
Emulgel Loaded with Flaxseed Extracts as New Therapeutic Approach in Wound Treatment

 , ,
, ,  ,
,  ,
,  , , ,
, , ,  , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Chemical Part
2.1.2. Microbiological Part
- -
- Agar-well diffusion test medium; deionized water (containing agar 13%), meat extract (3%), sodium chloride (10%), glucose (4%), dibasic potassium phosphate (1%) and meat peptone (5%); after preparation, the test medium was autoclaved.
- -
- Brain Heart Infusion (BHI) Broth; deionized water, BHI (3.7%, Biolife Italiana Srl, Milano, Italy).
- -
- Mueller Hinton Broth with 5% Blood; deionized water, Mueller Hinton Broth (2.2%, Biolife Italiana Srl, Italy), Horse Lysate Blood (5%, Allevamenti Blood di Fiastra Maddalena).
- -
- 5% Sheep Blood Agar; deionized water, Columbia Agar Base (4.4%, Microbiol Srl, Macchiareddu, Cagliari, Italy), Defibrinated Sheep Blood (5%, Allevamenti Blood di Fiastra Maddalena). Bacterial suspension at concentrations of 1 × 105 CFU/mL was used for the antimicrobial test.
2.1.3. Biochemical Part
2.2. Methods
2.2.1. Extraction Procedure
2.3. Extracts Characterization (D.E. and L.E.)
2.3.1. Chemical Analysis
2.3.2. Total Phenol Content and Antioxidant Activity
2.3.3. Antimicrobial Activity Assay
2.3.4. Cell Culture and Viability
2.3.5. Anti-Inflammatory Activity
2.4. Emulgel Preparation and Characterization
- -
- oil phase (O): L.E. 22.0 g, cetostearyl alcohol 6.0 g, cetomacrogol 1000 2.0 g
- -
- water phase (W): FG90 1% wt. solution 69.0 g, D.E. 1.0 g,
2.4.1. In Vivo Evaluation of the Formulation Skin-Feel
2.4.2. Viscosity Measurement
2.4.3. Scanning Electron Microscopy
3. Results
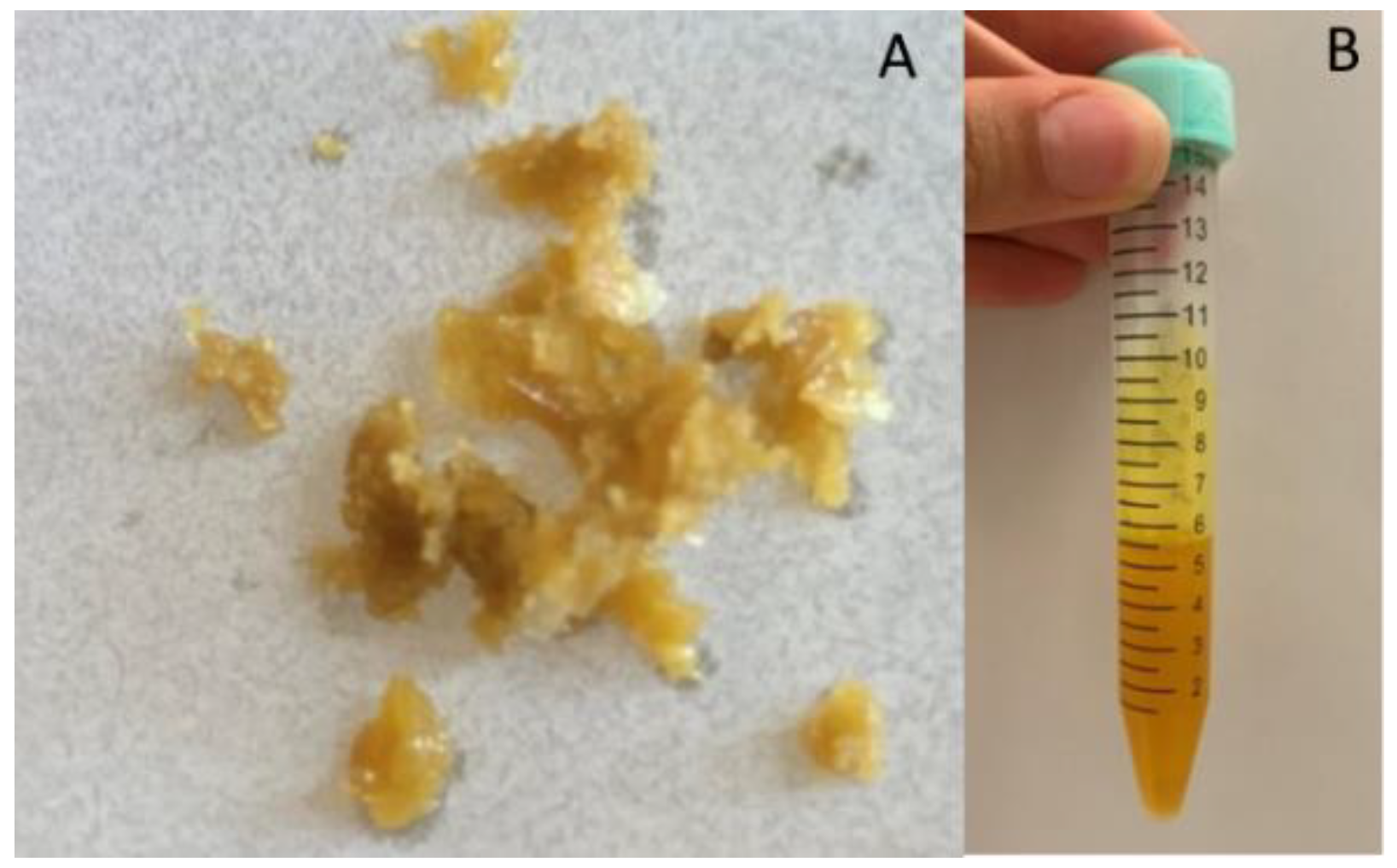
3.1. Dry and Liquid Extracts Preparation
3.2. Extracts Characterization
3.2.1. Chemical Analysis
3.2.2. Total Phenolic Content and Antioxidant Activity
3.2.3. Antimicrobial Activity
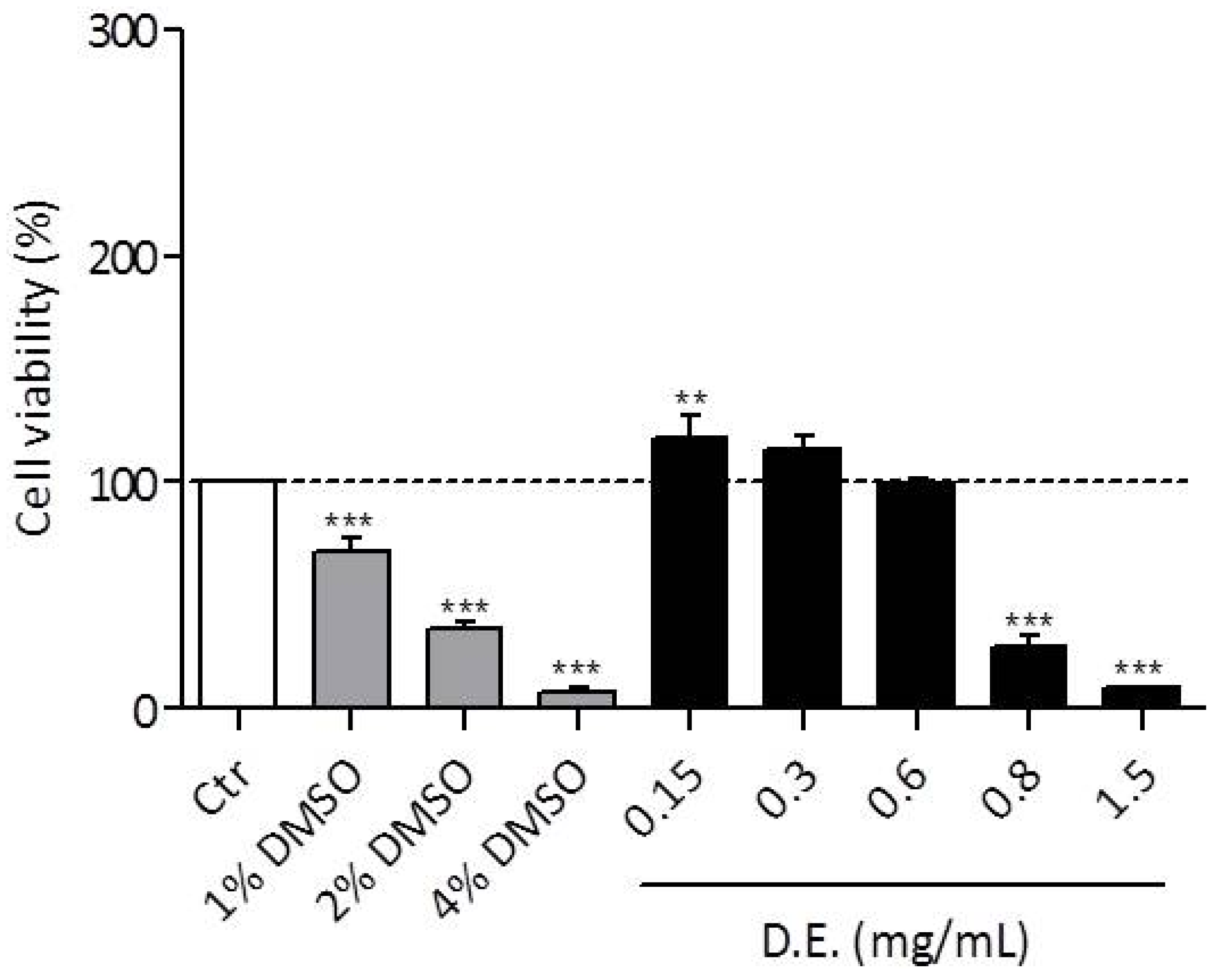
3.2.4. Cytotoxic and Anti-Inflammatory Activity of D.E. in LPS-Stimulated RAW 264.7 Cell Line
3.2.5. In Vitro Cytotoxic Effect on Keratinocytes
3.3. Formulation of D.E. and L.E.
- -
- Oil phase (O): vaseline 15 g, liquid paraffin 6 g, cetostearyl alcohol 7.2 g, cetomacrogol 1.8 g;
