Opin. Immunol. 2020, 64, 1–8. [Google Scholar] [CrossRef]
- Canton, J.; Blees, H.; Henry, C.M.; Buck, M.D.; Schulz, O.; Rogers, N.C.; Childs, E.; Zelenay, S.; Rhys, H.; Domart, M.C.; et al. The receptor DNGR-1 signals for phagosomal rupture to promote cross-presentation of dead-cell-associated antigens. Nat. Immunol. 2021, 22, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Tang-Huau, T.-L.; Gueguen, P.; Goudot, C.; Durand, M.; Bohec, M.; Baulande, S.; Pasquier, B.; Amigorena, S.; Segura, E. Human in vivo-generated mono-cytederived dendritic cells and macrophages cross-present antigens through a vacuolar pathway. Nat. Commun. 2018, 9, 2570. [Google Scholar] [CrossRef]
- Burgdorf, S.; Kautz, A.; Böhnert, V.; Knolle, P.A.; Kurts, C. Distinct Pathways of Antigen Uptake and Intracellular Routing in CD4 and CD8 T Cell Activation. Science 2007, 316, 612–616. [Google Scholar] [CrossRef]
- van Montfoort, N.; Camps, M.G.; Khan, S.; Filippov, D.V.; Weterings, J.J.; Griffith, J.M.; Geuze, H.J.; van Hall, T.; Verbeek, J.S.; Melief, C.J.; et al. Antigen storage compartments in ma-ture dendritic cells facilitate prolonged cytotoxic T lymphocyte cross-priming capacity. Proc. Natl. Acad. Sci. USA 2009, 106, 6730–6735. [Google Scholar] [CrossRef] [Green Version]
- Gardner, A.; Ruffell, B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Schlitzer, A.; McGovern, N.; Ginhoux, F. Dendritic cells and monocyte-derived cells: Two complementary and integrated functional systems. Semin. Cell Dev. Biol. 2015, 41, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sun, L.; Chen, Z. Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.-R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [Green Version]
- Goubau, D.; Schlee, M.; Deddouche, S.; Pruijssers, A.J.; Zillinger, T.; Goldeck, M.; Schuberth, C.; Van Der Veen, A.G.; Fujimura, T.; Rehwinkel, J.; et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nat. Cell Biol. 2014, 514, 372–375. [Google Scholar] [CrossRef] [Green Version]
- Nair-Gupta, P.; Baccarini, A.; Tung, N.; Seyffer, F.; Florey, O.; Huang, Y.; Banerjee, M.; Overholtzer, M.; Roche, P.; Tampé, R.; et al. TLR Signals Induce Phagosomal MHC-I Delivery from the Endosomal Recycling Compartment to Allow Cross-Presentation. Cell 2014, 158, 506–521. [Google Scholar] [CrossRef] [Green Version]
- Weimershaus, M.; Mauvais, F.-X.; Saveanu, L.; Adiko, C.; Babdor, J.; Abramova, A.; Montealegre, S.; Lawand, M.; Evnouchidou, I.; Huber, K.J.; et al. Innate Immune Signals Induce Antero-grade Endosome Transport Promoting MHC Class I Cross-Presentation. Cell Rep. 2018, 24, 3568–3581. [Google Scholar] [CrossRef] [Green Version]
- Pauwels, A.-M.; Härtlova, A.; Peltier, J.; Driege, Y.; Baudelet, G.; Brodin, P.; Beyaert, R.; Hoffmann, E. Spatiotemporal Changes of the Phagosomal Pro-teome in Dendritic Cells in Response to LPS Stimulation. Mol. Cell Proteom. 2019, 18, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Burgdorf, S.; Schölz, C.; Kautz, A.; Tampé, R.; Kurts, C. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat. Immunol. 2008, 9, 558–566. [Google Scholar] [CrossRef]
- Anderluzzi, G.; Schmidt, S.T.; Cunliffe, R.; Woods, S.; Roberts, C.W.; Veggi, D.; Ferlenghi, I.; O’Hagan, D.T.; Baudner, B.C.; Perrie, Y. Rational design of adjuvants for subunit vac-cines: The format of cationic adjuvants affects the induction of antigen-specific antibody responses. J. Control. Release 2021, 330, 933–944. [Google Scholar] [CrossRef]