- -
- Water phase (W): water 70 g.
- -
- Oil phase (O): L.E. 22 g, cetostearyl alcohol 6.0 g, cetomacrogol 2.0 g.
- -
- Water phase (W): water 69 g, D.E. 1.0 g.
- -
- oil phase (O): L.E. 22.0 g, cetostearyl alcohol 6.0 g, cetomacrogol 1000 2.0 g
- -
- water phase (W): FG90 1% wt. solution 69.0 g, D.E. 1.0 g.
3.4. Emulgel Characterization
3.4.1. Organoleptic Properties and Stability
3.4.2. Droplet Size Measurement
3.4.3. Antimicrobial Activity
3.4.4. Rheological Characterization
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jhala, A.J.; Hall, L.M. Flax (Linum usitatissimum L.): Current Uses and Future Applications. Aust. J. Basic Appl. Sci. 2010, 4, 4304–4312. [Google Scholar]
- Parikh, M.; Netticadan, T.; Pierce, G.N. Flaxseed: Its bioactive components and their cardiovascular benefits. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Shim, Y.Y.; Gui, B.; Arnison, P.G.; Wang, Y.; Reaney, M.J.T. Flaxseed (Linum usitatissimum L.) bioactive compounds and peptide nomenclature: A review. Trends Food Sci. Technol. 2014, 38, 5–20. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, J.; Qiu, C.; Ye, Y.; Guo, X.; Chen, G.; Li, T.; Wang, Y.; Fu, X.; Liu, R.H. Comparison of phytochemical profiles and health benefits in fiber and oil flaxseeds (Linum usitatissimum L.). Food Chem. 2017, 214, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Kitts, D.D.; Yuan, Y.V.; Wijewickreme, A.N.; Thompson, L.U. Antioxidant activity of the flaxseed lignin secoisolariciresinoldiglycoside and its mammalian lignan metabolites enterodiol and enterolactone. Mol. Cell. Biochem. 1999, 202, 91–100. [Google Scholar] [CrossRef]
- Marand, M.A.; Amjadi, M.S.; Marand, M.A.; Roufegarinejad, L.; Mahdi Jafari, S. Fortification of yogurt with flaxseed powder and evaluation of its fatty acid profile, physicochemical, antioxidant, and sensory properties. Powder Technol. 2020, 359, 76–84. [Google Scholar] [CrossRef]
- Kyselka, J.; Rabiej, D.; Dragoun, M.; Kreps, F.; Burčová, Z.; Němečková, I.; Smolová, J.; Bjelková, M.; Szydłowska-Czerniak, A.; Schmidt, Š.; et al. Antioxidant and antimicrobial activity of linseed lignans and phenolic acids. Eur. Food Res. Technol. 2017, 243, 1633–1644. [Google Scholar] [CrossRef]