- Khadke, S.; Roces, C.B.; Cameron, A.; Devitt, A.; Perrie, Y. Formulation and manufacturing of lymphatic targeting liposomes us-ing microfluidics. J. Control. Release 2019, 307, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Henriksen-Lacey, M.; Christensen, D.; Bramwell, V.W.; Lindenstrøm, T.; Agger, E.M.; Andersen, P.; Perrie, Y. Liposomal cationic charge and antigen adsorption are important properties for the efficient deposition of antigen at the injection site and ability of the vaccine to induce a CMI response. J. Control. Release 2010, 145, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.; Henriksen-Lacey, M.; Kamath, A.T.; Lindenstrøm, T.; Korsholm, K.S.; Christensen, J.P.; Rochat, A.F.; Lambert, P.H.; Andersen, P.; Siegrist, C.A.; et al. A cationic vaccine ad-juvant based on a saturated quaternary ammonium lipid have different in vivo distribution kinetics and display a distinct CD4 T cell-inducing capacity compared to its unsaturated analog. J. Control. Release 2012, 160, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Henriksen-Lacey, M.; Wilkhu, J.; Devitt, A.; Christensen, D.; Perrie, Y. Effect of Incorporating Cholesterol into DDA:TDB Liposomal Adjuvants on Bilayer Properties, Biodistribution, and Immune Responses. Mol. Pharm. 2014, 11, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, R.; Bramwell, V.W.; Kirby, D.J.; Perrie, Y. Pegylation of DDA:TDB liposomal adjuvants reduces the vaccine depot effect and alters the Th1/Th2 immune responses. J. Control. Release 2012, 158, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Ma, Y.; Wang, C.; Hai, L.; Yan, C.; Zhang, Y.; Liu, F.; Cai, L. PEGylated cationic liposomes robustly augment vaccine-induced immune responses: Role of lymphatic trafficking and biodistribution. J. Control. Release 2012, 159, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.T.; Olsen, C.L.; Franzyk, H.; Wørzner, K.; Korsholm, K.S.; Rades, T.; Andersen, P.; Foget, C.; Christensen, D. Comparison of two different PEGylation strate-gies for the liposomal adjuvant CAF09: Towards induction of CTL responses upon subcutaneous vaccine administration. Eur. J. Pharm. Biopharm. 2019, 140, 29–39. [Google Scholar] [CrossRef]
- Nijen Twilhaar, M.K.; Czentner, L.; Grabowska, J.; Affandi, A.J.; Lau, C.Y.J.; Olesek, K.; Kalay, K.; van Nostrum, C.F.; van Kooyk, Y.; Storm, G.; et al. Optimization of Liposomes for Antigen Targeting to Splenic CD169(+) Macrophages. Pharmaceutics 2020, 12, 1138. [Google Scholar] [CrossRef] [PubMed]
- Edgar, L.J.; Kawasaki, N.; Nycholat, C.M.; Paulson, J.C. Targeted Delivery of Antigen to Activated CD169+ Macrophages Induces Bias for Expansion of CD8+ T Cells. Cell Chem. Biol. 2019, 26, 131–136.e4. [Google Scholar] [CrossRef]
- Mohamed, M.; Abu Lila, A.S.; Shimizu, T.; Alaaeldin, E.; Hussein, A.; Sarhan, H.A.; Szebeni, J.; Ishida, T. PEGylated liposomes: Immunological responses. Sci. Technol. Adv. Mater. 2019, 20, 710–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oussoren, C.; Storm, G. Liposomes to target the lymphatics by subcutaneous administration. Adv. Drug Deliv. Rev. 2001, 50, 143–156. [Google Scholar] [CrossRef]
- Carstens, M.G.; Camps, M.G.; Henriksen-Lacey, M.; Franken, K.; Ottenhoff, T.H.; Perrie, Y.; Bouwstra, J.A.; Ossendorp, F.; Jiskoot, W. Effect of vesicle size on tissue localization and immunogenicity of liposomal DNA vaccines. Vaccine 2011, 29, 4761–4770. [Google Scholar] [CrossRef]
- Zhang, R.; Billingsley, M.; Mitchell, M.J. Biomaterials for vaccine-based cancer immunotherapy. J. Control. Release 2018, 292, 256–276. [Google Scholar] [CrossRef]
- Verbeke, R.; Lentacker, I.; Wayteck, L.; Breckpot, K.; Van Bockstal, M.; Descamps, B.; Vanhove, C.; De Smedt, S.C.; Dewitte, H. Co-delivery of nucleoside-modified mRNA and TLR agonists for cancer immunotherapy: Restoring the immunogenicity of immunosilent mRNA. J. Control. Release 2017, 266, 287–300. [Google Scholar] [CrossRef]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Hunter, A.C.; Andresen, T.L. Factors Controlling Nanoparticle Pharmacokinetics: An Integrated Analysis and Perspective. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 481–503. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Rosigkeit, S.; Meng, M.; Grunwitz, C.; Gomes, P.; Kreft, A.; Hayduk, N.; Heck, R.; Pickert, G.; Ziegler, K.; Abassi, Y.; et al. Monitoring Translation Activity of mRNA-Loaded Nanoparticles in Mice. Mol. Pharm. 2018, 15, 3909–3919. [Google Scholar] [CrossRef] [PubMed]
- Perrie, Y.; Crofts, F.; Devitt, A.; Griffiths, H.R.; Kastner, E.; Nadella, V. Designing liposomal adjuvants for the next generation of vaccines. Adv. Drug Deliv. Rev. 2016, 99, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, Y.; Nakamura, M.; Takeuchi, N.; Tamaki, K.; Shimizu, S.; Yoshiike, Y.; Taguchi, M.; Ohno, H.; Ozaki, K.-I.; Onishi, H. Effect of cationic lipid in cationic liposomes on siRNA delivery into the lung by intravenous injection of cationic lipoplex. J. Drug Target. 2019, 27, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Heuts, J.; Jiskoot, W.; Ossendorp, F.; van der Maaden, K. Cationic Nanoparticle-Based Cancer Vaccines. Pharmaceutics 2021, 13, 596. [Google Scholar] [CrossRef] [PubMed]
- Varypataki, E.M.; Benne, N.; Bouwstra, J.; Jiskoot, W.; Ossendorp, F. Efficient Eradication of Established Tumors in Mice with Cationic Liposome-Based Synthetic Long-Peptide Vaccines. Cancer Immunol. Res. 2017, 5, 222–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Beuckelaer, A.; Pollard, C.; Van Lint, S.; Roose, K.; Van Hoecke, L.; Naessens, T.; Udhayakumar, V.K.; Smet, M.; Sanders, N.; Lienenklaus, S.; et al. Type I Interferons Interfere with the Ca-pacity of mRNA Lipoplex Vaccines to Elicit Cytolytic T Cell Responses. Mol. Ther. 2016, 24, 2012–2020. [Google Scholar] [CrossRef] [Green Version]
- Broos, K.; Van der Jeught, K.; Puttemans, J.; Goyvaerts, C.; Heirman, C.; Dewitte, H.; Verbeke, R.; Lentacker, I.; Thielemans, K.; Breckpot, K. Particle-mediated Intravenous Delivery of Antigen mRNA Results in Strong Antigen-specific T-cell Responses Despite the Induction of Type I Interferon. Mol. Ther. Nucleic Acids 2016, 5, e326. [Google Scholar] [CrossRef] [Green Version]
- Sayour, E.J.; De Leon, G.; Pham, C.; Grippin, A.; Kemeny, H.; Chua, J.; Huang, J.; Sampson, J.H.; Sanchez-Perez, L.; Flores, C.; et al. Systemic activation of antigen-presenting cells via RNA-loaded nanoparticles. OncoImmunology 2016, 6, e1256527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.-R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011, 208, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Van Hoecke, L.; Roose, K.; Ballegeer, M.; Zhong, Z.; Sanders, N.N.; De Koker, S.; Saelens, X.; van Lint, S. The Opposing Effect of Type I IFN on the T Cell Response by Non-modified mRNA-Lipoplex Vaccines Is Determined by the Route of Administration. Mol. Ther. Nucleic Acids 2020, 22, 373–381. [Google Scholar] [CrossRef]
- Limmer, A.; Ohl, J.; Kurts, C.; Ljunggren, H.-G.; Reiss, Y.; Groettrup, M.; Momburg, F.; Arnold, B.; Knolle, P.A. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat. Med. 2000, 6, 1348–1354. [Google Scholar] [CrossRef] [Green Version]
- Hirosue, S.; Vokali, E.; Raghavan, V.R.; Rincon-Restrepo, M.; Lund, A.W.; Corthésy-Henrioud, P.; Capotosti, F.; Halin Winter, C.; Hugues, S.; Swartz, M.A. Steady-state antigen scav-enging, cross-presentation, and CD8+ T cell priming: A new role for lymphatic endothelial cells. J. Immunol. 2014, 192, 5002–5011. [Google Scholar] [CrossRef] [Green Version]
- Henriksen-Lacey, M.; Bramwell, V.W.; Christensen, D.; Agger, E.-M.; Andersen, P.; Perrie, Y. Liposomes based on dimethyldiocta-decylammonium promote a depot effect and enhance immunogenicity of soluble antigen. J. Control. Release 2010, 142, 180–186. [Google Scholar] [CrossRef]
- Varypataki, E.M.; Silva, A.L.; Barnier-Quer, C.; Collin, N.; Ossendorp, F.; Jiskoot, W. Synthetic long peptide-based vaccine formula-tions for induction of cell mediated immunity: A comparative study of cationic liposomes and PLGA nanoparticles. J. Control. Release 2016, 226, 98–106. [Google Scholar] [CrossRef]
- Heuts, J.; Varypataki, E.M.; van der Maaden, K.; Romeijn, S.; Drijfhout, J.W.; van Scheltinga, A.T.; Ossendorp, F.; Jiskoot, W. Cationic Liposomes: A Flexible Vaccine Delivery System for Physicochemically Diverse Antigenic Peptides. Pharm. Res. 2018, 35, 207. [Google Scholar] [CrossRef] [Green Version]