- Kaithwas, G.; Mukerjee, A.; Kumar, P.; Majumdar, D.K. Linum usitatissimum (linseed/flaxseed) fixed oil: Antimicrobial activity and efficacy in bovine mastitis. Inflammopharmacology 2011, 19, 45–52. [Google Scholar] [CrossRef]
- Kajla, P.; Sharma, A.; Sood, D.R. Flaxseed, a potential functional food source. Int. J. Food Sci. Technol. 2015, 52, 1857–1871. [Google Scholar] [CrossRef]
- Rafiee, S.; Nekouyian, N.; Hosseini, S.; Sarabandi, F.; Chavoshi-Nejad, M.; Mohsenikia, M.; Yadollah-Damavandi, S.; Seifaee, A.; Jangholi, E.; Eghtedari, D.; et al. Effect of Topical Linum usitatissimum on Full Thickness Excisional Skin Wounds. Trauma Mon. 2017, 22, e39045. [Google Scholar] [CrossRef] [Green Version]
- Pagano, C.; Marinozzi, M.; Baiocchi, C.; Beccari, T.; Calarco, P.; Ceccarini, M.R.; Chielli, M.; Orabona, C.; Orecchini, E.; Ortenzi, R.; et al. Bioadhesive Polymeric Films Based on Red Onion Skins Extract for Wound Treatment: An Innovative and Eco-Friendly Formulation. Molecules 2020, 25, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagano, C.; Perioli, L.; Baiocchi, C.; Bartoccini, A.; Beccari, T.; Blasi, F.; Calarco, P.; Ceccarini, M.R.; Cossignani, L.; di Michele, A.; et al. Preparation and characterization of polymeric microparticles loaded with Moringa oleifera leaf extract for exuding wound treatment. Int. J. Pharm. 2020, 587, 119700. [Google Scholar] [CrossRef] [PubMed]
- Pagano, C.; Perioli, L.; Blasi, F.; Bastianini, M.; Chiesi, C.; Cossignani, L. Optimisation of phenol extraction from wine using layered double hydroxides and technological evaluation of the bioactive-rich powder. Int. J. Food Sci. Technol. 2017, 52, 2582–2588. [Google Scholar] [CrossRef]
- Pollini, L.; Tringaniello, C.; Ianni, F.; Blasi, F.; Manes, J.; Cossignani, L. Impact of ultrasound extraction parameters on the antioxidant properties of Moringa oleifera leaves. Antioxidants 2020, 9, 277. [Google Scholar] [CrossRef]