- Cruz, L.J.; Rueda, F.; Cordobilla, B.; Simón, L.; Hosta, L.; Albericio, F.; Domingo, J.C.; Rigau, L.H. Targeting Nanosystems to Human DCs via Fc Receptor as an Effective Strategy to Deliver Antigen for Immunotherapy. Mol. Pharm. 2010, 8, 104–116. [Google Scholar] [CrossRef]
- Bae, J.; Parayath, N.; Ma, W.; Amiji, M.; Munshi, N.; Anderson, K.C. BCMA peptide-engineered nanoparticles enhance induction and function of antigen-specific CD8(+) cytotoxic T lymphocytes against multiple myeloma: Clinical applications. Leukemia 2020, 34, 210–223. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, H.; Yang, Y.; Jia, W.; Su, T.; Che, Y.; Feng, Y.; Yuan, X.; Wang, X. Mannose-Modified Liposome Co-Delivery of Human Papillomavirus Type 16 E7 Peptide and CpG Oligodeoxynucleotide Adjuvant Enhances Antitumor Activity Against Established Large TC-1 Grafted Tumors in Mice. Int. J. Nanomed. 2020, 15, 9571–9586. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, J.; Affandi, A.; van Dinther, D.; Twilhaar, M.N.; Olesek, K.; Hoogterp, L.; Ambrosini, M.; Heijnen, D.; Klaase, L.; Hidalgo, A.; et al. Liposome induction of CD8+ T cell responses depends on CD169+ macrophages and Batf3-dependent dendritic cells and is enhanced by GM3 inclusion. J. Control. Release 2021, 331, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Aarntzen, E.H.J.G.; De Vries, I.J.M.; Lesterhuis, W.J.; Schuurhuis, D.; Jacobs, J.F.M.; Bol, K.; Schreibelt, G.; Mus, R.; De Wilt, J.H.W.; Haanen, J.B.A.G.; et al. Targeting CD4+ T-Helper Cells Improves the Induction of Antitumor Responses in Dendritic Cell–Based Vaccination. Cancer Res. 2013, 73, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, C.; Chernyak, N.; Dominguez, D.; Cole, L.; Zhang, B.; Mirkin, C.A. RNA-Based Immunostimulatory Liposomal Spherical Nucleic Acids as Potent TLR7/8 Modulators. Small 2018, 14, e1803284. [Google Scholar] [CrossRef]
- Persano, S.; Guevara, M.L.; Li, Z.; Mai, J.; Ferrari, M.; Pompa, P.P.; Shen, H. Lipopolyplex potentiates anti-tumor immunity of mRNA-based vaccination. Biomaterials 2017, 125, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erikçi, E.; Gursel, M.; Gürsel, İ. Differential immune activation following encapsulation of immunostimulatory CpG oligodeox-ynucleotide in nanoliposomes. Biomaterials 2011, 32, 1715–1723. [Google Scholar] [CrossRef]
- Bayyurt, B.; Tincer, G.; Almacioglu, K.; Alpdundar, E.; Gursel, M.; Gursel, I. Encapsulation of two different TLR ligands into lipo-somes confer protective immunity and prevent tumor development. Journal Control. Release 2017, 247, 134–144. [Google Scholar] [CrossRef]
- Bal, S.; Hortensius, S.; Ding, Z.; Jiskoot, W.; Bouwstra, J.A. Co-encapsulation of antigen and Toll-like receptor ligand in cationic liposomes affects the quality of the immune response in mice after intradermal vaccination. Vaccine 2011, 29, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Kwong, B.; Liu, H.; Irvine, D.J. Induction of potent anti-tumor responses while eliminating systemic side effects via liposomeanchored combinatorial immunotherapy. Biomaterials 2011, 32, 5134–5147. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Crowe, W.N.; Wang, L.; Lu, Y.; Petty, W.J.; Habib, A.A.; Zhao, D. An inhalable nanoparticulate STING agonist synergizes with radiotherapy to confer long-term control of lung metastases. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.; Duan, S.; Ye, F.; Hou, X.; Li, X.; Zhao, J.; Yu, X.; Hu, Z.; Tang, Z.; Mo, F.; et al. The enhanced antitumor-specific immune response with mannose- and CpG-ODN-coated liposomes delivering TRP2 peptide. Theranostics 2018, 8, 1723–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, M.C.; Crespo, M.P.; Abraham, W.; Moynihan, K.D.; Szeto, G.L.; Chen, S.H.; Melo, M.B.; Mueller, S.; Irvine, D.J. Nanoparticulate STING agonists are po-tent lymph node-targeted vaccine adjuvants. J. Clin. Investig. 