- Pollini, L.; Rocchi, R.; Cossignani, L.; Mañes, J.; Compagnone, D.; Blasi, F. Phenol profiling and nutraceutical potential of Lycium spp. leaf extracts obtained with ultrasound and microwave assisted techniques. Antioxidants 2019, 8, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balouiri, M.; Sadiki, M.; Ibnsouda, S.K. Methods for in vitro evaluating antimicrobial activity: A review. J. Pharm Anal. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Ceccarini, M.R.; Vannini, S.; Cataldi, S.; Moretti, M.; Villarini, M.; Fioretti, B.; Albi, E.; Beccari, T.; Codini, M. In Vitro Protective Effects of Lycium barbarum Berries Cultivated in Umbria (Italy) on Human Hepatocellular Carcinoma Cells. BioMed Res. Int. 2016, 2016, 7529521. [Google Scholar]
- Pagano, C.; Perioli, L.; Latterini, L.; Nocchetti, M.; Ceccarini, M.R.; Marani, M.; Ramella, D.; Ricci, M. Folic acid-layered double hydroxides hybrids in skin formulations: Technological, photochemical and in vitro cytotoxicity on human keratinocytes and fibroblasts. Appl. Clay Sci. 2019, 168, 382–395. [Google Scholar] [CrossRef]
- Teh, S.S.; Bekhit, A.E.D.; Birch, J. Antioxidative polyphenols from defatted oilseed cakes: Effect of solvents. Antioxidants 2014, 3, 67–80. [Google Scholar] [CrossRef]
- Deng, Q.; Yu, X.; Ma, F.; Xu, J.; Huang, F.; Huang, Q.; Sheng, F. Comparative analysis of the in-vitro antioxidant activity and bioactive compounds of flaxseed in China according to variety and geographical origin. Int. J. Food Prop. 2017, 20, S2708–S2722. [Google Scholar] [CrossRef] [Green Version]
- Tawaha, K.; Alali, F.; Gharaibeh, M.; Mohammad, M.; El-Elimat, T. Antioxidant activity and total phenolic content of selected Jordanian species. Food Chem. 2007, 104, 1372–1378. [Google Scholar] [CrossRef]
- Hoel, T.; Casals, J.B.; Eng, J. In vitro antimicrobial susceptibility testing of rapidly growing mycobacteria using the tablet diffusion method: Resistance pattern of Norwegian Mycobacterium fortuitum and Mycobacterium chelonae isolates. APMIS 1993, 101, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Istituto Superiore di Sanità. Macrogol Cetostearile Etere Crema e Unguento Base; Farmacopea Ufficiale Italiana XII Ed; Ist. Poligrafico dello Stato: Rome, Italy, 2008; p. 1203. [Google Scholar]