2015, 125, 2532–2546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garu, A.; Moku, G.; Gulla, S.K.; Chaudhuri, A. Genetic Immunization With In Vivo Dendritic Cell-targeting Liposomal DNA Vaccine Carrier Induces Long-lasting Antitumor Immune Response. Mol. Ther. 2016, 24, 385–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomann, J.-S.; Heurtault, B.; Weidner, S.; Brayé, M.; Beyrath, J.; Fournel, S.; Schuber, F.; Frisch, B. Antitumor activity of liposomal ErbB2/HER2 epitope peptide-based vaccine constructs incorporating TLR agonists and mannose receptor targeting. Biomaterials 2011, 32, 4574–4583. [Google Scholar] [CrossRef]
- Shen, K.-Y.; Liu, H.-Y.; Yan, W.-L.; Wu, C.-C.; Lee, M.-H.; Leng, C.-H.; Liu, S.J. Liposomal TLR9 Agonist Combined with TLR2 Ago-nist-Fused Antigen Can Modulate Tumor Microenvironment through Dendritic Cells. Cancers 2020, 12, 810. [Google Scholar] [CrossRef] [Green Version]
- Grabowska, J.; Stolk, D.A.; Nijen Twilhaar, M.K.; Ambrosini, M.; Storm, G.; van der Vliet, H.J.; de Gruijl, T.D.; van Kooyk, Y.; den Haan, J.M.M. Liposomal Nanovaccine Con-taining α-Galactosylceramide and Ganglioside GM3 Stimulates Robust CD8(+) T Cell Responses via CD169(+) Macrophages and Cdc1. Vaccines 2021, 9, 56. [Google Scholar] [CrossRef]
- Boks, M.A.; Bruijns, S.C.M.; Ambrosini, M.; Kalay, H.; van Bloois, L.; Storm, G.; de Gruil, T.D.; van Kooyk, Y. In situ Delivery of Tumor Antigen- and Adju-vant-Loaded Liposomes Boosts Antigen-Specific T-Cell Responses by Human Dermal Dendritic Cells. J. Investig. Dermatol. 2015, 135, 2697–2704. [Google Scholar] [CrossRef] [Green Version]
- Boks, M.A.; Ambrosini, M.; Bruijns, S.C.; Kalay, H.; van Bloois, L.; Storm, G.; Garcia-Vallejo, J.J.; van Kooyk, Y. MPLA incorporation into DC-targeting glycoliposomes favours anti-tumour T cell responses. J. Control. Release 2015, 216, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.B.; Sivananthan, S.J.; Duthie, M.S.; Vergara, J.; Guderian, J.A.; Moon, E.; Coblentz, D.; Reed, S.G.; Carter, D. A nanoliposome delivery system to synergisti-cally trigger TLR4 AND TLR7. J. Nanobiotechnol. 2014, 12, 17. [Google Scholar] [CrossRef] [Green Version]
- Koshy, S.T.; Cheung, A.S.; Gu, L.; Graveline, A.R.; Mooney, D.J. Liposomal Delivery Enhances Immune Activation by STING Ago-nists for Cancer Immunotherapy. Adv. Biosyst. 2017, 1, 1600013. [Google Scholar] [CrossRef]
- Nakamura, T.; Miyabe, H.; Hyodo, M.; Sato, Y.; Hayakawa, Y.; Harashima, H. Liposomes loaded with a STING pathway ligand, cyclic di-GMP, enhance cancer immunotherapy against metastatic melanoma. J. Control. Release 2015, 216, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef]
- den Haan, J.M.; Lehar, S.M.; Bevan, M.J. CD8(+) but not CD8(−) dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 2000, 192, 1685–1696. [Google Scholar] [CrossRef]
- Savina, A.; Peres, A.; Cebrian, I.; Carmo, N.; Moita, C.; Hacohen, N.; Moita, L.F.; Amigorena, S. The small GTPase Rac2 controls phagosomal alkaliniza-tion and antigen crosspresentation selectively in CD8(+) dendritic cells. Immunity 2009, 30, 544–555. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.L.; Zhan, Y.; Proietto, A.I.; Prato, S.; Wu, L.; Heath, W.R.; Villadangos, J.A.; Lew, A.M. Selective suicide of cross-presenting CD8+ dendritic cells by cytochrome c injection shows functional heterogeneity within this subset. Proc. Natl. Acad. Sci. USA 2008, 105, 3029–3034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Diamond, M.S.; et al. Batf3 Deficiency Reveals a Critical Role for CD8α+ Dendritic Cells in Cytotoxic T Cell Immunity. Science 2008, 322, 1097–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nierkens, S.; Tel, J.; Janssen, E.; Adema, G.J. Antigen cross-presentation by dendritic cell subsets: One general or all sergeants? Trends Immunol. 2013, 34, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberkampf, M.; Guillerey, C.; Mouriès, J.; Rosenbaum, P.; Fayolle, C.; Bobard, A.; Savina, A.; Ogier-Denis, E.; Enninga, J.; Amigorena, S.; et al. Mitochondrial reactive oxygen species reg-ulate the induction of CD8(+) T cells by plasmacytoid dendritic cells. Nat. Commun. 2018, 9, 2241. [Google Scholar] [CrossRef] [Green Version]
- Mouries, J.; Moron, G.; Schlecht-Louf, G.; Escriou, N.; Dadaglio, G.; Leclerc, C. Plasmacytoid dendritic cells efficiently cross-prime naive T cells in vivo after TLR activation. Blood 2008, 112, 3713–3722. [Google Scholar] [CrossRef]
- Backer, R.; Van Leeuwen, F.; Kraal, G.; Haan, J.M.M.D. CD8– dendritic cells preferentially cross-presentSaccharomyces cerevisiae antigens. Eur. J. Immunol. 2008, 38, 370–380. [Google Scholar] [CrossRef]
- den Haan, J.M.M.; Bevan, M.J. Constitutive versus activation-dependent cross-presentation of immune complexes by CD8(+) and CD8(-) dendritic cells in vivo. J. Exp. Med. 2002, 196, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Bosteels, C.; Neyt, K.; Vanheerswynghels, M.; van Helden, M.J.; Sichien, D.; Debeuf, N.; De Prijck, S.; Bosteels, V.; Vandamme, N.; Martens, L.; et al. Inflammatory Type 2 cDCs Acquire Features of cDC1s and Macrophages to Orchestrate Immunity to Respiratory Virus Infection. Immunity 2020, 52, 1039–1056.e9. [Google Scholar] [CrossRef] [PubMed]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Amigorena, S. Chapter One—Cross-Presentation in Mouse and Human Dendritic Cells. In Advances in Immunology; Alt, F.W., Ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 1–31. [Google Scholar]
- Ahrens, S.; Zelenay, S.; Sancho, D.; Hanč, P.; Kjær, S.; Feest, C.; Fletcher, G.; Durkin, C.; Postigo, A.; Skehel, M.; et al. F-Actin Is an Evolutionarily Conserved Damage-Associated Molecular Pattern Recognized by DNGR-1, a Receptor for Dead Cells. Immunity 2012, 36, 635–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jongbloed, S.L.; Kassianos, A.; McDonald, K.J.; Clark, G.; Ju, X.; Angel, C.; Chen, C.-J.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachem, A.; Güttler, S.; Hartung, E.; Ebstein, F.; Schaefer, M.; Tannert, A.; Salama, A.; Movassaghi, K.; Opitz, C.; Mages, H.W.; et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J. Exp. Med. 2010, 207, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Crozat, K.; Guiton, R.; Contreras, V.; Feuillet, V.; Dutertre, C.A.; Ventre, E.; Vu Manh, T.P.; Baranek, T.; Storset, A.K.; Marvel, J.; et al. The XC chemokine receptor 1 is a conserved selec-tive marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J. Exp. Med. 2010, 207, 1283–1292. [Google Scholar] [CrossRef]
- Colletti, N.J.; Liu, H.; Gower, A.C.; Alekseyev, Y.O.; Arendt, C.W.; Shaw, M.H. TLR3 Signaling Promotes the Induction of Unique Human BDCA-3 Dendritic Cell Populations. Front. Immunol. 2016, 7, 88. [Google Scholar] [CrossRef] [Green Version]
- Zelenay, S.; Keller, A.M.; Whitney, P.G.; Schraml, B.U.; Deddouche, S.; Rogers, N.C.; Schulz, O.; Sancho, D.; Reis e Sousa, C. The dendritic cell receptor DNGR-1 con-trols endocytic handling of necrotic cell antigens to favor cross-priming of CTLs in virus-infected mice. J. Clin. Investig. 2012, 122, 1615–1627. [Google Scholar] [CrossRef] [Green Version]
- Cytlak, U.; Resteu, A.; Pagan, S.; Green, K.; Milne, P.; Maisuria, S.; McDonald, D.; Hulme, G.; Filby, A.; Carpenter, B.; et al. Differential IRF8 Transcription Factor Requirement Defines Two Pathways of Dendritic Cell Development in Humans. Immunity 2020, 53, 353–370.e8. [Google Scholar] [CrossRef]
- Villani, A.C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef] [Green Version]
- Bourdely, P.; Anselmi, G.; Vaivode, K.; Ramos, R.N.; Missolo-Koussou, Y.; Hidalgo, S.; Tosselo, J.; Nuñez, N.; Richer, W.; Vincent-Salomon, A.; et al. Transcriptional and Functional Analy-sis of CD1c(+) Human Dendritic Cells Identifies a CD163(+) Subset Priming CD8(+)CD103(+) T Cells. Immunity 2020, 53, 335–352.e8. [Google Scholar] [CrossRef]