- Tin, S.; Sakharkar, K.R.; Lim, C.S.; Sakharkar, M.K. Activity of Chitosans in combination with antibiotics in Pseudomonas aeruginosa. Int. J. Biol. Sci. 2009, 5, 153160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asli, A.; Brouillette, E.; Ster, C.; Ghinet, M.G.; Brzezinski, R.; Lacasse, P.; Jacques, M.; Malouin, F. Antibiofilm and antibacterial effects of specific chitosan molecules on Staphylococcus aureus isolates associated with bovine mastitis. PLoS ONE 2017, 12, e0176988. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Ahmed, S. A review on chitosan and its nanocomposites in drug delivery. Int. J. Biol. Macromol. 2018, 109, 273–286. [Google Scholar] [CrossRef]
- Perioli, L.; Ambrogi, V.; Pagano, C.; Scuota, S.; Rossi, C. FG90 chitosan as a new polymer for metronidazole mucoadhesive tablets for vaginal administration. Int. J. Pharm. 2009, 377, 120–127. [Google Scholar] [CrossRef]
- Filimon, M.N.; Popescu, R.; Sinitean, A.; Dumitrescu, G. The Assessment of Chitosan Solutions Effects on Bacterial Strains. Rev. Chim. 2018, 69, 1485–1488. [Google Scholar] [CrossRef]
- Matica, M.A.; Aachmann, F.L.; Tøndervik, A.; Sletta, H.; Ostafe, V. Chitosan as a Wound Dressing Starting Material: Antimicrobial Properties and Mode of Action. Int. J. Mol. Sci. 2019, 20, 5889. [Google Scholar] [CrossRef] [Green Version]
- Goy, R.C.; de Britto, D.; Assis, O.B.G. A review of the antimicrobial activity of chitosan. Polimeros 2009, 19, 241–247. [Google Scholar] [CrossRef]
- Friedman, A.J.; Phan, J.; Schairer, D.O.; Champer, J.; Qin, M.; Pirouz, A.; Blecher-Paz, K.; Oren, A.; Liu, P.T.; Modlin, R.L.; et al. Antimicrobial and Anti-Inflammatory Activity of Chitosan–Alginate Nanoparticles: A Targeted Therapy for Cutaneous Pathogens. J. Investig. Dermatol. 2013, 133, 1231–1239. [Google Scholar] [CrossRef] [Green Version]
- No Time to Wait: Securing the Future from Drug-Resistant Infections; Report to the secretary general of the United Nations. 2019. Available online: https://www.who.int/antimicrobial-resistance/interagency-coordination-group/final-report/en (accessed on 15 July 2021).
- WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed. Report OMS February 2017. Available online: https://www.who.int/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 15 July 2021).