- Santegoets, S.J.; Duurland, C.L.; Jordanova, E.J.; van Ham, V.J.; Ehsan, I.; Loof, N.M.; Narang, V.; Dutertre, C.A.; Ginhoux, F.; van Egmond, S.L.; et al. CD163(+) cytokine-producing cDC2 stim-ulate intratumoral type 1 T cell responses in HPV16-induced oropharyngeal cancer. J. Immunother. Cancer 2020, 8, e001053. [Google Scholar] [CrossRef] [PubMed]
- See, P.; Dutertre, C.-A.; Chen, J.; Günther, P.; McGovern, N.; Irac, S.E.; Gunawan, M.; Beyer, M.; Händler, K.; Duan, K.; et al. Mapping the human DC lineage through the integration of high-dimensional techniques. Science 2017, 356, eaag3009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Affandi, A.J.; Grabowska, J.; Olesek, K.; Lopez Venegas, M.; Barbaria, A.; Rodríguez, E.; Mulder, P.P.G.; Pijffers, H.J.; Ambrosini, M.; Kalay, H.; et al. Selective tumor antigen vaccine de-livery to human CD169(+) antigen-presenting cells using ganglioside-liposomes. Proc. Natl. Acad. Sci. USA 2020, 117, 27528–27539. [Google Scholar] [CrossRef]
- Tacken, P.J.; De Vries, I.J.M.; Torensma, R.; Figdor, C. Dendritic-cell immunotherapy: From ex vivo loading to in vivo targeting. Nat. Rev. Immunol. 2007, 7, 790–802. [Google Scholar] [CrossRef]
- Stolk, D.A.; de Haas, A.; Vree, J.; Duinkerken, S.; Lübbers, J.; Van De Ven, R.; Ambrosini, M.; Kalay, H.; Bruijns, S.; Van Der Vliet, H.J.; et al. Lipo-Based Vaccines as an Approach to Target Dendritic Cells for Induction of T- and iNKT Cell Responses. Front. Immunol. 2020, 11, 990. [Google Scholar] [CrossRef] [PubMed]
- Wamhoff, E.-C.; Schulze, J.; Bellmann, L.; Rentzsch, M.; Bachem, G.; Fuchsberger, F.F.; Rademacher, J.; Hermann, M.; Del Frari, B.; van Dalen, R.; et al. A Specific, Glycomimetic Langerin Lig-and for Human Langerhans Cell Targeting. ACS Cent. Sci. 2019, 5, 808–820. [Google Scholar] [PubMed] [Green Version]
- Schulze, J.; Rentzsch, M.; Kim, D.; Bellmann, L.; Stoitzner, P.; Rademacher, C. A Liposomal Platform for Delivery of a Protein Antigen to Langerin-Expressing Cells. Biochemistry 2019, 58, 2576–2580. [Google Scholar] [CrossRef] [PubMed]
- Klauber, T.C.; Laursen, J.M.; Zucker, D.; Brix, S.; Jensen, S.S.; Andresen, T.L. Delivery of TLR7 agonist to monocytes and dendritic cells by DCIR targeted liposomes induces robust production of anti-cancer cytokines. Acta Biomater. 2017, 53, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueda, F.; Eich, C.; Cordobilla, B.; Domingo, P.; Acosta, G.; Albericio, F.; Cruz, L.J.; Domingo, J.C. Effect of TLR ligands co-encapsulated with multiepitopic antigen in nanoliposomes targeted to human DCs via Fc receptor for cancer vaccines. Immunobiology 2017, 222, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Masterman, K.A.; Haigh, O.L.; Tullett, K.M.; Leal-Rojas, I.M.; Walpole, C.; Pearson, F.E.; Cebon, J.; Schmidt, C.; O’Brien, L.; Rosendahl, N.; et al. Human CLEC9A antibodies deliver NY-ESO-1 antigen to CD141(+) dendritic cells to activate naïve and memory NY-ESO-1-specific CD8(+) T cells. J. Immunother. Cancer 2020, 8, e000691. [Google Scholar] [CrossRef] [PubMed]
- Pearson, F.E.; Tullett, K.M.; Leal-Rojas, I.M.; Haigh, O.L.; Masterman, K.-A.; Walpole, C.; Bridgeman, J.S.; McLaren, J.E.; Ladell, K.; Miners, K.; et al. Human CLEC9A antibodies deliver Wilms’ tumor 1 (WT1) antigen to CD141 + dendritic cells to activate naïve and memory WT1-specific CD8 + T cells. Clin. Transl. Immunol. 2020, 9, e1141. [Google Scholar] [CrossRef]
- Ghinnagow, R.; De Meester, J.; Cruz, L.J.; Aspord, C.; Corgnac, S.; Macho-Fernandez, E.; Soulard, D.; Fontaine, J.; Chaperot, L.; Charles, J.; et al. Co-delivery of the NKT agonist α-galactosylceramide and tumor antigens to cross-priming dendritic cells breaks tolerance to self-antigens and promotes antitumor responses. OncoImmunology 2017, 6, e1339855. [Google Scholar] [CrossRef]
- Mebius, R.E.; Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef]
- Akiyama, H.; Miller, C.; Patel, H.V.; Hatch, S.C.; Archer, J.; Ramirez, N.-G.P.; Gummuluru, S. Virus particle release from glycosphin-golipid-enriched microdomains is essential for dendritic cell-mediated capture and transfer of HIV-1 and henipavirus. J. Virol. 2014, 88, 8813–8825. [Google Scholar] [CrossRef] [Green Version]
- Puryear, W.B.; Yu, X.; Ramirez, N.P.; Reinhard, B.M.; Gummuluru, S. HIV-1 incorporation of host-cell-derived glycosphingolipid GM3 allows for capture by mature dendritic cells. Proc. Natl. Acad. Sci. USA 2012, 109, 7475–7480. [Google Scholar] [CrossRef] [Green Version]
- Puryear, W.B.; Akiyama, H.; Geer, S.D.; Ramirez, N.P.; Yu, X.; Reinhard, B.M.; Gummuluru, S. Interferon-Inducible Mechanism of Dendritic Cell-Mediated HIV-1 Dissemination Is Dependent on Siglec-1/CD169. PLOS Pathog. 2013, 9, e1003291. [Google Scholar] [CrossRef] [Green Version]
- Saunderson, S.C.; Dunn, A.C.; Crocker, P.; McLellan, A.D. CD169 mediates the capture of exosomes in spleen and lymph node. Blood 2014, 123, 208–216. [Google Scholar] [CrossRef]
- Crocker, P.R.; Paulson, J.C.; Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007, 7, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Macauley, M.; Crocker, P.R.; Paulson, J.C. Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol. 2014, 14, 653–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabowska, J.; Lopez-Venegas, M.A.; Affandi, A.J.; Haan, J.M.M.D. CD169+ Macrophages Capture and Dendritic Cells Instruct: The Interplay of the Gatekeeper and the General of the Immune System. Front. Immunol. 2018, 9, 2472. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Bandara, A.; Akiyama, H.; Eshaghi, B.; Stelter, D.; Keyes, T.; Straub, J.E.; Gummuluru, S.; Reinhard, B.M. Membrane-wrapped nanoparticles probe divergent roles of GM3 and phosphatidylserine in lipid-mediated viral entry pathways. Proc. Natl. Acad. Sci. USA 2018, 115, E9041–E9050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Feizpour, A.; Ramirez, N.-G.P.; Wu, L.; Akiyama, H.; Xu, F.; Gummuluru, S.; Reinhard, B.M. Glycosphingolipid-functionalized nanoparticles recapitu-late CD169-dependent HIV-1 uptake and trafficking in dendritic cells. Nat. Commun. 2014, 5, 4136. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Xu, F.; Ramirez, N.-G.P.; Kijewski, S.D.G.; Akiyama, H.; Gummuluru, S.; Reinhard, B.M. Dressing up Nanoparticles: A Membrane Wrap to Induce Formation of the Virological Synapse. ACS Nano 2015, 9, 4182–4192. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.C.; Kawasaki, N.; Nycholat, C.M.; Han, S.; Pilotte, J.; Crocker, P.R.; Paulson, J.C. Antigen Delivery to Macrophages Using Liposomal Nanoparticles Targeting Sialoadhesin/CD169. PLoS ONE 2012, 7, e39039. [Google Scholar] [CrossRef]
- Leylek, R.; Alcántara-Hernández, M.; Lanzar, Z.; Lüdtke, A.; Perez, O.A.; Reizis, B.; Idoyaga, J. Integrated Cross-Species Analysis Identi-fies a Conserved Transitional Dendritic Cell Population. Cell Rep. 2019, 29, 3736–3750.e8. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nijen Twilhaar, M.K.; Czentner, L.; van Nostrum, C.F.; Storm, G.; den Haan, J.M.M. Mimicking Pathogens to Augment the Potency of Liposomal Cancer Vaccines. Pharmaceutics 2021, 13, 954. https://doi.org/10.3390/pharmaceutics13070954
Nijen Twilhaar MK, Czentner L, van Nostrum CF, Storm G, den Haan JMM. Mimicking Pathogens to Augment the Potency of Liposomal Cancer Vaccines. Pharmaceutics. 2021; 13(7):954. https://doi.org/10.3390/pharmaceutics13070954
Chicago/Turabian StyleNijen Twilhaar, Maarten K., Lucas Czentner, Cornelus F. van Nostrum, Gert Storm, and Joke M. M. den Haan. 2021. "Mimicking Pathogens to Augment the Potency of Liposomal Cancer Vaccines" Pharmaceutics 13, no. 7: 954. https://doi.org/10.3390/pharmaceutics13070954
APA StyleNijen Twilhaar, M. K., Czentner, L., van Nostrum, C. F., Storm, G., & den Haan, J. M. M. (2021). Mimicking Pathogens to Augment the Potency of Liposomal Cancer Vaccines. Pharmaceutics, 13(7), 954. https://doi.org/10.3390/pharmaceutics13070954