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Growth Conditions |
|---|---|
| Gram + bacteria | |
| Staphylococcus epidermidis WDCM 00036 | 37 °C for 24 ± 2 h |
| Enterococcus faecalis WDCM 00087 | 37 °C for 24 ± 2 h |
| Bacillus subtilis WDCM 00003 | 30 °C for 24 ± 2 h |
| Staphylococcus aureus WDCM 00034 | 37 °C for 24 ± 2 h |
| Streptococcus pyogenes ATCC 19615 | 37 °C for 24–48 h |
| Gram − bacteria | |
| Pseudomonas aeruginosa WDCM 00025 | 25 °C for 24–48 h |
| Klebsiella pneumoniae WDCM 00097 | 37 °C for 24 ± 2 h |
| Proteus mirabilis WDCM 00023 | 37 °C for 24 ± 2 h |
| Escherichia coli WDCM 00013 | 37 °C for 24 ± 2 h |
| Yeast | |
| Candida CM 00054 albicans WD | 25 °C for 24–72 h |
| Extract | TPC (Mean ± SD) mg GAE/g Dry Flaxseed Flour | FRAP (Mean ± SD) µmol Fe2+/g Dry Flaxseed Flour | ABTS (Mean ± SD) mg TE/g Dry Flaxseed Flour |
|---|---|---|---|
| D.E. | 1.94 ± 0.09 | 15.73 ± 3.10 | 5.25 ± 0.35 |
| L.E. | 1.62 ± 0.01 | 11.69 ± 0.21 | 0.62 ± 0.04 |
| Strains | D.E. 100 mg/mL (mm) | D.E. 150 mg/mL (mm) | Marketed D.E. 100 mg/mL (mm) | Marketed D.E. 150 mg/mL (mm) | L.E. (mm) | Marketed Flaxseed Oil (mm) |
|---|---|---|---|---|---|---|
| Gram + | ||||||
| S. epidermidis | - | - | - | 16 | - | - |
| E. faecalis | - | - | - | - | - | - |
| B. subtilis | - | - | - | - | - | - |
| S. aureus | - | 18 | - | 15 | - | - |
| S. pyogenes | 20 | 20 | 19 | 22 | 20 | - |
| Gram − | ||||||
| P. aeruginosa | - | - | - | - | - | - |
| K. pneumoniae | - | - | - | - | - | - |
| P. mirabilis | - | - | - | - | - | - |
| E. coli | - | - | - | - | - | - |
| Yeast | ||||||
| C. albicans | - | - | - | - | - | - |
| Strain | D.E. (150 mg/mL) | L.E. (0.87 mg/mL) | FG90 (1% wt.) | Emulgel | Base Cream (Control) |
|---|---|---|---|---|---|
| S. aureus WDCM 00034 | 18 | - | - | 24 | - |
| S. pyogenes ATCC 19615 | 20 | 20 | 25 | 36 | - |
| P. aeruginosa WDCM 00025 | - | - | 20 | 31 | - |
| K. pneumonia WDCM 00097 | - | - | 23 | 27 | - |
| E. coli WDCM 00013 | - | - | 20 | 26 | - |
| Sample | MIC (mg/mL) | MBC (mg/mL) |
|---|---|---|
| Ciprofloxacin (control) | 1 μg/mL | 1 μg/mL |
| D.E. | 0.59 | 1.17 |
| L.E. | 0.22 | 0.44 |
| FG90 | 0.30 | 0.30 |
| Emulgel | 5.20 | 5.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagano, C.; Baiocchi, C.; Beccari, T.; Blasi, F.; Cossignani, L.; Ceccarini, M.R.; Orabona, C.; Orecchini, E.; Di Raimo, E.; Primavilla, S.; et al. Emulgel Loaded with Flaxseed Extracts as New Therapeutic Approach in Wound Treatment. Pharmaceutics 2021, 13, 1107. https://doi.org/10.3390/pharmaceutics13081107
Pagano C, Baiocchi C, Beccari T, Blasi F, Cossignani L, Ceccarini MR, Orabona C, Orecchini E, Di Raimo E, Primavilla S, et al. Emulgel Loaded with Flaxseed Extracts as New Therapeutic Approach in Wound Treatment. Pharmaceutics. 2021; 13(8):1107. https://doi.org/10.3390/pharmaceutics13081107
Chicago/Turabian StylePagano, Cinzia, Claudio Baiocchi, Tommaso Beccari, Francesca Blasi, Lina Cossignani, Maria Rachele Ceccarini, Ciriana Orabona, Elena Orecchini, Enrico Di Raimo, Sara Primavilla, and et al. 2021. "Emulgel Loaded with Flaxseed Extracts as New Therapeutic Approach in Wound Treatment" Pharmaceutics 13, no. 8: 1107. https://doi.org/10.3390/pharmaceutics13081107
APA StylePagano, C., Baiocchi, C., Beccari, T., Blasi, F., Cossignani, L., Ceccarini, M. R., Orabona, C., Orecchini, E., Di Raimo, E., Primavilla, S., Salvini, L., Michele, A. D., Perioli, L., & Ricci, M. (2021). Emulgel Loaded with Flaxseed Extracts as New Therapeutic Approach in Wound Treatment. Pharmaceutics, 13(8), 1107. https://doi.org/10.3390/pharmaceutics13081107